Патент на изобретение №2315762
|
||||||||||||||||||||||||||
(54) СПОСОБ СИНТЕЗА ХИРАЛЬНЫХ N-АРИЛПИПЕРАЗИНОВ
(57) Реферат:
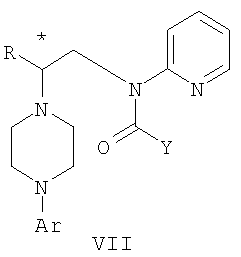
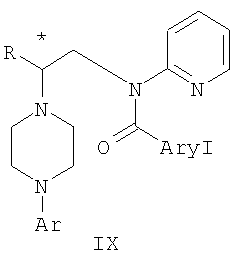
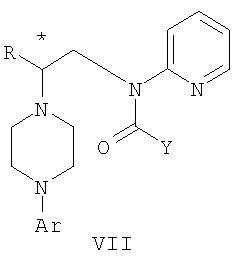
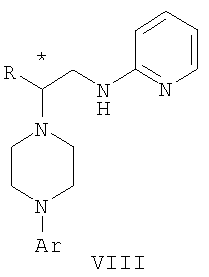
Изобретение относится к новому способу получения соединения формулы VII, формулы VIII, формулы IX, которые действуют на 5НТ рецепторы центральной нервной системы.
где R представляет собой C1-С3 алкил, Y представляет собой фрагмент, выбранный из группы, включающей C1-С6 алкокси, C1-С6 алкил, и Ar представляет собой 2,3-дигидробензодиоксин-5-ил,
где R представляет собой C1-С3 алкил, Ar представляет собой 2,3-дигидробензодиоксин-5-ил, и * указывает хиральный центр,
где R и Ar определены выше, Ary1 представляет собой C6-С12 ароматическую группу, необязательно замещенную и содержащую до трех заместителей, независимо выбранных из группы, включающей атомы галогенов, алкил, алкокси, алкоксикарбонил, нитро, амино, алкиламино, диалкиламино, галогеналкил, дигалогеналкил, тригалогеналкил, нитрил и амидо-заместители, каждый из которых содержит не более шести атомов углерода. Технический результат – практическое осуществление синтеза N-арилпиперазинов. 3 н. и 8 з.п. ф-лы.
Область техники Данное изобретение относится к способам синтеза хиральных замещенных N-арилпиперазинов для получения соединений, которые связываются с 5НТ рецепторами центральной нервной системы, и их промежуточных продуктов. Предпосылки изобретения Некоторые N,N
Возможен также “перевернутый” вариант данного химического уравнения. В таком подходе анилиновый аналог иприта взаимодействует с алкиламином, как показано, например, в публикации J. Labeled Compounds and RadioPharm. (1986) Vol.XXIV, №4, 351. Однако большая коммерческая доступность гидрохлорида бис(2-хлорэтил)амина по сравнению с доступностью анилиновых производных иприта делает данный подход менее привлекательным.
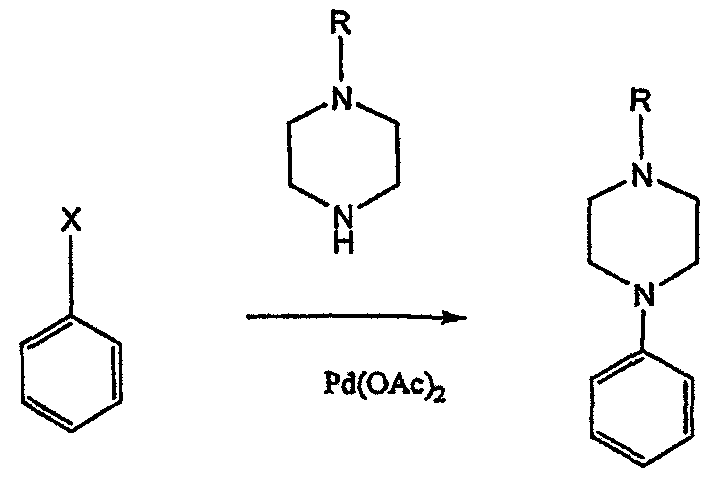
Асимметричные арилпиперазины также получают сочетанием пиперазинов с арилтрифлатами или -бромидами. В публикации Tetrahedron Letters (1998), 39, 2219 указывается, что выходы в данном способе в значительной степени зависят от (арильного) субстрата и обычно являются низкими.
Главным объектом данного изобретения является получение пиперазинов, в которых хиральный центр находится на атоме азота. Некоторые способы получения хиральных N-пиперазинов описаны в литературе. Одним из таких известных способов является разделение рацемической смеси, недостатком которого является то, что половина вещества является отходом процесса. Другим известным способом является замещение арилпиперазином уходящей группы, присоединенной к хиральному центру. Однако одним из недостатков такого способа является то, что замещение уходящей группы пространственно затруднено. Повышение реакционной способности уходящей группы создает другие трудности при осуществлении данного способа: в публикации JP 01125357 указывается, что бензил-(S)-бромпропионат взаимодействует с 1-бензилпиперазином с получением в качестве ожидаемого продукта замещения (R)-изомера. Карбонильная группа при активации процесса замещения повышает также склонность соседнего хирального центра к рацемизации в условиях реакции. В публикации WO 95/33743 описывается альтернативный способ, в котором устранена проблема рацемизации при активации применением хирального циклического сульфамата в качестве реакционно-способного агента алкилирования.
