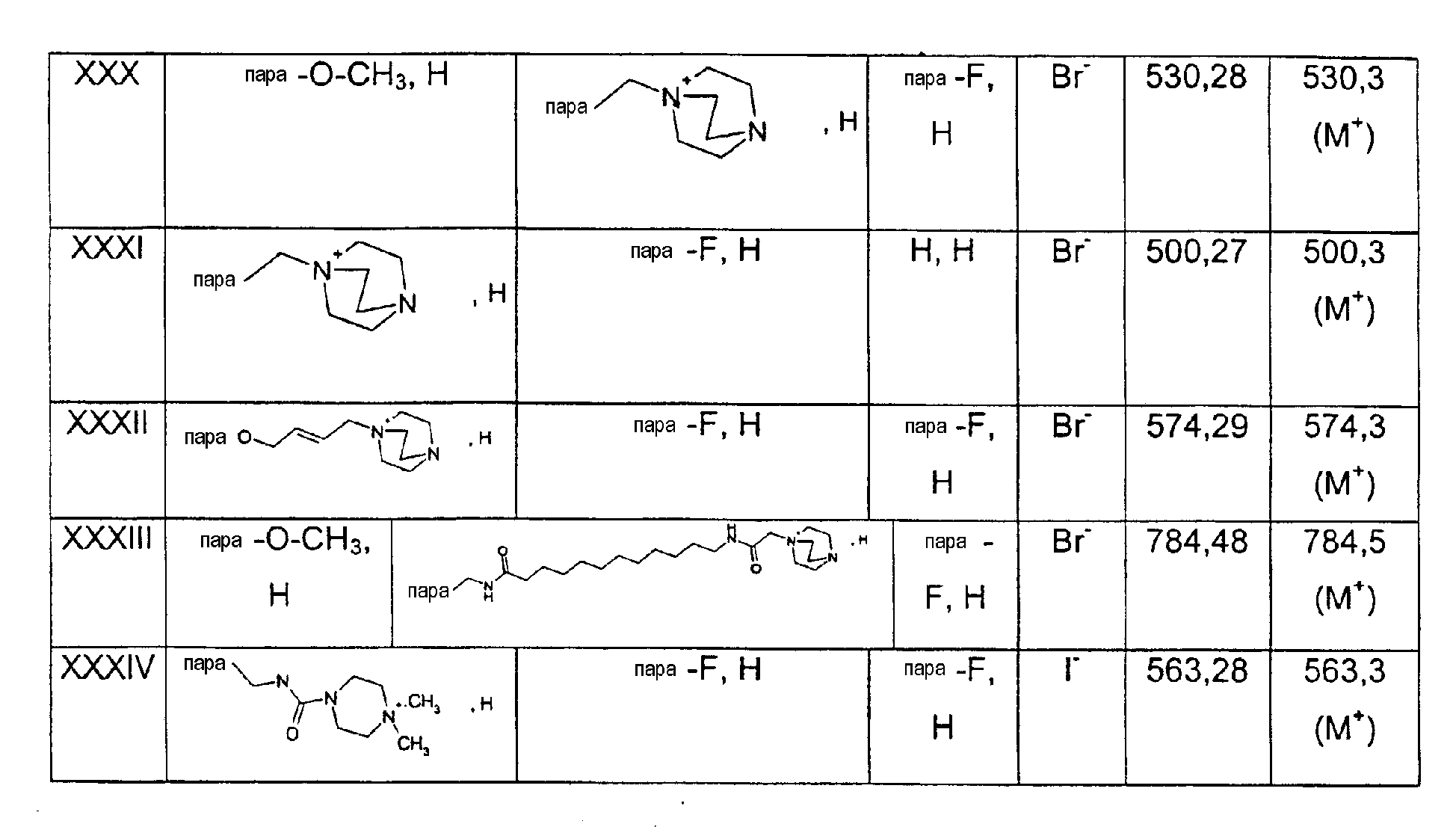
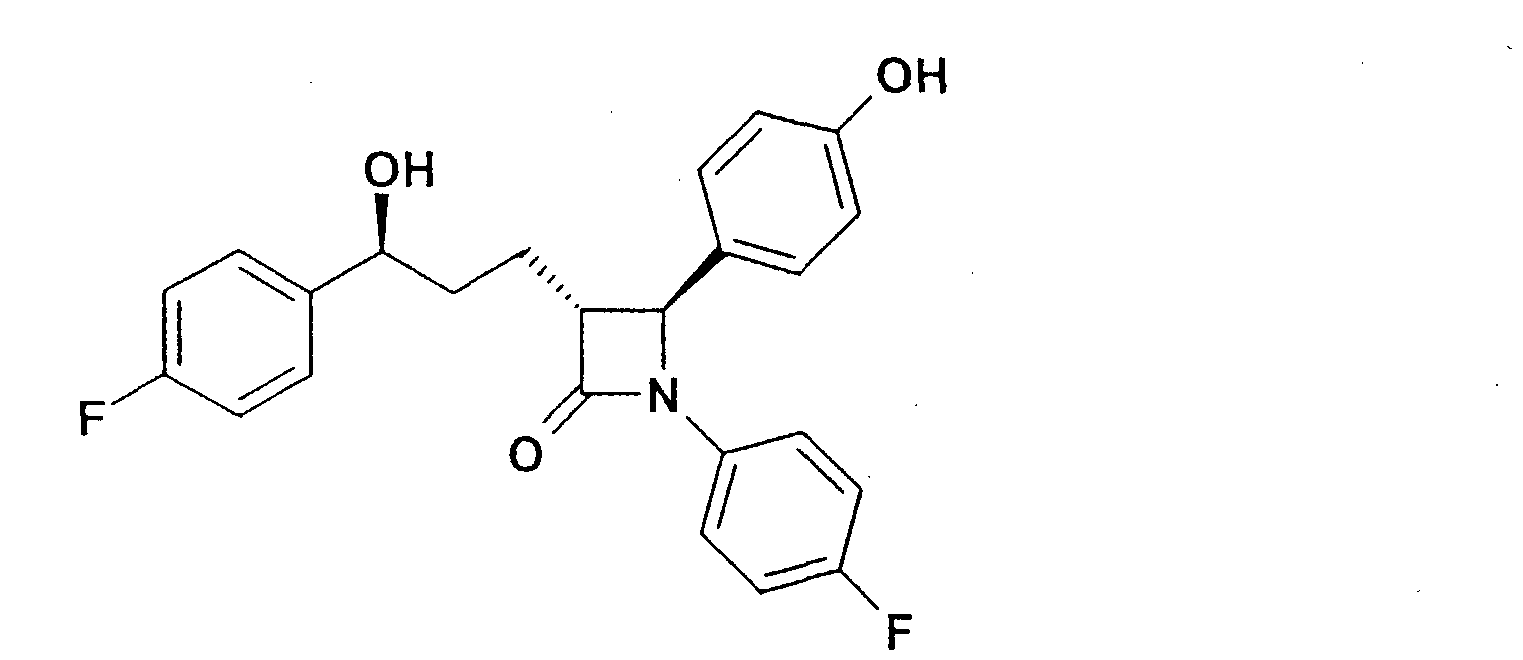
Патент на изобретение №2315753
|
||||||||||||||||||||||||||||||||||
(54) КАТИОНОЗАМЕЩЕННЫЕ ДИФЕНИЛАЗЕТИДИНОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ, И ИХ ПРИМЕНЕНИЕ
(57) Реферат:
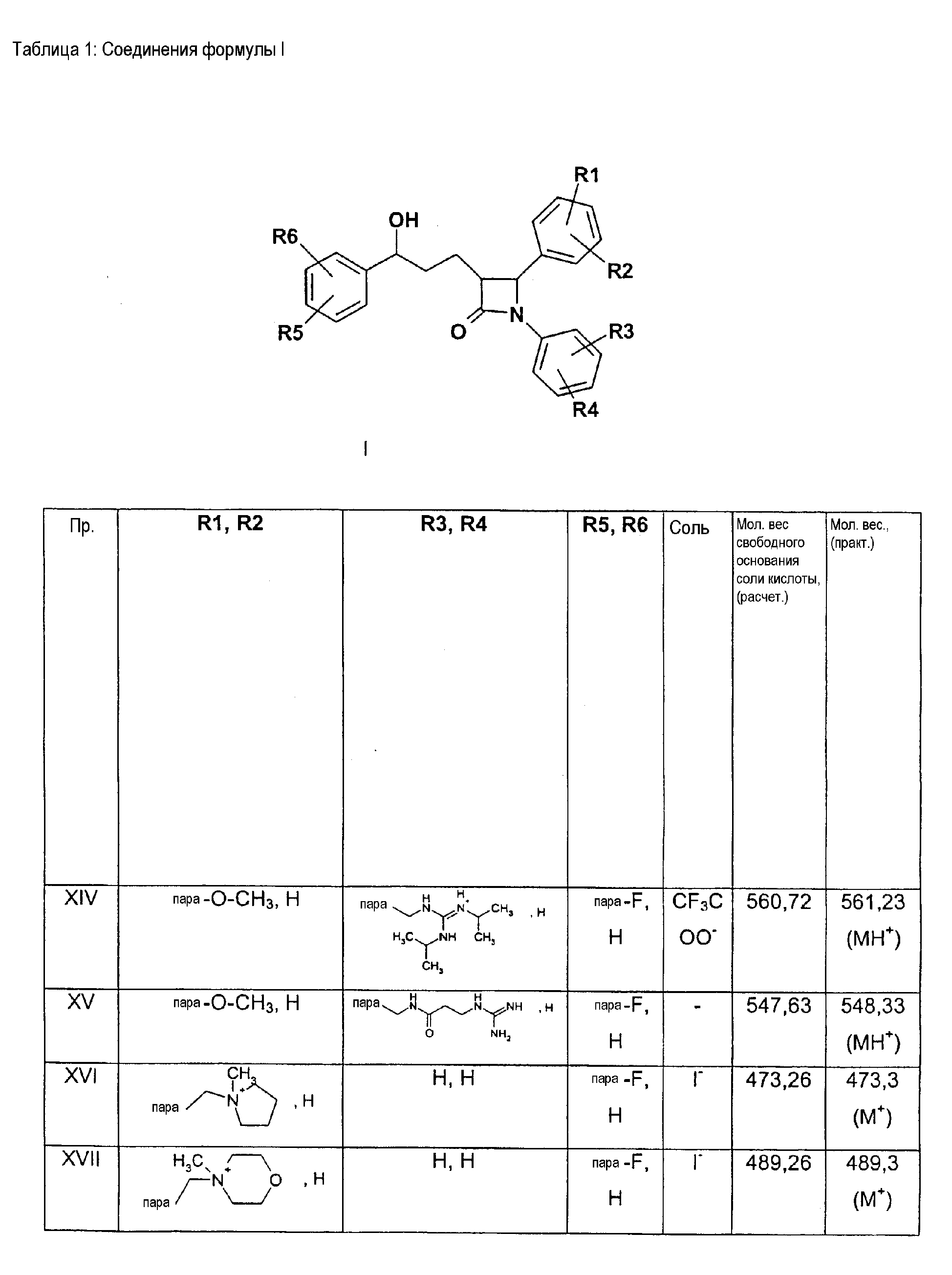
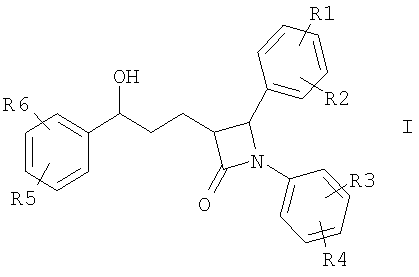
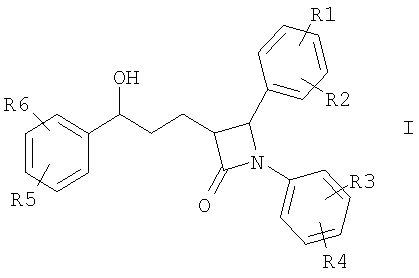
Описываются соединения формулы I
где R1-R5 независимо друг от друга (С0-С30)-алкилен-(LAG)n, где n равно 1, и причем один или несколько С-атомов алкиленового остатка могут быть заменены -О-, -(С=0)-, -С=СН-, -NH-; H, F, Cl, Br, I, (С1-С6)-алкил, O-(С1-С6)-алкил; (LAG)n означает моно- или трициклический триалкиламмонийалкильный остаток или LAG может означать -(CH2)0-10-C(=NH)(NH2), -(CH2)0-10-C(=NH)(NHOH) или -NR7-C(=NR8)(NR9R10). Описываются лекарственное средство для снижения уровня холестерина в сыворотке и способ его получения. Указанные соединения пригодны, например, в качестве гиполипидемических средств. 3 н. и 3 з.п. ф-лы, 1 табл.
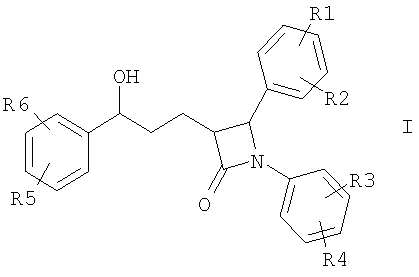
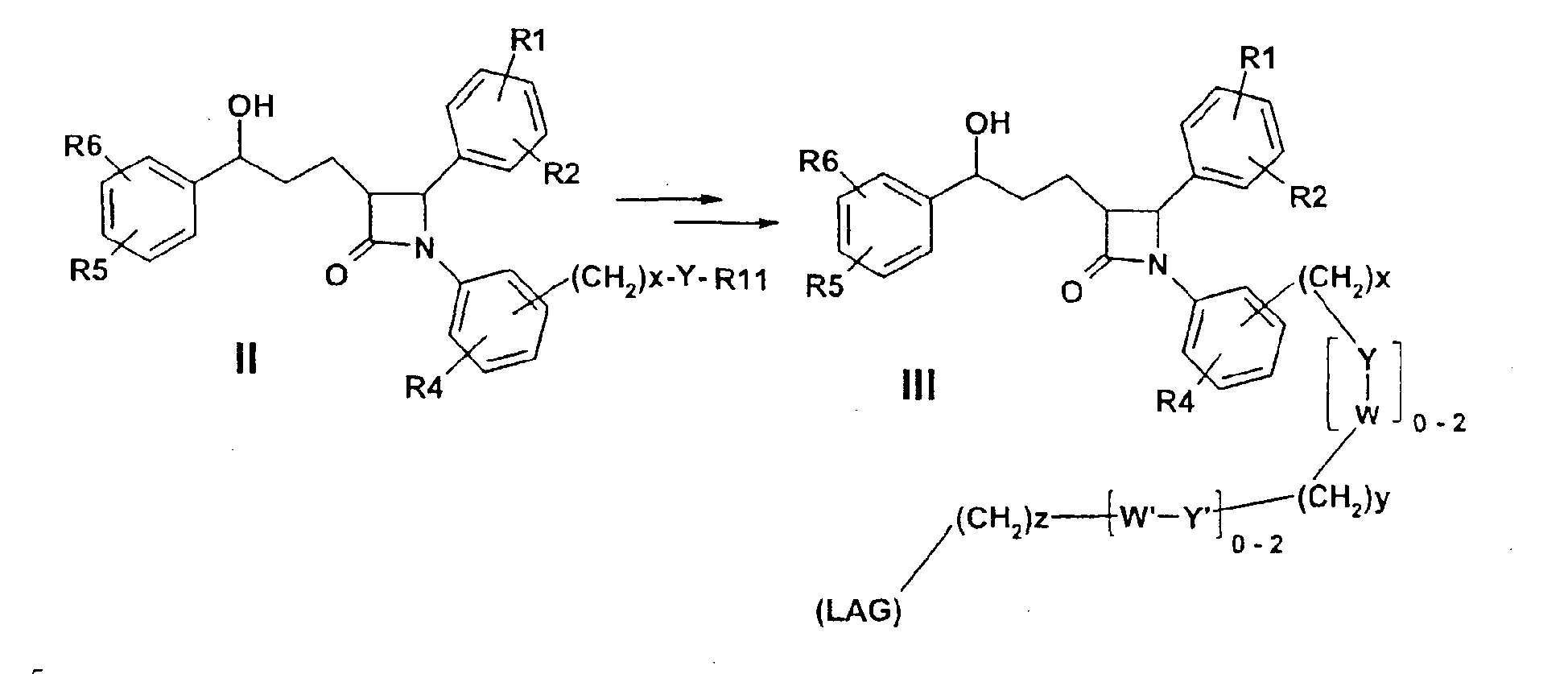
Изобретение относится к катионозамещенным дифенилазетидинонам, их физиологически переносимым солям, а также физиологически функциональным производным. Дифенилазетидиноны (такие как, например, эзетимиб), а также их применение для лечения гиперлипидемии, а также артериосклероза и гиперхолестеринемии уже были описаны [см. Drugs of the Future 2000, 25(7):679-685 и патент США 5756470]. Задачей изобретения является получение других соединений, пригодных для применения в терапевтических целях и обладающих гиполипидемическим действием. В частности, задача заключается в получении соединений, которые в отличие от соединений, описанных в уровне техники, всасываются в очень незначительной степени. Под очень незначительной всасываемостью понимается интерстинольная резорбция менее 10%, предпочтительно меньше или равная 5%. Новые соединения должны, в частности, иметь меньшую всасываемость, чем эзетимиб. При меньшей всасываемости фармацевтически активные вещества обнаруживают, как правило, заметно меньшие побочные эффекты. Изобретение относится поэтому к соединениям формулы I
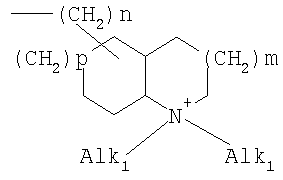
где означают R1, R2, R3, R4, R5, R6 независимо друг от друга (С0-С30)-алкилен-(LAG)n, причем n может быть от 1 до 5 и причем один или несколько С-атомов алкиленового остатка могут быть заменены -S(O)n– с n=0-2, -О-, -(С=O)-, -(C=S)-, -СН=СН-, -С=С-, -N((C1-С6)-алкил)-, -N(фенил)-, -N((C1-C6)-алкилфенил)-, -N(CO-(CH2)1-10-СООН)- или -NH-; Н, F, Cl, Br, I, CF3, NO2, N3, CN, COOH, COO(C1-C6)алкил, CONH2, CONH(С1-С6)алкил, CON[(C1-C6)алкил]2, (C1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, О-(C1-С6)-алкил, причем в алкильном остатке один, несколько или все водороды могут быть заменены фтором; C(=NH)(NH2), РО3Н2, SO3Н, SO2-NH2, SO2NH(C1-C6)-алкил, SO2N[(C1-С6)-алкил]2, S-(C1-C6)-алкил, S-(CH2)n-фенил, SO-(C1-C6)-алкил, SO-(CH2)n-фенил, SO2-(C1-C6)-алкил, SO2-(CH2)n-фенил, причем n может быть от 0 до 6, а фенильный остаток может быть до двух раз замещен F, Cl, Br, ОН, CF3, NO2, CN, OCF3, О-(C1-С6)-алкилом, (C1-С6)-алкилом, NH2; NH2, NH-(C1-C6)-алкил, N((C1-C6)-алкил)2, NH(С1-С7)-ацил, фенил, О-(CH2)n-фенил, причем n может быть от 0 до 6, причем фенильное кольцо может быть от одного до трех раз замещено F, Cl, Br, I, ОН, CF3, NO2, CN, OCF3, О-(C1-C6)-алкилом, (C1-С6)-алкилом, NH2, NH(C1-C6)-алкилом, N((C1-С6)-алкил)2, SO2-СН3, СООН, СОО-(C1-С6)-алкилом, CONH2; (LAG)n – моно-, би- или трициклический остаток триалкиламмония, моно-, би- или трициклический триалкиламмонийалкильный остаток, -(СН2)0-10-С(=NH)(NH2), -(CH2)0-10-C(=NH)(NHOH) или -NR7-C(=NR8)(NR9R10), и R7, R8, R9 и R10 независимо друг от друга могут быть Н, (C1-С6)-алкилом, (C1-C6)-алкилфенилом, фенилом, (С3-C8)-циклоалкилом, а n может быть от 0 до 5; причем всегда, по меньшей мере, один из остатков R1-R6 должен означать (С0-С30)-алкилен-(LAG)n, n может быть от 1 до 5 и причем один или несколько С-атомов алкиленового остатка могут быть заменены -S(O)n-, где n=0-2, -О-, -(С=O)-, -(C=S)-, -CH=CH-, -С а также их фармацевтически приемлемые соли. Предпочтительны соединения формулы I, в которых, по меньшей мере, один из остатков R1-R6 имеет