Хотя циклические сульфаматы легко взаимодействуют с пиперазинами, получение самих сульфаматов является многостадийным процессом. Например, когда R представляет собой 2-пиридил, необходимо осуществить четыре стадии химического разделения или превращения. В публикации Acta Pol. Pharm., 56 (1), 41-47; 1999 сообщается, что хиральные аминокислоты взаимодействуют с N-метил-N,N-бис(2-хлорэтаном). Функциональная группа карбоновой кислоты делает хиральный центр склонным к рацемизации как при проведении реакции, так и на последующих стадиях выполнения синтеза. В соответствии с другим подходом в публикации J. Med. Chem. 30 (10), 1779-87; 1987 описывается, что хиральные бензиламины взаимодействуют с различными производными ипритов, как N-алкил-, так и N-арил-производными. Применяемые хиральные амины были получены разделением. В публикации WO94/24115 описывается взаимодействие В настоящее время большинство способов синтеза N-арил-N Краткое описание изобретения Данное изобретение включает способ получения соединения формулы VII
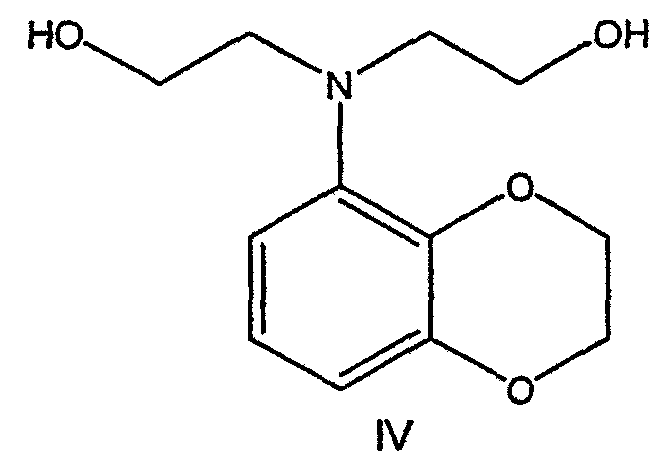
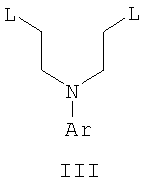
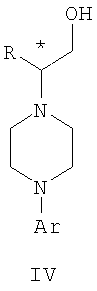
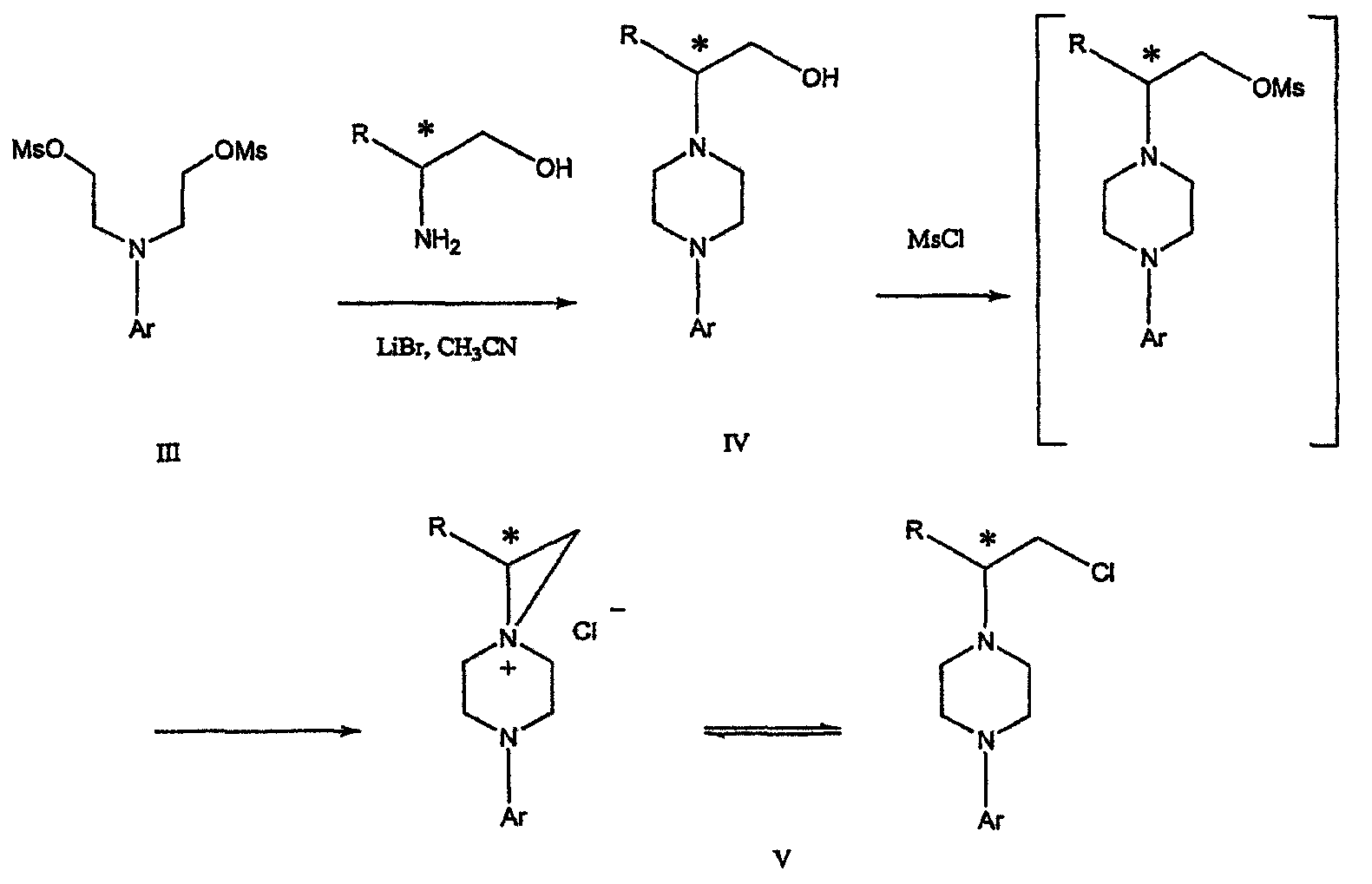
где R представляет собой С1-С3 алкил, Y представляет собой С1-С6 алкокси, С1-С6 алкил, С3-С7 циклоалкил или С3-С7 циклоалкокси, Ar представляет собой 2,3-дигидробензодиоксин-5-ил или фенил, необязательно замещенный и содержащий до трех заместителей, независимо выбранных из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила, указанный способ включает: а) взаимодействие соединения формулы III с хиральным 2-амино-1-(С3-С5)алканолом в полярном апротонном растворителе для получения соединения формулы IV
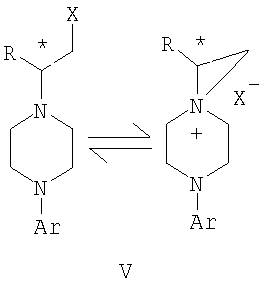
где L представляет собой уходящую группу, выбранную из Cl, Br, мезилата и тозилата, и * указывает хиральный центр; b) превращение соединения формулы IV в соединение формулы V
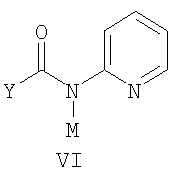
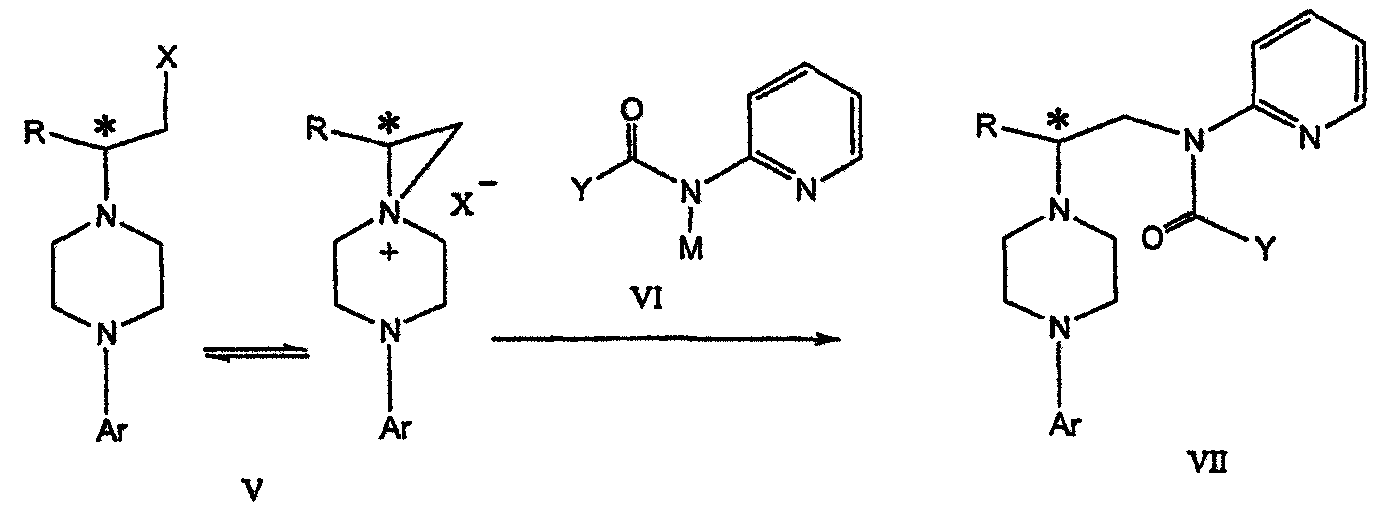