значение (С0-С30)-алкилен-(LAG), причем один или несколько С-атомов алкиленового остатка могут быть заменены -О-, -(С=O)-, -N((C1-С6)-алкил)-, -N(CO-(CH2)1-10-COOH)- или -NH-. Особенно предпочтительны соединения формулы I, в которых один из остатков R1 или R3 имеет значение (С0-С30)-алкилен-(LAG), причем один или несколько С-атомов алкиленового остатка могут быть заменены -О-, -(С=O)-, -N(СН3)- или -NH-. Еще более предпочтительны соединения формулы I, в которых один из остатков R1 или R3 имеет значение -(СН2)0-1-Y-W-(С0-С25)-алкилен-Y Далее, предпочтительны соединения формулы I, в которых группа LAG является бициклическим триалкиламмонийалкильным остатком. Под моно-, би- или трициклическим остатком триалкиламмония понимаются, например, остатки
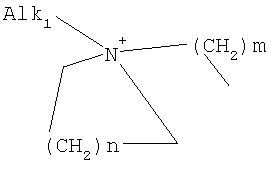
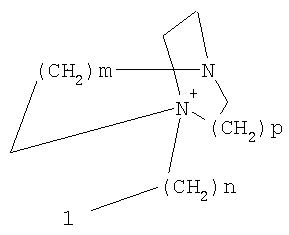
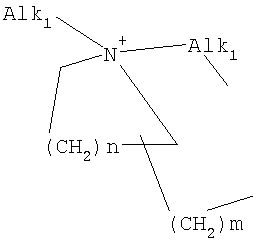
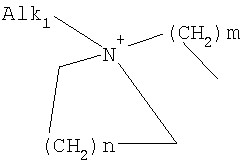
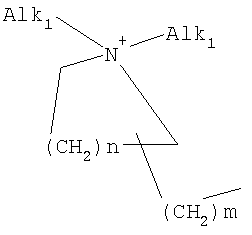
причем n, m и р независимо друг от друга могут быть 0-10, и одна или несколько СН2-групп независимо друг от друга могут быть заменены О, S(О)n с n=0-2, NH, N-(C1-С10)-алкилом, N-фенилом или N-СН2-фенилом. Под моно- или бициклическим триалкиламмонийалкильным остатком понимаются, например, такие остатки, как
или причем n, m и р независимо друг от друга могут быть 0-10, и одна или несколько СН2-групп независимо друг от друга могут быть заменены О, S(O)n, причем n может быть от 0 до 2, NH, N-(C1-C10)-алкилом, N-фенилом или N-СН2-фенилом, a Alk1 означает линейный или разветвленный алкильный остаток с 1-20 атомами углерода. Фармацевтически приемлемые соли благодаря своей высокой растворимости в воде по сравнению с исходными или основными соединениями особенно подходят для применения в медицине. Эти соли должны иметь фармацевтически переносимый анион или катион. Подходящими фармацевтически переносимыми солями соединений согласно изобретению, образующимися при добавлении кислот, являются соли неорганических кислот, таких как соляная кислота, бромистоводородная, фосфорная, метафосфорная, азотная, сульфоновая и серная кислоты, а также органических кислот, таких как, например, уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изетионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, янтарная, p-толуолсульфоновая, винная и трифторуксусная кислоты. Для медицинских целей особенно предпочтительно применяют соляную кислоту. Подходящими фармацевтически переносимыми солями оснований являются аммонийные соли, соли щелочных металлов (такие как соли натрия и калия) и соли щелочно-земельных металлов (такие как соли магния и кальция). Соли с фармацевтически непереносимым анионом также охватываются рамками изобретения как полезные промежуточные продукты для получения или очистки фармацевтически переносимых солей и/или для нетерапевтических применений, например для применений in vitro. Используемое в описании понятие “физиологически функциональное производное” означает любое физиологически переносимое производное соединения согласно изобретению, например сложный эфир, который при введении млекопитающему, такому как, например, человек, способен (прямо или косвенно) образовывать такое соединение или его активный метаболит. Другим объектом данного изобретения являются пролекарства соединений согласно изобретению. Такие пролекарства могут быть метаболизированы in vivo в соединение согласно изобретению. Сами пролекарства могут быть активными или неактивными. Соединения согласно изобретению могут также находиться в различных полиморфных формах, например, таких как аморфные и кристаллические полиморфные формы. Все полиморфные формы соединений согласно изобретению охватываются рамками изобретения и являются дальнейшим объектом изобретения. Далее все ссылки на “соединение(я) по формуле I” относятся к соединению(ям) формулы I, как она описана выше, а также к их солям, сольватам и описанным физиологически функциональным производным. Соединения формулы I и их фармацевтически приемлемые соли и физиологически функциональные производные представляют собой идеальные лекарственные средства для лечения нарушений липидного обмена, в частности гиперлипидемии. Соединения формулы I подходят также для воздействия на уровень холестерина в сыворотке, а также для профилактики и лечения артериосклеротических проявлений. Соединение(я) формулы (I) могут приниматься также в комбинации с другими активными веществами. Количество соединения формулы I, которое требуется для достижения желаемого биологического эффекта, зависит от ряда факторов, например конкретного выбранного соединения, планируемого применения, типа приема и клинического состояния пациента. Обычно суточная доза составляет от 0,1 мг до 100 мг (обычно от 0,1 мг до 50 мг) в день на килограмм веса тела, например 0,1-10 мг/кг/сутки. Таблетки или капсулы могут содержать, например, от 0,01 до 100 мг, обычно от 0,02 до 50 мг. В случае фармацевтически переносимых солей вышеуказанные весовые количества относятся к весу образующегося из соли иона дифенилацетидинона. Для профилактики или лечения указанных выше состояний соединения формулы (I) могут применяться непосредственно в виде соединения, однако предпочтительно они предлагаются с приемлемым носителем в виде одной фармацевтической композиции. Носитель должен быть переносимым, в том смысле, что должен быть совместимым с другими компонентами композиции и не должен вредить здоровью пациента. Носитель может быть твердым веществом, или жидкостью, или тем и другим и предпочтительно формироваться с соединением в виде разовой дозы, например, такой как таблетка, которая может содержать от 0,05% до 95 вес.% активного вещества. Также могут иметься и другие фармацевтически активные вещества, в том числе другие соединения формулы (I). Фармацевтические композиции согласно изобретению могут быть получены по одному из известных фармацевтических методов, которые в основном состоят в том, чтобы смешать компоненты с фармакологически переносимой основой и/или вспомогательными веществами. Фармацевтические композиции согласно изобретению являются пригодными для орального и перорального (например, сублингвального) применения, хотя самый подходящий способ введения в каждом отдельном случае зависит от вида и тяжести излечиваемого заболевания и от типа фактически применяемого соединения формулы (I). Изобретением охватываются также дражированные лекарственные формы и дражированные лекарственные формы пролонгированного действия. Предпочтительны препаративные формы, устойчивые к действию кислот и желудочного сока. Подходящие устойчивые к действию желудочного сока покрытия включают ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы и анионные полимеры метакриловой кислоты и метилового эфира метакриловой кислоты. Фармацевтические соединения, подходящие для орального приема, могут находиться в виде отдельных дозированных форм, таких как, например, капсулы, облатки, сосательные таблетки или таблетки, которые содержат определенное количество соединения формулы (I); таких как порошок или гранулят; таких как раствор или суспензия в водной или неводной жидкости, или как эмульсия “масло-в-воде” или “вода-в-масле”. Эти композиции могут, как уже упоминалось, быть приготовлены любым подходящим фармацевтическим способом, включающим стадию, на которой активное вещество и носитель (который может состоять из одного или нескольких дополнительных компонентов) приводятся в контакт. Обычно композиции получают путем равномерного и однородного смешения активного вещества с жидким и/или тонкодисперсным твердым носителем, после чего продукт, если требуется, формуется. Так, например, таблетку можно изготовить путем прессования или формования порошка или гранулята соединения, возможно, с одним или несколькими дополнительными компонентами. Прессованные таблетки могут быть получены таблетированием соединения в свободнотекучей форме, такой как, например, порошок или гранулят, возможно, смешанный с вяжущим, смазкой, инертным разбавителем и/или одним (несколькими) поверхностно-активным веществом/диспергирующим агентом, в подходящем аппарате. Формованные таблетки могут быть получены формованием порошкообразного соединения, смоченного инертным жидким разбавителем, в подходящем аппарате. Фармацевтические композиции, подходящие для перорального (сублингвального) приема, включают сосательные таблетки, содержащие соединение формулы (I) вместе с вкусовым веществом, обычно сахарозой и гуммиарабиком или трагантом, и пастилки, которые содержат соединение в инертной основе, такое как желатин и глицерин, или сахарозу и гуммиарабик. В качестве