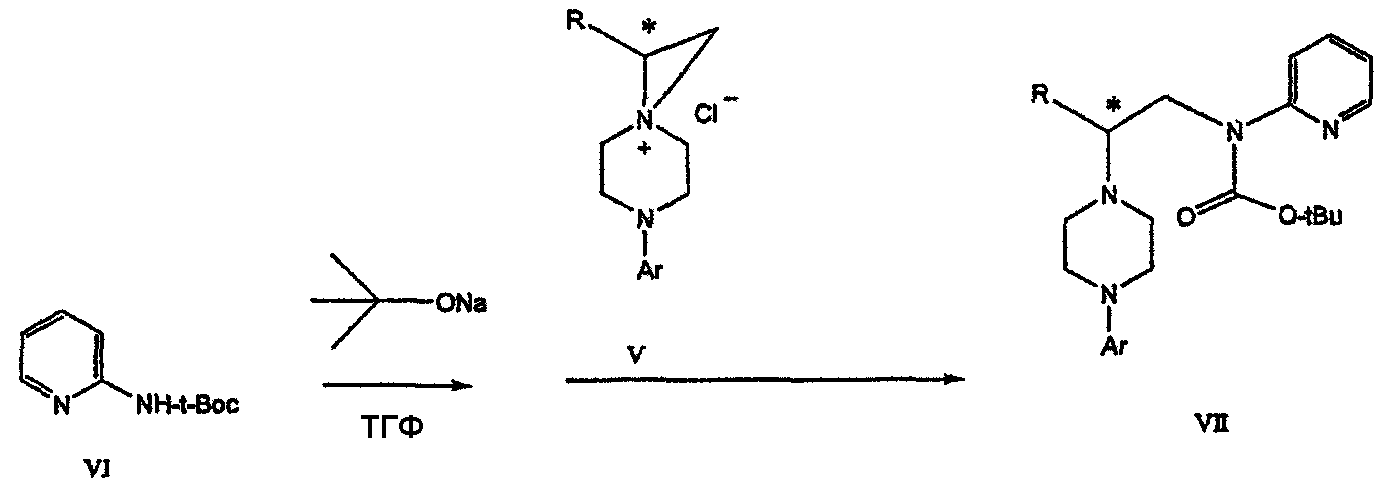
где Х представляет сбой Cl, Br, трифлат, тозилат или мезилат; и с) обработку соединения формулы V соединением формулы VI в апротонном растворителе
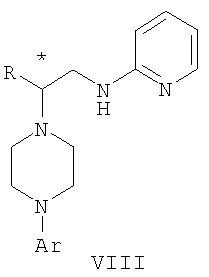
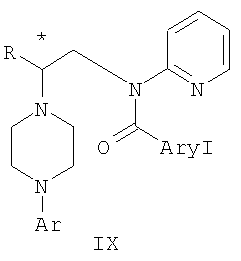
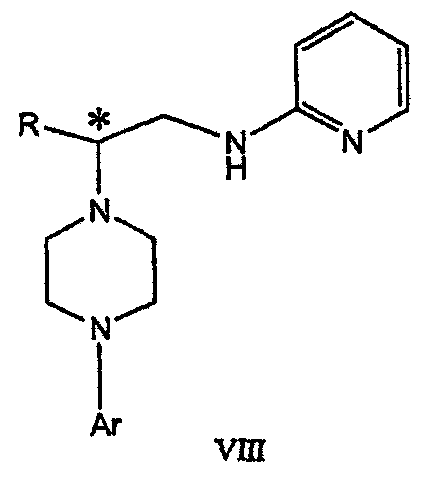
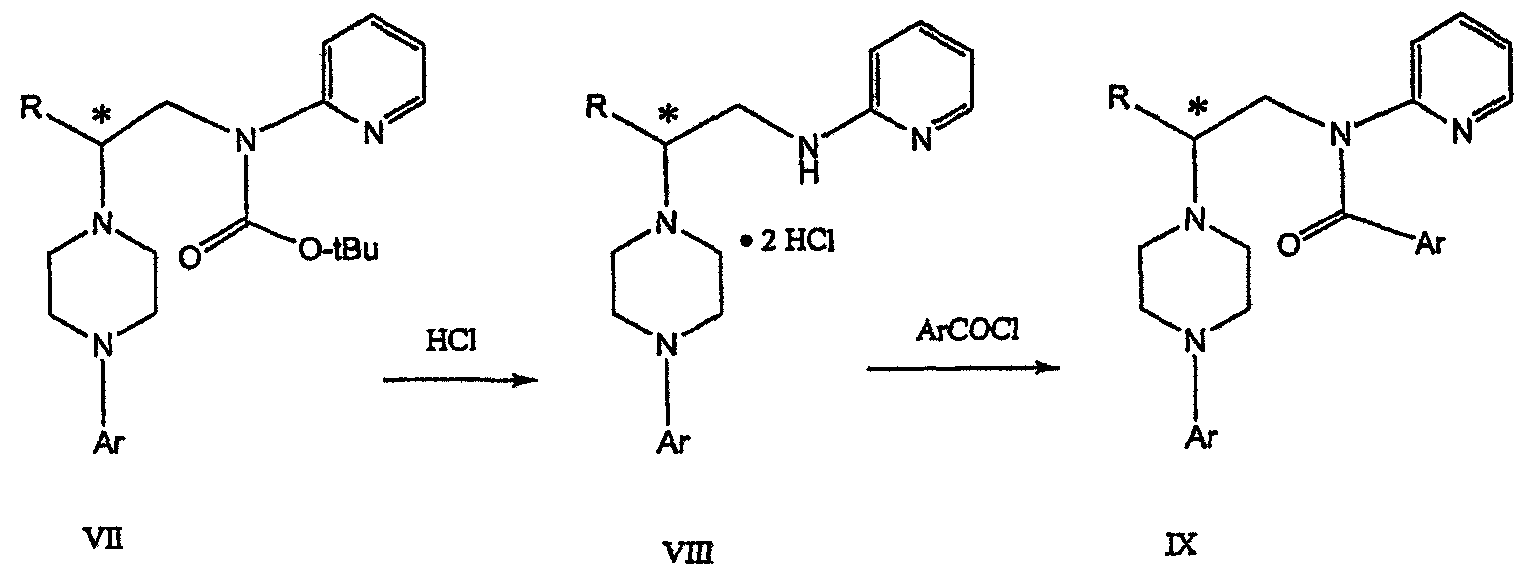
где М представляет собой щелочной металл (например, Na, Li, K), и Y представляет собой фрагмент, выбранный из группы, включающей С1-С6 алкокси, С1-С6 алкил, С3-С7 циклоалкил и С3-С7 циклоалкокси. Данное изобретение включает также способ получения соединения формулы IX, включающий стадии (а), (b) и (c), описанные выше, а также дополнительные стадии: (d) обработку соединения формулы VII протонной кислотой с получением соединения формулы VIII
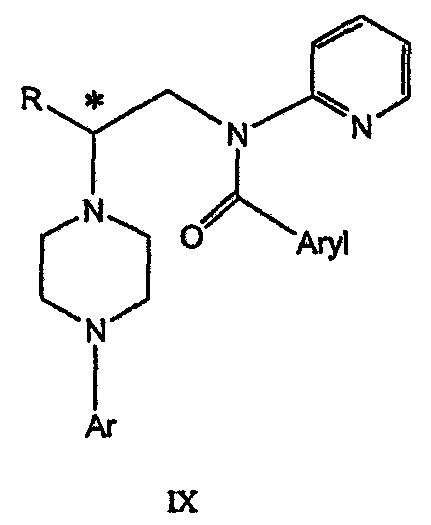
(е) обработку соединения формулы VIII ароильным соединением, выбранным из ароилхлорида, ароилбромида и ароилангидрида, в присутствии основания для получения соединения формулы IX
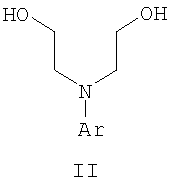
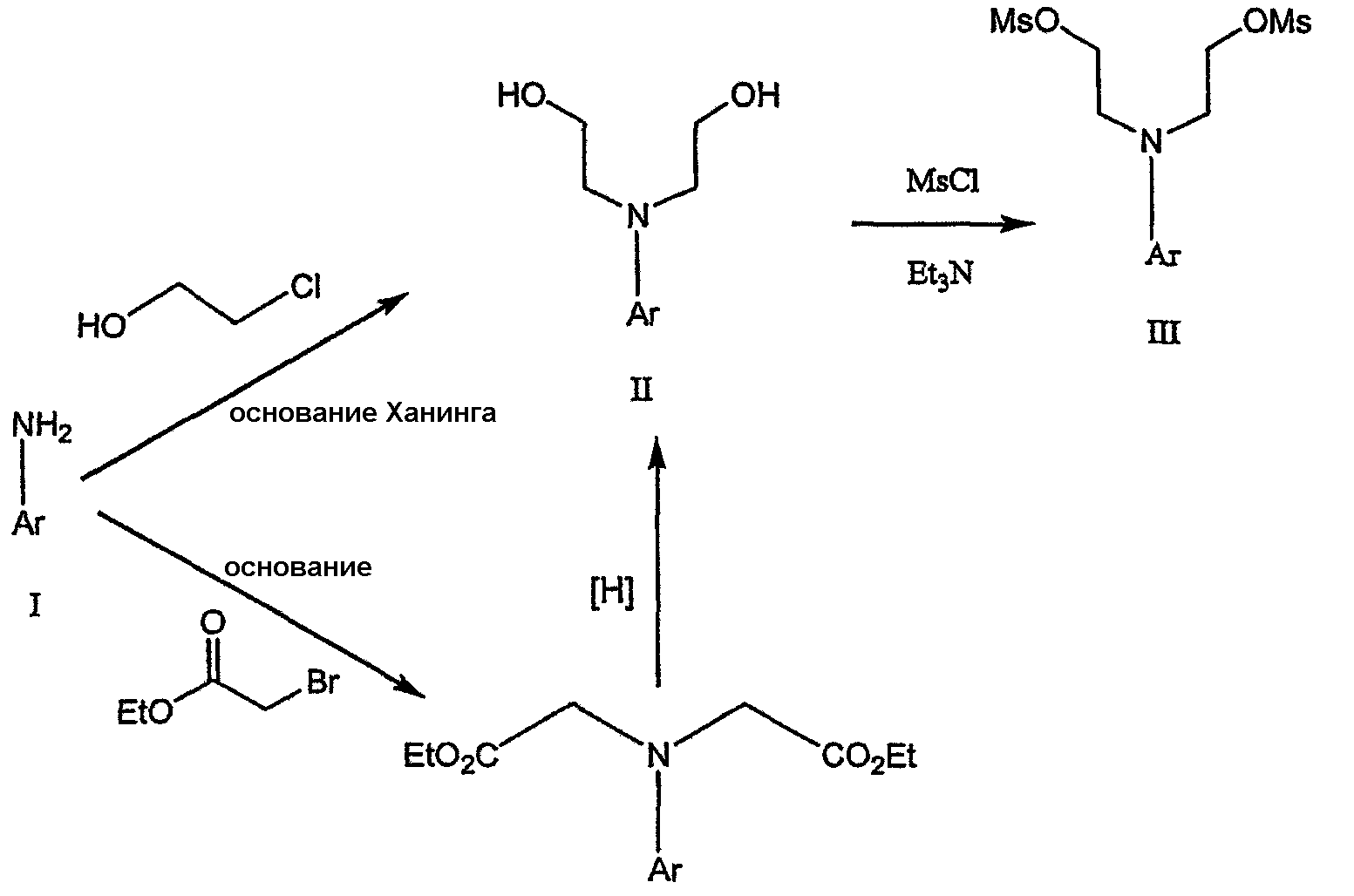
где Ary1 представляет собой С6-С12 ароматическую группу, необязательно замещенную и содержащую до трех заместителей, независимо выбранных из группы, включающей атомы галогенов, алкил, алкокси, алкоксикарбонил, нитро, амино, алкиламино, диалкиламино, галогеналкил, дигалогеналкил, тригалогеналкил, нитрил и амидо-заместители, каждый из которых содержит не более шести атомов углерода. Подробное описание изобретения Предпочтительное воплощение данного изобретения представляет собой способ получения N-арилпиперазинов с хиральными N В предпочтительном воплощении данного изобретения синтез начинается с получения димезилатного производного формулы III сначала диалкилированием анилина формулы I хлорэтанолом для получения диола формулы II. Альтернативно, диол формулы II получают диалкилированием анилина алкилгалогенацетатом с последующим восстановлением. Две гидроксильные группы обычно превращаются в подходящие удаляемые группы, такие как мезилатные удаляемые группы:
Димезилат взаимодействует с хиральным 2-амино-1-пропанолом (аланинол) для получения целевого пиперазина. В других воплощениях данного изобретения хиральное амино-производное представляет собой 2-амино-1-бутанол, 2-амино-1-пентанол или 2-амино-3-метил-1-бутанол. Удаляемые группы, которые могут использоваться при практическом осуществлении данного изобретения, не являются мезилатными группами, и представляют собой тозилат, хлор или бром. Хиральность аминного компонента остается неизменной в данном способе. Спиртовая группа, которая не требует защиты в процессе циклизации, сохраняется для дальнейших структурных преобразований. Полученный первичный спирт затем активируется для замещения, например, обработкой метансульфонилхлоридом или -бромидом. Считается, что данная реакция приводит к получению мезилата, который является переходным промежуточным продуктом и приводит к получению соединения формулы V.
В предпочтительном воплощении данного изобретения соединение формулы V взаимодействует с анионом, полученным из 2-(t-Boc)-аминопиридина, для введения аминофенильной боковой цепи и получения соединения формулы VII.
Данное изобретение включает также применение вместо трет-бутокси-группы других групп; подходящими группами являются С1-С6 алкокси, С1-С6 алкил, С3-С7 циклоалкил и С3-С7 циклоалкокси. Когда такая группа представляет собой одну из упомянутых выше циклических групп, один или несколько атомов углерода могут находиться вне кольца, и указанная группа может представлять собой, например, циклогексилметокси или этилциклопентил. Соединение формулы VII может далее реагировать с получением соединений формул VIII и IX. Предпочтительно, защитная t-Вос-группа удаляется с помощью HCl/EtOH для получения амина формулы VIII в виде HCl соли. Соль может использоваться сразу для введения свободной функциональной NH группы. Хотя в воплощении, представленном ниже, показано ацилирование ароилхлоридами, в практике данного изобретения могут использоваться и другие ацил-производные.