дальнейших активных веществ для комбинированных препаратов подходят все антидиабетические средства, которые названы в Красном списке 2001, глава 12. Они могут быть скомбинированы с соединениями согласно изобретению формулы I, в частности, для улучшения синергетического действия. Применение комбинации действующих веществ может осуществляться или путем раздельного введения действующих веществ, или в виде комбинированного препарата, в котором несколько действующих веществ находятся в одной фармацевтической готовой лекарственной форме. Антидиабетические средства включают инсулин и производные инсулина, такие как, например, Lantus® или HMR 1964, производные ГПП-1, например, раскрытые в заявке WO 98/08871, принадлежащей Novo Nordisk A/S, а также действующие орально гипогликемические действующие вещества. Действующие орально гипогликемические вещества предпочтительно включают сульфонилмочевину, бигуадин, меглитинид, оксадиазолидиндион, тиазолидиндион, ингибиторы глюкозидазы, антагонисты глюкагона, агонисты ГПП-1, активаторы калиевых каналов, такие как, например, раскрытые в заявках WO 97/26265 и WO 99/03861, принадлежащих Novo Nordisk A/S, сенсибилизаторы инсулина, ингибиторы ферментов печени, которые участвуют в стимулировании глюконеогенеза и/или глюкогенолиза, модуляторы усвоения глюкозы, соединения, изменяющие жировой обмен, такие как антигиперлипидемические действующие вещества и антилипидемические действующие вещества, соединения, снижающие потребление продуктов питания, агонисты PPAR и PXR, и действующие вещества, которые воздействуют на АТФ-зависимые калиевые каналы бета-клеток. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором ГМГ-КоА-редуктазы, таким как симвастатин, флювастатин, правастатин, ловастатин, аторвастатин, серивастатин, розувастатин. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором всасывания холестерина, таким как, например, эзетимиб, тиквезид, памаквезид. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с агонистом PPAR-гамма, таким как, например, розиглитазон, пиоглитазон, JTT-501, GI 262570. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с агонистом PPAR-альфа, таким как, например, GW 9578, GW 7647. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации со смешанным агонистом PPAR альфа/гамма, таким как, например, GW 1536, AVE 8042, AVE 8134, AVE 0847. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с фибратом, таким как, например, фенофибрат, клофибрат, безафибрат. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором MTP, таким как, например, Bay 13-9952, BMS-201038, R-103757. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором всасывания желчной кислоты, таким как, например, HMR 1453. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с CETP-ингибитором, таким как, например, Bay 194789. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с полимерным поглотителем желчной кислоты, таким как, например, холестирамин, колесолвам. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с индуктором LDL-рецепторов, таким как, например, HMR1171, HMR1586. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором ACAT, таким как, например, авасимиб. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с антиоксидантом, таким как, например, OPC-14117. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором липопротеинлипазы, таким как, например, NO-1886. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором АТФ-цитратлиазы, таким как, например, SB-204990. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором скваленсинтетазы, таким как, например, BMS-188494. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с антагонистом липопротеина(ов), таким как, например, CI-1027 или никотиновая кислота. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с ингибитором липазы, таким как, например, орлистат. В одном способе осуществления изобретения соединения формулы I вводятся в комбинации с инсулином. В одном способе осуществления соединения формулы I вводятся в комбинации с сульфонилмочевиной, такой как, например, толбутамид, глибенкламид, глипизид или гликлазид. В одном способе осуществления соединения формулы I вводятся в комбинации с бигуанидом, таким как, например, метформин. Еще в одном способе осуществления соединения формулы I вводятся в комбинации с меглитинидом, таким как, например, репаглинид. В одном способе осуществления соединения формулы I вводятся в комбинации с тиазолидиндионом, таким как, например, троглитазон, циглитазон, пиоглитазон, розиглитазон, или с соединениями, раскрытыми в патенте WO 97/41097, принадлежащем Исследовательскому центру им Dr.Reddy, в частности, 5-[[4-[(3,4-дигидро-3-метил-4-оксо-2-хиназолинилметокси]фенил]метил]-2,4-тиазолидиндион. В одном способе осуществления соединения формулы I вводятся в комбинации с ингибитором В одном способе осуществления соединения формулы I вводятся в комбинации с действующим веществом, которое воздействует на АТФ-зависимые калиевые каналы бета-клеток, такие как, например, толбутамид, глибенкламид, глипизид, глиазид или репаглинид. В одном способе осуществления соединения формулы I вводятся в комбинации с более чем одним из названных выше соединений, например в комбинации с сульфонилмочевиной и метформином, сульфонилмочевиной и акарбозой, репаглинидом и метформином, инсулином и сульфонилмочевиной, инсулином и метформином, инсулином и троглитазоном, инсулином и ловастатином, и т.д. В еще одном способе осуществления соединения формулы I вводятся в комбинации с агонистами CART, агонистами NPY, например агонистами MC-3 или MC-4, агонистами орексина, агонистами H3, агонистами TNF, агонистами CRF, антагонистами CRF-BP, агонистами урокортина, В одном способе осуществления изобретения другим действующим веществом является лептин. В одном способе осуществления другим действующим веществом является дексамфетамин или амфетамин. В одном способе осуществления другим действующим веществом является фенфлурамин или дексфенфлурамин. В еще одном способе осуществления другим действующим веществом является сибутрамин. В одном способе осуществления другим действующим веществом является орлистат. В одном способе осуществления другим действующим веществом является мазиндол или фентермин. В одном способе осуществления соединения формулы I вводятся в комбинации с балластными веществами, предпочтительно нерастворимыми балластными веществами, такими как, например, Каромакс®. Комбинация с Каромаксом® может быть осуществлена или в одной лекарственной форме, или путем раздельного приема соединения формулы I и Каромакса®. При этом Каромакс® может приниматься также в виде продукта питания, такого как, например, выпечка или дольки мюсли. Комбинация соединения формулы I с Каромаксом® отличается, помимо улучшения активности, в частности, в отношении снижения уровня LDL-холестерина, также своей улучшенной переносимостью. Разумеется, что все подходящие комбинации соединений согласно изобретению с одним или несколькими указанными выше соединениями и, по выбору, одним или несколькими дополнительными фармакологически действующими веществами должны рассматриваться как подпадающие под объем притязаний настоящего изобретения. Объектом изобретения являются, кроме того, как смеси стереоизомеров формулы I, так и чистые стереоизомеры формулы I, как также смеси диастереомеров формулы I, так и чистые диастереомеры. Разделение смесей осуществляется хроматографическим путем. Предпочтительны как рацемические, так и чисто энантиомерные соединения формулы I следующей структуры:
В качестве аминозащищенных групп предпочтительно приводятся остаток бензилоксикарбонила (Z), отщепляемый каталитическим гидрированием, остаток 2-(3,5-диметилоксифенил)пропил(2)оксикарбонила (Ddz) или тритила (Trt), отщепляемые слабыми кислотами, остаток t-бутилкарбамата (BOC), отщепляемый такими кислотами, как 3M соляная кислота, и остаток 9-флуоренилметилоксикарбонила (Fmoc), отщепляемый вторичными аминами. Изобретение относится, кроме того, к способу получения производных дифенилазетидинона формулы I
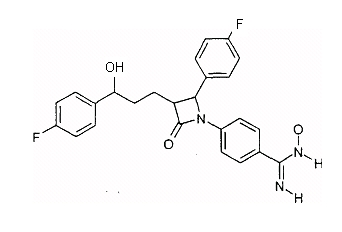
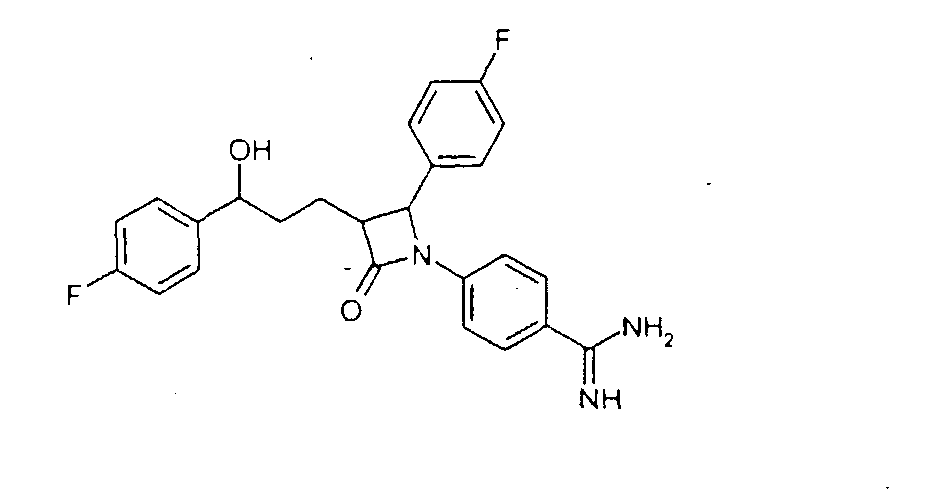
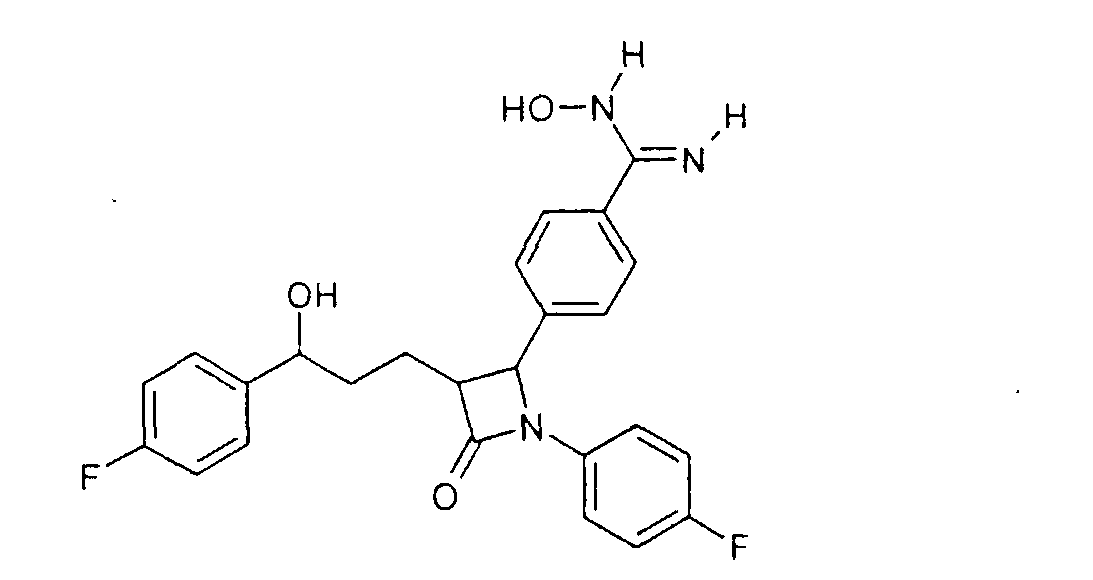