Поскольку в данном синтезе хиральный заместитель вводится на стадии образования пиперазина, все что для этого необходимо – это хиральный амин. Реакция неожиданно очень эффективна даже при наличии близкорасположенной свободной гидроксильной группы (например, III
В дальнейшем гидроксильная группа может использоваться для введения аминопиридильной функциональной группы реакцией замещения. На первый взгляд и из предшествующего уровня сразу трудно оценить, насколько серьезно побочные реакции, описанные выше, могут затруднять такое замещение. Многое зависит от специфического агента алкилирования. В публикации WO 9703982 описано, что аминопиридин VIa в особых условиях может подвергаться взаимодействию с соединениями общей формулы Va, где Х представляет собой удаляемую группу, для получения VIIa. При разработке данного изобретения заявителями было обнаружено, что анион алкилацил-соединений (т.е. VIb) при взаимодействии с V (X = Cl) приводит к получению значительного количества (20%) нежелательного продукта алкилирования азота пиридильного кольца с получением соединения Х. В предпочтительном воплощении данного изобретения Y представляет собой алкокси-группу. Данное изобретение обеспечивает практическое осуществление синтеза N-арилпиперазинов, где хиральный заместитель вводится на стадии образования пиперазинового кольца, а 2-аминопиридильный заместитель вводится с помощью реакции замещения. Применение t-Boc-2-аминопиридина, VI, как описано в данном изобретении, приводит к значительному снижению количества (<7%) побочного продукта и повышению доли желательного соединения VII. Как показано в предыдущем разделе, защитная t-Boc-группа легко удаляется, и после этого свободный амин может подвергаться ацилированию. В данном описании и в прилагаемой формуле изобретения, если не указано другого значения, термин “галоген” относится к F, Cl и Br, термины “алкил”, “алкан”, “алканол” и “алкокси” включают алкильные группы с прямой и разветвленной цепью. Приведенные далее примеры представлены для иллюстрации некоторых воплощений данного изобретения, но не должны рассматриваться как ограничивающие область данного изобретения. Пример 1 2-[(2,3-Дигидробензо[1,4]диоксин-5-ил)-(2-гидроксиэтил)амино]этанол (II) 2,3-Дигидробензо[1,4]диоксин-5-иламин (31,1 г, 0,2 моль) смешивают с 2-хлорэтанолом (210 мл, 3,1 моль) и основанием Ханига (105 мл, 0,6 моль). Полученный темный раствор нагревают до 120°С и перемешивают при этой температуре при постоянном контроле состава реакционной массы с помощью ВЭЖХ. Спустя 12,5 часа реакцию останавливают. Добавляют этилацетат (300 мл) и раствор промывают разбавленным раствором соли (1×250 мл), затем раствором соли (2×75 мл). Водные слои объединяют, значение рН доводят до 7 добавлением К2СО3 и раствор снова промывают этилацетатом (2×100 мл). Органические слои объединяют и экстрагируют 2N HCl (3×150 мл). Полученный водный раствор нейтрализуют твердым К2СО3 до рН 7 и экстрагируют этилацетатом (3×100 мл). Органическую фазу сушат MgSO4, концентрируют и добавляют толуол (2×50) для удаления остаточного количества хлорэтанола. Получают 39,6 г (80%) неочищенного продукта в виде темного масла с чистотой 94% (площ.) (ЖХ-МС). 1Н ЯМР (CDCl3), Пример 2 2-[(2,3-Дигидробензо[1,4]диоксин-5-ил)-(2-метансульфонилоксиэтил)амино]этиловый эфир метансульфоновой кислоты (III) К раствору соединения II (39,6 г, 0,165 моль) и триэтиламина (69 мл, 0,5 моль) в метиленхлориде (250 мл), охлаждаемому на ледяной бане до 5°С, добавляют раствор мезилхлорида (38 мл, 0,5 моль) в метиленхлориде (50 мл). Добавление проводят в течение 0,5 часа при температуре не более 18°С. Ледяную баню удаляют и полученную суспензию перемешивают при комнатной температуре в течение 1 часа. В это время ТСХ и ВЭЖХ показывают исчезновение исходных веществ. Реакционную смесь промывают водой (1×150 мл) и 5% водным раствором NaHCO3 (1×150 мл), сушат MgSO4 и концентрируют с получением III в виде масла красного цвета, выход неочищенного продукта составляет 67,0 г (102%). 1Н ЯМР (CDCl3), Пример 3 2-[(2,3-Дигидробензо[1,4]диоксин-5-ил)пиперазин-1-ил]пропан-1-ол (IV) Димезилат III (67,0 г, 0,17 моль), D-аланинол (14,0 г, 0,19 моль), бромид лития (31,0 г, 0,35 моль) и карбонат калия (74,8 г, 0,54 моль) смешивают с ацетонитрилом (750 мл). Полученную суспензию кипятят с обратным холодильником (82°С) в течение 27 часов, контролируя состав реакционной смеси ВЭЖХ. Реакционную смесь охлаждают, фильтруют и выделенный осадок промывают ацетонитрилом. Маточную жидкость концентрируют до небольшого объема, фильтруют через 200 см3 силикагеля и элюируют 1,5 л 10% МеОН в EtOAc. Растворитель удаляют на роторном испарителе, остаток снова растворяют в EtOAc (200 мл). Полученный раствор промывают водой (2×50 мл), сушат MgSO4 и концентрируют с получением IV в виде густого золотистого масла, которое постепенно кристаллизуется; выход 29,4 г (63%), чистота 88,3% (площ.) (ЖХ-МС). Температура плавления 91-92°С. 1Н ЯМР (CDCl3), Пример 4 6-(2,3-Дигидробензо[1,4]диоксин-5-ил)-1-метил-6-аза-3-азониаспиро[2,5]октан хлорид (V) Неочищенный продукт IV (29,4 г, 0,106 моль) и триэтиламин (16,2 мл, 0,116 моль) растворяют в CH2Cl2 (150 мл) и к полученному раствору добавляют раствор мезилхлорида (8,6 мл, 0,111 моль) в CH2Cl2 (50 мл) при охлаждении до 5-15°С в течение 0,5 часа. Перемешивание продолжают при комнатной температуре в течение ночи, получая прозрачный красный раствор. Полученный раствор промывают водой (1×100 мл) и 5% водн. NaHCO3 (1×100 мл). Объединенные водные слои снова промывают CH2Cl2 (2×50 мл). Органические слои сушат MgSO4 и концентрируют, получая 31,6 г (101%) продукта V в виде густого красного масла. 1Н ЯМР (CDCl3), Пример 5 трет-Бутиловый эфир {2-[4-(2,3-дигидробензо[1,4]диоксин-5-ил)пиперазин-1-ил]пропил}пиридин-2-илкарбаминовой кислоты (VII) t-Boc-2-аминопиридин (24,7 г, 0,127 моль) и трет-бутоксид натрия (19,3 г, 0,2 моль) смешивают с ТГФ (250 мл) и перемешивают в течение 0,5 часа при комнатной температуре. К смеси добавляют хлорид V (31,6 г, 0,106 моль) в ТГФ (100 мл), затем твердый К2СО3 (23,4 г, 0,17 моль). Реакционную смесь нагревают до температуры кипения (68°С) и кипятят с обратным холодильником, контролируя состав смеси ТСХ (EtOAc/гексан 3:2, об./об.). Исходные вещества полностью исчезают через 97 часов. Реакционную смесь охлаждают, разбавляют EtOAc (400 мл), промывают водой (3×150 мл) и раствором соли (1×100 мл). Водные слои подвергают обратной экстракции EtOAc (2×75 мл). Объединенный органический раствор сушат MgSO4 и концентрируют, получая 49 г неочищенного масла, содержащего (ЖХ-МС) 67,9% VII (выход – 69%) и 10,8% V. 1Н ЯМР (CDCl3), Пример 6 {2-[4-(2,3-Дигидробензо[1,4]диоксин-5-ил)пиперазин-1-ил]пропил}пиридин-2-иламин (VIII) Соединение VII в виде неочищенного масла (49,0 г, 0,106 моль) растворяют в этаноле (150 мл) и к полученному раствору добавляют 1N раствор HCl в этаноле (212 мл). Полученный раствор кипятят с обратным холодильником в течение 18 часов, затем концентрируют в вакууме до небольшого объема (100 мл) до тех пор, пока не начинает кристаллизоваться продукт. К полученной суспензии медленно небольшими порциями добавляют эфир (100 мл) и смесь перемешивают при комнатной температуре в течение 2 часов. Бледно-серые кристаллы фильтруют и промывают смесью этанол/эфир (50:50), получая 22,2 г соединения VIII (49% для трех стадий). Чистота (ЖХ-МС) равна 97,9%. Продукт перекристаллизовывают из метанола (150 мл) и эфира (200 мл) с получением 19,3 г VIII с чистотой 99%. 