Y может означать S, О, (C=O), (C=S), CH=CH, C R11 может означать H или, если Y=(C=O) или (C=S), OH; W, Y x, y и z могут независимо друг от друга означать от 0 до 10. Связь -(CH2)x-Y-R11 в соединении II может альтернативно быть на одном из двух других фенильных колец. Способ получения соединения формулы I отличается тем, что, например, амин или гидроксисоединение формулы II подвергают взаимодействию с агентом алкилирования или ацилирования, который предпочтительно в омега-положении имеет дополнительную функциональную группу, возможно, в защищенной форме. Она будет применяться (после снятия защиты) для соединения (LAG), например, с образованием простой эфирной связи, амино- или амидной связи. Следующие далее примеры служат для разъяснения изобретения, не ограничивая его описанными в примерах продуктами и способами осуществления. Пример I 4-{1-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}-N-гидроксибензамидин (3)
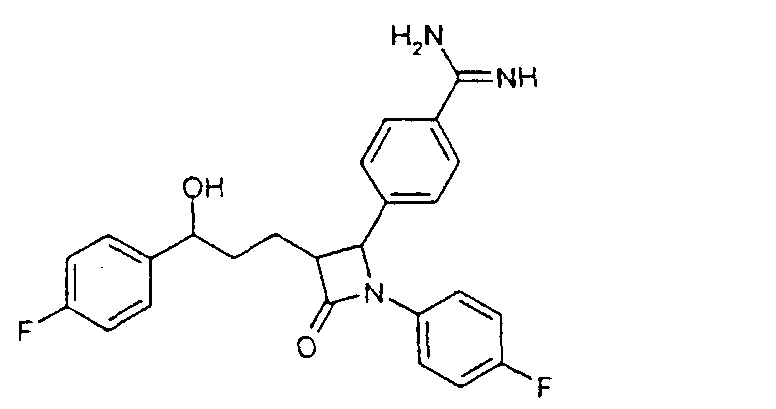
a) 4-[5-(4-Фторфенил)-1-(4-фторфениламино)-5-гидрокси-2-(2-оксо-4-фенилоксазолидин-3-карбонил)пентил]бензонитрил (1) 2,5 г 3-[5-(4-фторфенил)-5-гидроксипентаноил]-4-фенилоксазолидин-2-она растворяли в 30 мл дихлорметана в атмосфере аргона, добавляли 3,9 г 4-[(4-фторфенилимино)метил]бензонитрила и охлаждали до -10°C. К смеси добавляли 6,4 мл диизопропилэтиламина и в течение 30 мин 4,05 мл триметилсилилхлорида, так чтобы температура не превышала -5°C. При этой температуре перемешивали еще 1 час и затем охлаждали до -25°C. После этого медленно добавляли 0,8 мл тетрахлорида титана. Темную смесь в течение ночи перемешивали при температуре от -25 до -30°C, после чего разлагали 35 мл 7%-го раствора винной кислоты и перемешивали 1 час при комнатной температуре. Наконец, добавляли 15 мл 20%-го раствора гидрокарбоната натрия и снова перемешивали 1 час. После разделения фаз органическую фазу промывали 30 мл воды, сушили над сульфатом магния и концентрировали до примерно 10 мл. После добавления 2 мл бис-триметилсилилацетамида нагревали 30 мин с обратным холодильником, после чего концентрировали в вакууме. Остаток кристаллизовали смесью этилацетат/гептан. Отсасывали и сушили в вакууме. Получили продукт с молекулярным весом 653,81 (C37H37F2N3O4Si); МС (ESI+): 654,3 (M+H+), 582,2 (M + H+ – Si(CH3)3). b) {1-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензонитрил (2) 2 г 4-[5-(4-фторфенил)-1-(4-фторфениламино)-5-гидрокси-2-(2-оксо-4-фенилоксазолидин-3-карбонил)пентил]бензонитрила (1) растворяли в 20 мл метил-трет-бутилового эфира и нагревали примерно 1 час 40°C с 100 мг тетрабутиламмонийфторидтригидрата и 1,3 мл бис-триметилсилилацетамида. Проводили реакцию на тонкослойной хроматограмме. По окончании взаимодействия сначала добавляли 0,2 мл ледяной уксусной кислоты, перемешивали 30 мин и концентрировали. Остаток разлагали 20 мл смеси изопропанол/2N серная кислота =10:1 и перемешивали 1 час. После добавления на кончике ложки твердого гидрокарбоната натрия снова концентрировали в вакууме, адсорбировали этилацетатом, органическую фазу промывали водой, сушили и после удаления растворителя чистили остаток с помощью хроматографии на колонках (SiO2, CH2Cl2/метанол =100:1). Получили продукт с молекулярным весом 418,45 (C25H20F2N2О2); МС (DCI+): 419 (M+H+). c) 4-{1-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}-N-гидроксибензамидин (3) К раствору из 200 мг {1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензонитрила и 0,45 мл триэтиламина в 15 мл изопропанола добавляли 199 мг гидроксиламмонийгидрохлорида и перемешивали 12 часов при комнатной температуре. Реакционный раствор дважды экстрагировали смесью этиловый эфир уксусной кислоты/вода. Органическую фазу сушили над сульфатом магния и концентрировали. Получили продукт с молекулярным весом 451,48 (C25H23F2N3O3); МС (ESI) 452,10 (M+H+) Пример II 4-{1-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензамидин (4)
100 мг 4-{1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}-N-гидроксибензамидина (3) растворяли в 100 мл тетрагидрофурана и гидрировали 2 мл концентрированного аммиака над никелем Ренея 30 часов при давлении водорода 75 бар и температуре 25°C. После добавления сульфата магния реакционный раствор фильтровали. Фильтрат концентрировали и разделяли с помощью ВЭЖХ (Knauer, колонка Eurospher-100-10-C18, вода (0,1% трифторуксусной кислоты)/ацетонитрил (0,1% трифторуксусной кислоты)=80/20 Пример III 4-[3-[3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]-N-гидроксибензамидин (9)
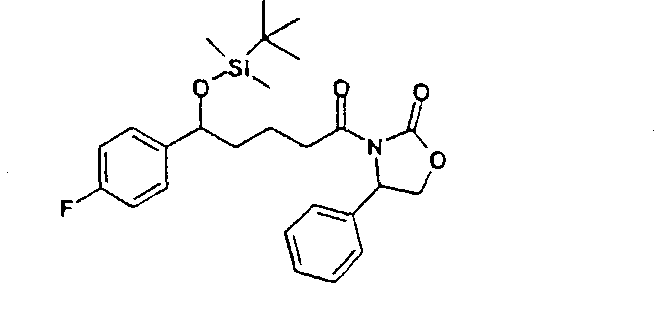
a) 3-[5-(трет-Бутилдиметилсиланилокси)-5-(4-фторфенил)пентаноил]-4-фенилоксазолидин-2-он (5)
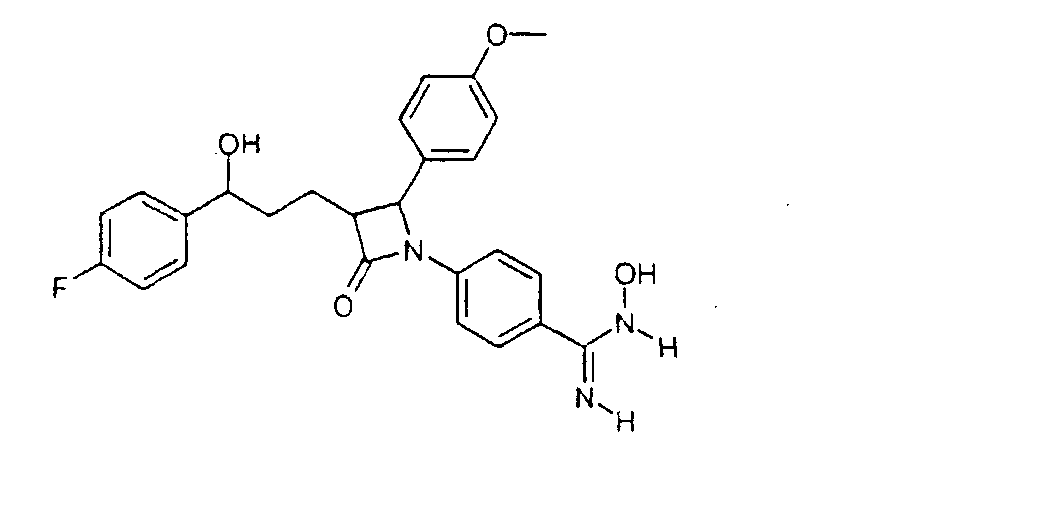
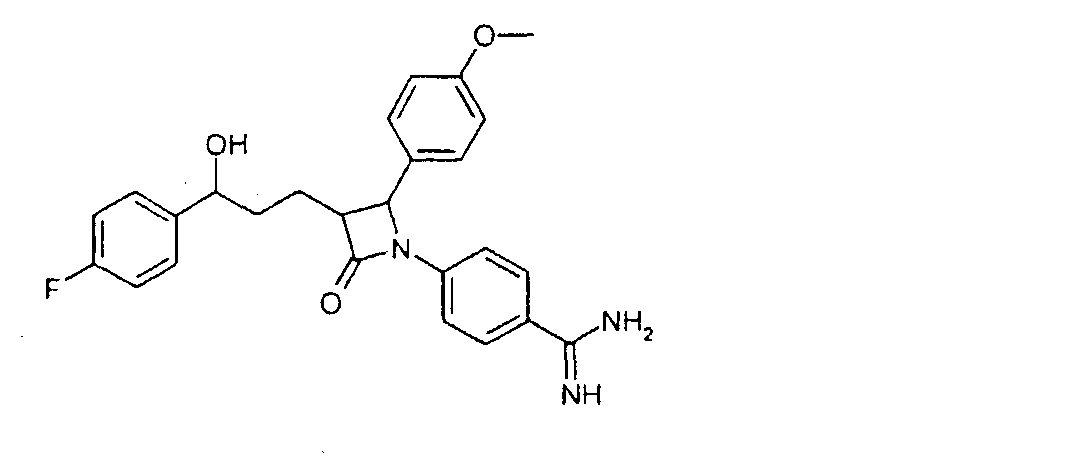
27 г 3-[5-(4-фторфенил)-5-гидроксипентаноил]-4-фенилоксазолидин-2-она растворяли с 13,6 г трет-бутилдиметилсилилхлорида и 10,2 г имидазола в 36 мл диметилформамида и 90 мин перемешивали при 60°C. По окончании реакции смесь растворяли в этиловом эфире уксусной кислоты и дважды промывали водой. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Получили 3-[5-(трет-бутилдиметилсиланилокси)-5-(4-фторфенил)пентаноил]-4-фенилоксазолидин-2-он (5) с молекулярным весом 471,65 (C26H34FNO4Si); МС (ESI): 340,28 (MH+ – HOSi(CH3)2C(CH3)3). b) 4-[5-(трет-Бутилдиметилсиланилокси)-5-(4-фторфенил)-1-(4-метоксифенил)-2-(2-оксо-4-фенилоксазолидин-3-карбонил)пентиламино]бензонитрил (6) 16,2 г 3-[5-(трет-бутилдиметилсиланилокси)-5-(4-фторфенил)пентаноил]-4-фенилоксазолидин-2-она растворяли в 350 мл дихлорметана. К раствору добавляли 19,8 мл основания Хенига и 10,14 г 4-[(4-метоксифенилимино)метил]бензонитрила и охлаждали до -10°C. К охлажденному раствору добавили 8,52 мл триметилсилилтрифлата и перемешивали 30 мин при -10°C. Раствор охлаждали теперь до -30°C и добавляли 44 мл раствора тетрахлорида титана. Реакционную смесь перемешивали 2 часа при температуре от -30 до -40°C. После этого раствор оставляли нагреваться до комнатной температуры, промывали реакционный раствор последовательно 200 мл 2N серной кислоты, 300 мл 20%-го раствора гидросульфита натрия и насыщенного раствора поваренной соли. Органическую фазу сушили над сульфатом магния, концентрировали в вакууме и остаток чистили над силикагелем смесью н-гептан/этиловый эфир уксусной кислоты 3/1. Получили 4-[5-(трет-бутилдиметилсиланилокси)-5-(4-фторфенил)-1-(4-метоксифенил)-2-(2-оксо-4-фенилоксазолидин-3-карбонил)пентиламино]бензонитрил (6) с молекулярным весом 707,93 (C41Н46FN3O5Si); МС (ESI): 590,51 (MH+-C7H5N2). c) 4-[3-[3-(трет-Бутилдиметилсиланилокси)-3-(4-фторфенил)пропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензонитрил (7) 13,2 г 4-[5-(трет-бутилдиметилсиланилокси)-5-(4-фторфенил)-1-(4-метоксифенил)-2-(2-оксо-4-фенилоксазолидин-3-карбонил)пентиламино]бензонитрила растворяли в 380 мл метил-трет-бутилового эфира, смешивали с 18,6 мл N,O-бис(триметилсилил)ацетамида и 1,86 мл 1M раствора тетрабутиламмонийфторида в тетрагидрофуране и перемешивали 2 часа при комнатной температуре. По окончании реакции добавляли 10 мл уксусной кислоты, концентрировали реакционную смесь в вакууме и чистили осадок над силикагелем смесью толуол/этиловый эфир уксусной кислоты 50/1. Получили 4-[3-[3-(трет-бутилдиметилсиланилокси)-3-(4-фторфенил)пропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензонитрила (7) с молекулярным весом 544,75 (С32Н37FN2O3Si); МС (ESI): 545,56 (M + H+). d) 4-[3-[3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензонитрил (8) 3,5 г 4-[3-[3-(трет-бутилдиметилсиланилокси)-3-(4-фторфенил)пропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензонитрила растворяли в 65 мл тетрагидрофурана, смешивали с 0,74 мл уксусной кислоты и 8,03 мл 1M раствора тетрабутиламмонийфторида в тетрагидрофуране и 2 часа перемешивали при комнатной температуре. После этого добавляли 4,82 мл раствора тетрабутиламмонийфторида и перемешивали еще 3 часа при температуре обратного холодильника. Охлажденную реакционную смесь концентрировали в вакууме и остаток чистили с помощью хроматографии над силикагелем смесью н-гептан/этиловый эфир уксусной кислоты 2/1. Получили 4-[3-[3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензонитрил (8) с молекулярным весом 430,48 (C26H23FN2O3); МС (ESI): 431,24 (M + H+). e) 4-[3-[3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]-N-гидроксибензамидин (9) К раствору из 200 мг 4-[3-[3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензонитрила (8) и 0,45 мл триэтиламина в 15 мл изопропанола добавляли 199 мг гидроксиламмонийгидрохлорида и 12 часов перемешивали при комнатной температуре. Реакционный раствор дважды экстрагировали смесью этиловый эфир уксусной кислоты/вода. Органическую фазу сушили над сульфатом магния и концентрировали. Получили продукт с молекулярным весом 463,51 (C26H26F1N3O4); МС (ESI) 464,19 (M + H+). Пример IV 4-[3-[3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензамидин (10)