1Н ЯМР (CD3OD), Пример 7 4-Циано-N-{2-[4-(2,3-дигидробензо[1,4]диоксин-5-ил)пиперазин-1-ил]пропил}-N-пиридин-2-илбензамид (IXa) Соединение VIII (18,7 г, 0,044 моль) добавляют к раствору К2СО3 (21,2 г, 0,15 моль) в 75 мл воды, смешанной с 90 мл EtOAc, при 0-5°С. Полученную двухфазную систему перемешивают в течение 0,5 часа до растворения всех твердых веществ. Затем в течение 15 минут добавляют раствор п-цианобензоилхлорида (8,0 г, 0,048 моль) в EtOAc (35 мл) при 5-7°С. Охлаждающую баню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Полноту протекания реакции определяют с помощью ТСХ. Органический слой отделяют и промывают водой (1×50 мл) и раствором соли (1×50 мл). Объединенные водные слои снова промывают EtOAc (1×60 мл). Объединенные EtOAc фракции сушат MgSO4 и фильтруют, затем кипятят с обратным холодильником в течение 0,5 часа с древесным углем Darco (2 г) и фильтруют через целит. Маточную жидкость разбавляют гептаном (90 мл), смешивают с силикагелем (20 г) и оставляют на 2 часа. Силикагель отфильтровывают, фильтрат концентрируют, получая свободное основание IXa в виде густого масла с чистотой (ЖХ) 94,5%. Полученное масло растворяют в EtOAc (100 мл) и обрабатывают 37 мл 1,2 N раствором HCl в EtOAc при 20-25°С. Гидрохлорид, выпавший в осадок в виде твердого белого вещества, выделяют фильтрованием и сушат в вакууме при 50°С, получая 20,8 г IX (91% на этой стадии, 19,4% на семи стадиях, исходя из I). 1Н ЯМР (CD3OD), ПРИМЕР 8 Алкилирование бензодиоксананилина до сложного диэфира
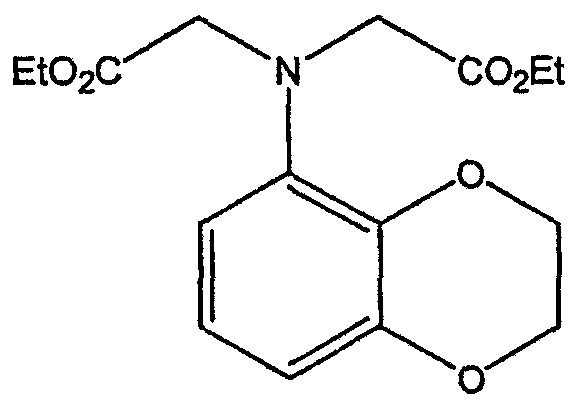
Смесь бензодиоксананилина (3,0 г, 20 ммоль), этилбромацетата (7,5 мл, 68 ммоль), основания Ханига (12,5 мл, 72 ммоль) и NaI (0,3 г, 2,0 ммоль) в толуоле (30 мл) кипятят с обратным холодильником в течение 23 часов. После этого реакционную смесь охлаждают до комнатной температуры и добавляют воду (25 мл). Разделяют два слоя. Водный слой экстрагируют толуолом (25 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая 6,5 г (100%) сложного диэфира в виде коричневого масла. 1Н ЯМР (CDCl3), ПРИМЕР 9 Восстановление бензодиоксанового диэфира до двухатомного спирта
Смесь диэфира (24 г, 74,3 ммоль) в ТГФ (240 мл) охлаждают до 0-5°С, затем медленно добавляют гранулы LAH (9,9 г, 260 ммоль), поддерживая температуру реакции ниже 10°С. После добавления LAH охлаждающую баню убирают и перемешивание продолжают в течение ночи. После перемешивания в течение 18 часов реакционную смесь охлаждают до 0±5°С на охлаждающей бане со смесью сухой лед/IPA. К реакционной смеси медленно добавляют воду (10 мл), затем 15% водный раствор гидроксида натрия (10 мл) и снова воду (30 мл). Полученную смесь перемешивают в течение 30 минут и затем фильтруют. Твердый осадок промывают ТГФ (100 мл). Фильтрат концентрируют в вакууме, получая 14,5 г (81%) двухатомного спирта формулы IV в виде густого прозрачного масла с чистотой 98% (площ.) (ЖХ-МС). 1Н ЯМР (CDCl3), Множество видоизменений данного изобретения, не отраженных в данном описании, будут понятны квалифицированному специалисту. Данное изобретение не ограничивается представленными в описании иллюстративными воплощениями, но включает все объекты, входящие в объем прилагаемой формулы изобретения, и их эквиваленты.
Формула изобретения
1. Способ получения соединения формулы VII
где R представляет собой C1-С3 алкил, Y представляет собой фрагмент, выбранный из группы, включающей C1-С6 алкокси, C1-С6 алкил, и Ar представляет собой 2,3-дигидробензодиоксин-5-ил, указанный способ включает следующие стадии: а) взаимодействие соединения формулы III
где L представляет собой удаляемую группу, выбранную из Cl, Br, мезилата, и Ar определен выше, с хиральным 2-амино-1-(С3-С5)алканолом в полярном апротонном растворителе для получения соединения формулы IV
где R и Ar определены выше, и * указывает хиральный центр; b) превращение соединения формулы IV в соединение формулы V
где Х представляет сбой Cl, или Br, R и Ar определены выше; и с) обработку соединения формулы V соединением формулы VI в апротонном растворителе
где М представляет собой щелочной металл, Y определен выше, с получением соединения формулы VII. 2. Способ получения соединения формулы VIII
где R представляет собой C1-С3 алкил, Ar представляет собой 2,3-дигидробензодиоксин-5-ил, и * указывает хиральный центр, в указанном способе проводят обработку протонной кислотой соединения формулы VII
где Y представляет собой фрагмент, выбранный из группы, включающей C1-С6 алкокси, C1-С6 алкил, и R и Ar определены выше. 3. Способ получения соединения формулы IX
где R и Ar определены выше, Ary1 представляет собой C6-С12 ароматическую группу, необязательно замещенную и содержащую до трех заместителей, независимо выбранных из группы, включающей атомы галогенов, алкил, алкокси, алкоксикарбонил, нитро, амино, алкиламино, диалкиламино, галогеналкил, дигалогеналкил, тригалогеналкил, нитрил и амидо-заместители, каждый из которых содержит не более шести атомов углерода, включающий обработку соединения формулы VIII
где R представляет собой C1-С3 алкил, Ar представляет собой 2,3-дигидробензодиоксин-5-ил, и * указывает хиральный центр, ароилхлоридом, ароилбромидом или ароилангидридом в присутствии основания. 4. Способ по п.3, где Ary1 представляет собой 4-цианофенил. 5. Способ по п.1, где соединение формулы III получают а) взаимодействием соединения формулы I ArNH2 где Ar представляет собой 2,3-дигидробензодиоксин-5-ил, с 2-хлорэтанолом в присутствии триалкиламинного основания для получения соединения формулы II
где Ar определен выше, и b) превращением соединения формулы II в соединение формулы III. 6. Способ по любому из пп.1 или 2, где Y представляет собой C1-С6 алкоксигруппу. 7. Способ по любому из пп.1-3, где Ar представляет собой 2,3-дигидробензодиоксин-5-ил. 8. Способ по п.1, где хиральный 2-амино-1-(С3-С5)алканол представляет собой D-2-амино-1-пропанол и R представляет собой метил. 9. Способ по п.6, где Y представляет собой трет-бутокси. 10. Способ по п.9, где Ar представляет собой 2,3-дигидробензодиоксин-5-ил. 11. Способ по любому из пп.9-10, где хиральный 2-амино-1-(С3-С5)алканол представляет собой D-2-амино-1-пропанол и R представляет собой метил.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 11.03.2009
Извещение опубликовано: 27.07.2010 БИ: 21/2010
|
||||||||||||||||||||||||||
 -дизамещенные пиперазины, в частности, содержащие N-арилзаместитель, воздействуют на центральную нервную систему (например, связываются с 5НТ рецепторами). В публикациях J. Med. Chem. (1995), 38(20), 4044-55 и JP 61152655 описывается традиционный подход к синтезу арилпиперазинового ядра, который включает взаимодействие анилинов с бис(дихлорэтил)амином. Пиперазины, представляющие собой продукт такого взаимодействия, далее модифицируют алкилированием полученного вторичного амина.
-дизамещенные пиперазины, в частности, содержащие N-арилзаместитель, воздействуют на центральную нервную систему (например, связываются с 5НТ рецепторами). В публикациях J. Med. Chem. (1995), 38(20), 4044-55 и JP 61152655 описывается традиционный подход к синтезу арилпиперазинового ядра, который включает взаимодействие анилинов с бис(дихлорэтил)амином. Пиперазины, представляющие собой продукт такого взаимодействия, далее модифицируют алкилированием полученного вторичного амина.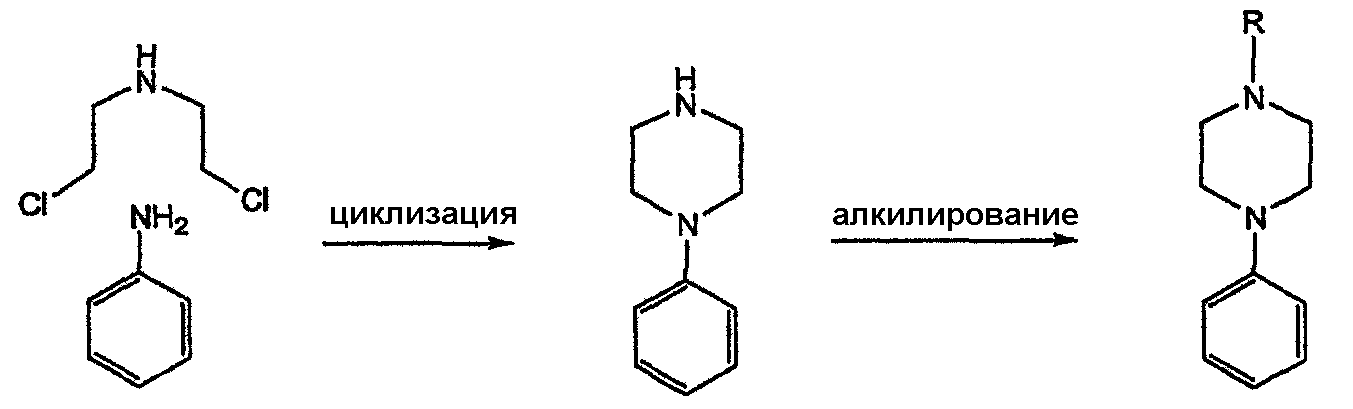
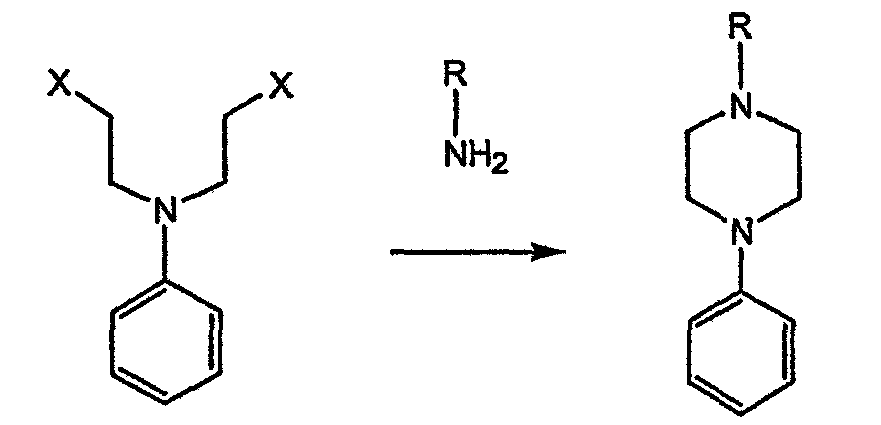
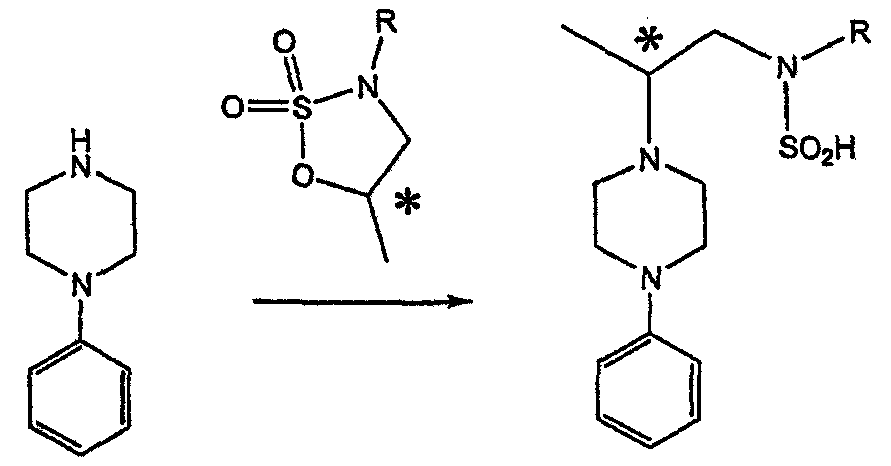
 -алкил (и арил)окси-хиральных аминов с ипритами с получением пиперазиновых производных.
-алкил (и арил)окси-хиральных аминов с ипритами с получением пиперазиновых производных. -атом относительно азота, поскольку основан на хиральных алкилирующих агентах, для получения которых необходимо осуществление многостадийного синтеза.
-атом относительно азота, поскольку основан на хиральных алкилирующих агентах, для получения которых необходимо осуществление многостадийного синтеза.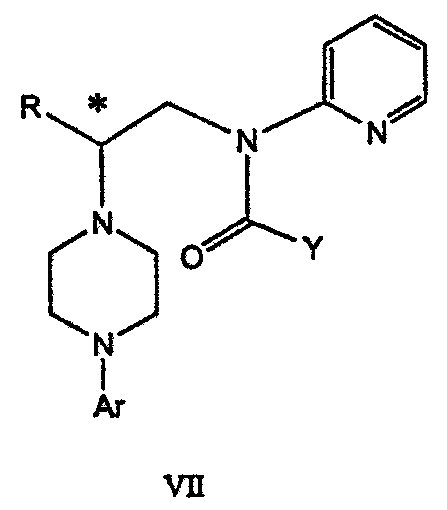
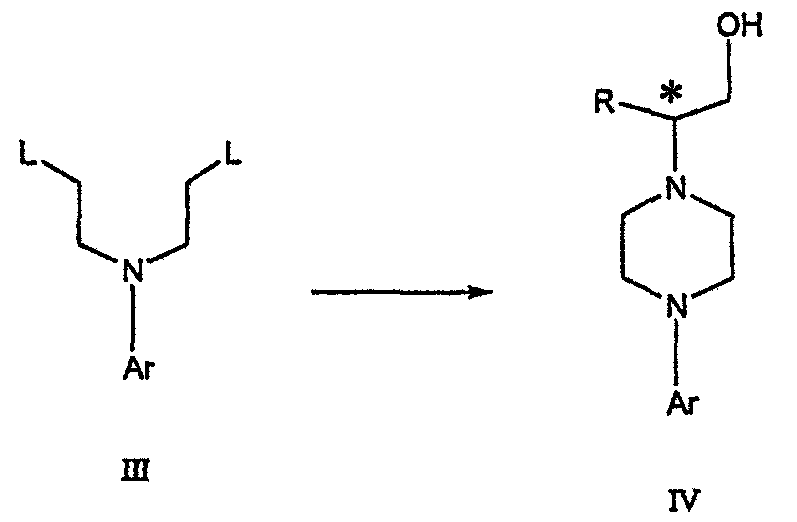
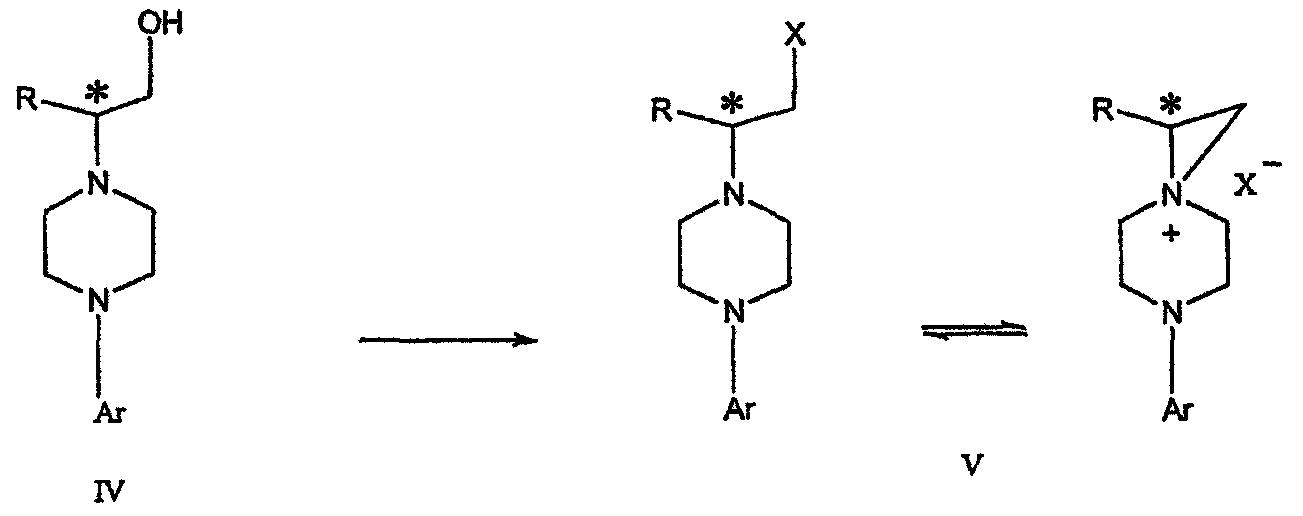
 IV).
IV).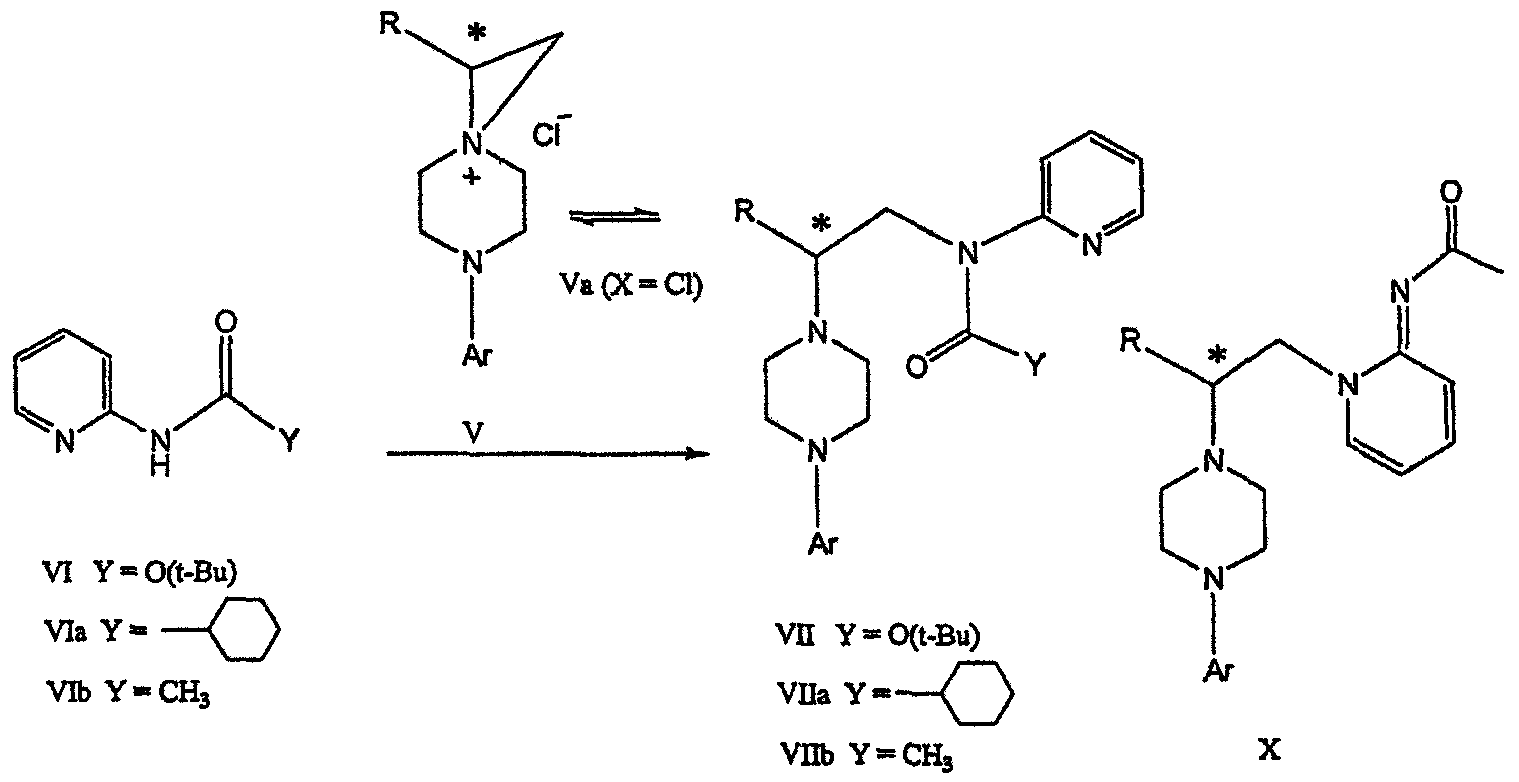
 : 6,88-6,70 (м, 3Н), 4,34-4,22 (м, 4Н), 3,54 (т, J=7,5 Гц, 4Н), 3,18 (т, J=7,5 Гц, 4Н).
: 6,88-6,70 (м, 3Н), 4,34-4,22 (м, 4Н), 3,54 (т, J=7,5 Гц, 4Н), 3,18 (т, J=7,5 Гц, 4Н).