40 мг 4-[3-[3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]-N-гидроксибензамидина растворяли в 10 мл тетрагидрофурана и гидрировали 1 мл концентрированного аммиака над никелем Ренея 6,5 часов при 25°C. После добавления сульфата магния реакционный раствор фильтровали. Фильтрат концентрировали и разделяли с помощью ВЭЖХ (Knauer Eurospher-100-10-C18, вода (0,1% трифторуксусной кислоты)/ацетонитрил (0,1% трифторуксусной кислоты)=80/20 Пример V 4-{2-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-1-ил}-N-гидроксибензамидин (12)
a) 4-[3-[3-(4-Фторфенил)-3-гидроксипропил]-2-(4-фторфенил)-4-оксоазетидин-1-ил]бензонитрил (11) Соединение (11) получали аналогично соединению примера IIId, с той разницей, что вместо 4-[(4-метоксифенилимино)метил]бензонитрила использовался 4-[(4-фторбензилиден)амино]бензонитрил. b) 4-{2-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-1-ил}-N-гидроксибензамидин (12) К раствору из 280 мг 4-[3-[3-(4-фторфенил)-3-гидроксипропил]-2-(4-фторфенил)-4-оксоазетидин-1-ил]бензонитрила (11) и 0,65 мл триэтиламина в 15 мл изопропанола добавляли 279 мг гидроксиламмонийгидрохлорида и перемешивали 12 часов при комнатной температуре. Реакционный раствор дважды экстрагировали смесью этиловый эфир уксусной кислоты/вода. Органическую фазу сушили над сульфатом магния и концентрировали. Получили продукт с молекулярным весом 451,48 (C25H23F2N3О3); МС (ESI) 452,10 (M+H+). Пример VI 4-{2-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-1-ил}бензамидин (13)
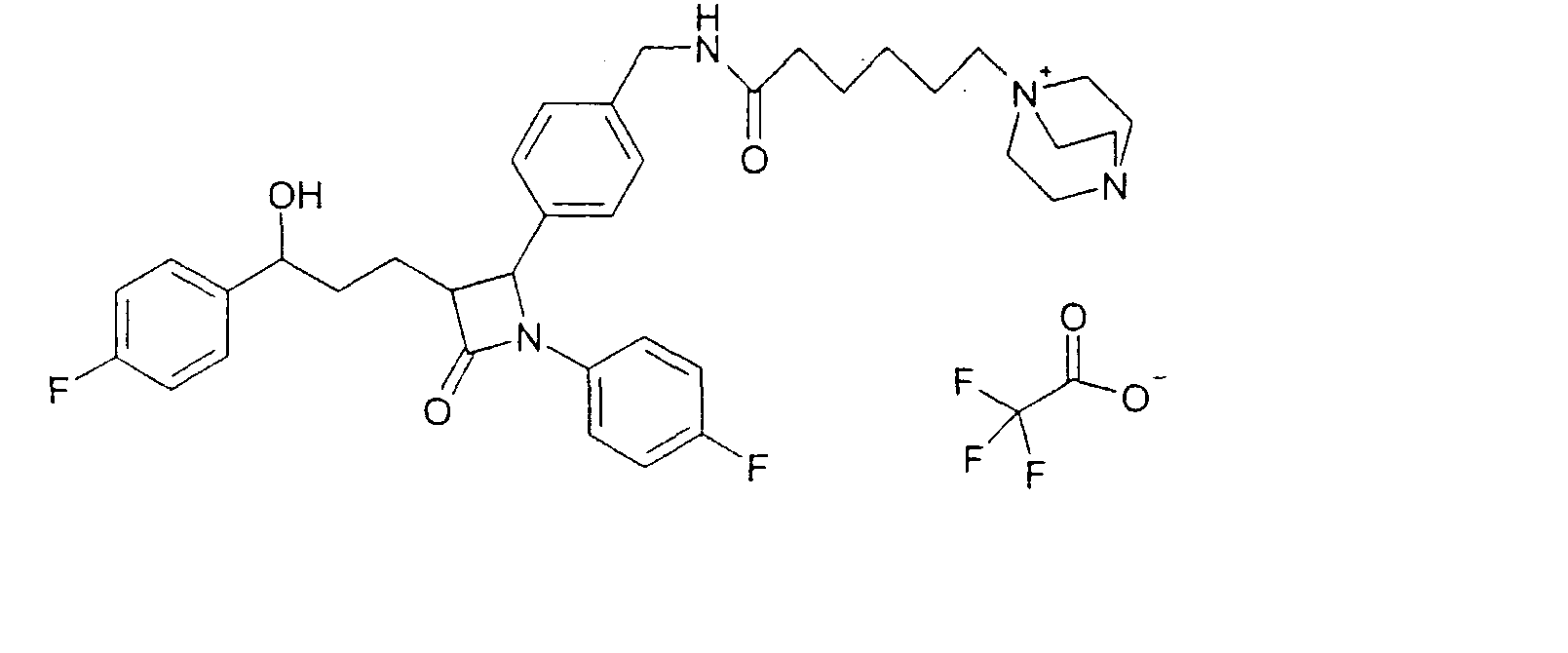
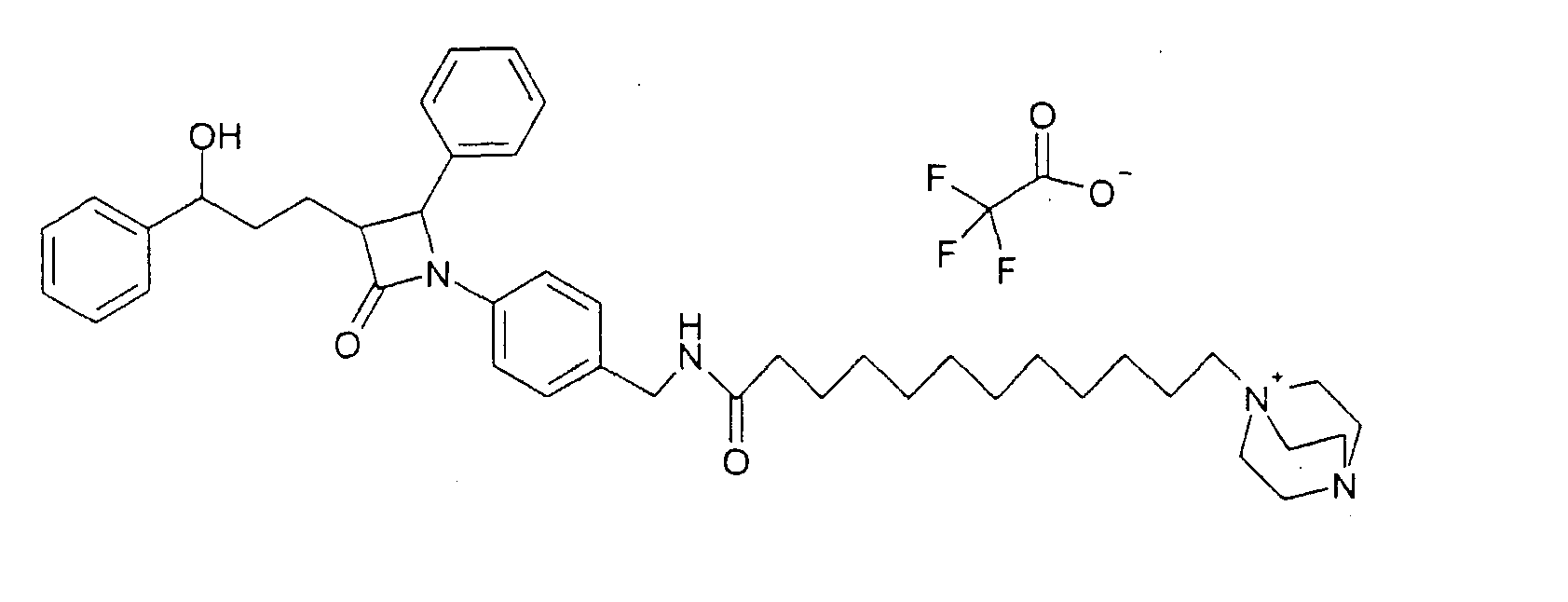
290 мг 4-{2-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-1-ил}-N-гидроксибензамидина (12) растворяли в 15 мл тетрагидрофурана и гидрировали 1,5 мл концентрированного аммиака 6,5 часов при 25°C над никелем Ренея. После добавления сульфата магния реакционный раствор фильтровали. Фильтрат концентрировали. Получили продукт с молекулярным весом 435,48 (C25H23F2N3O2); МС (ESI) 436,18 (M + H+) Пример VII Трифторацетат 1-[5-(4-{1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензилкарбамоил)пентил]-4-аза-1-азониабицикло[2.2.2]октана (15)
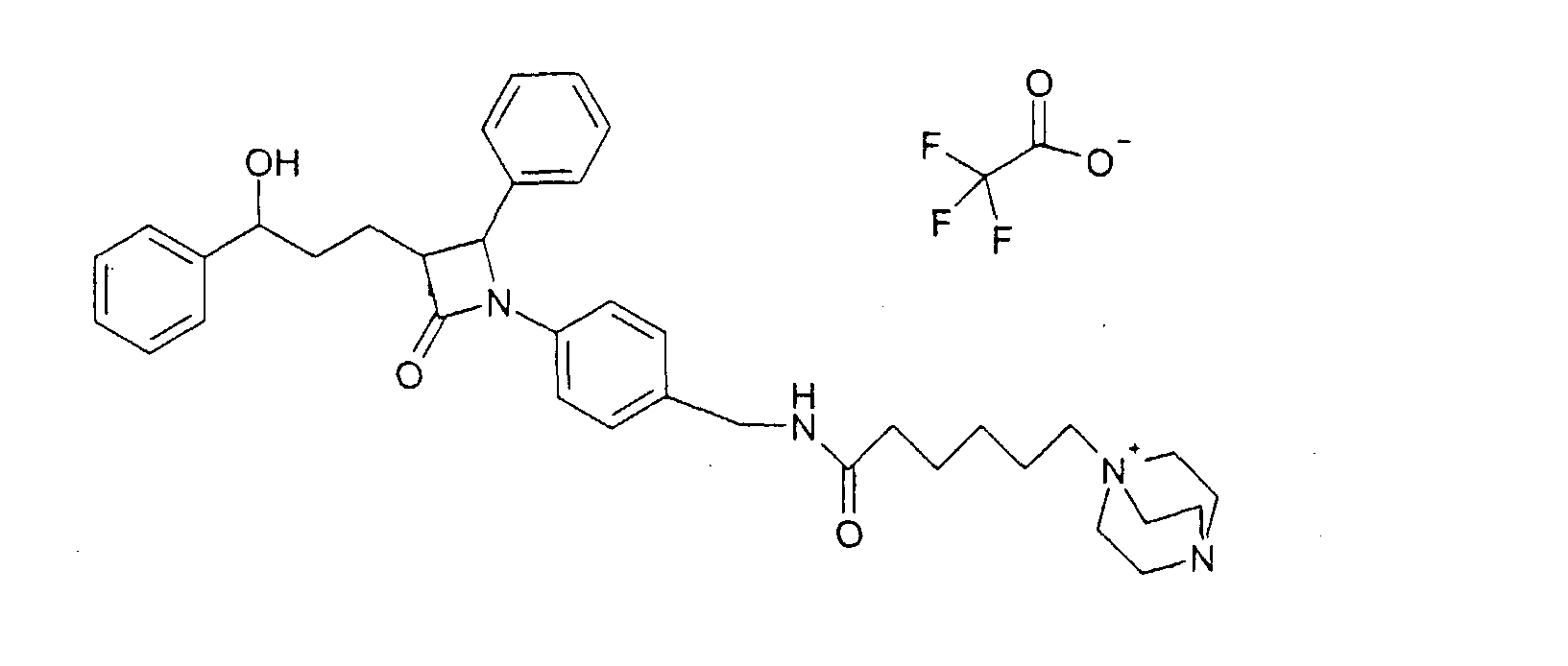
a) Бромид 1-(5-карбоксипентил)-4-аза-1-азониабицикло[2.2.2]октана (14) К раствору 1,5 г 1,4-диазабицикло[2.2.2]октана в 10 мл диметилсульфоксида добавляли при 70°C 1,0 г 6-бромгексановой кислоты в 5 мл диметилсульфоксида. Через 1 час прибавляли 100 мл воды и сушили вымораживанием. Остаток обрабатывали ацетоном. Остаток содержал продукт с молекулярным весом 227,33 (катион: C12H23N2O2 +); МС (ESI) 227,1 (M+). b) Трифторацетат 1-[5-(4-{1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензилкарбамоил)пентил]-4-аза-1-азониабицикло[2.2.2]октана (15) К раствору из 76 мг бромида 1-(5-карбоксипентил)-4-аза-1-азониабицикло[2.2.2]октана (14), 64 мкл диизопропилкарбодиимида и 56 мг гидроксибензотриазола в 2 мл диметилформамида добавляли раствор из 70 мг 4-(4-аминометилфенил)-1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]азетидин-2-она и 23 мкл триэтиламина в 0,5 мл диметилформамида и перемешивали 12 часов при комнатной температуре. Реакционный раствор концентрировали и разделяли с помощью ВЭЖХ (Knauer, колонка Eurospher-100-10-C18, вода (0,1% трифторуксусной кислоты)/ацетонитрил (0,1% трифторуксусной кислоты)=80/20 Пример VIII Трифторацетат 1-[5-(4-{3-[3-гидрокси-3-фенилпропил]-2-оксо-4-фенилазетидин-1-ил}бензилкарбамоил)пентил]-4-аза-1-азониабицикло[2.2.2]октана (17)
a) 1-(4-Аминометилфенил)-3-[3-гидрокси-3-фенилпропил]-4-фенилазетидин-2-он (16) Соединение (16) получали, как описано в примере IIIa-d, с той разницей, что вместо 4-[(4-метоксифенилимино)метил]бензонитрила применяли 4-(бензилиденамино)бензонитрил, а вместо 3-[5-(4-фторфенил)-5-гидроксипентаноил]-4-фенилоксазолидин-2-она применяли 3-[5-фенил-5-гидроксипентаноил]-4-фенилоксазолидин-2-он, и что продукт IIId подвергали восстановлению с никелем Ренея. b) Трифторацетат 1-[5-(4-{3-[3-гидрокси-3-фенилпропил]-2-оксо-4-фенилазетидин-1-ил}бензилкарбамоил)пентил]-4-аза-1-азониабицикло[2.2.2]октана (17) Синтез осуществляли аналогично примеру VIIb, исходя из 60 мг 1-(4-аминометилфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-фенилазетидин-2-она. Получили продукт с молекулярным весом 595,81 (катион: C37H47N4O3); МС (ESI) 595,36 (M+). Пример IX Трифторацетат 1-[11-(4-{1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензилкарбамоил)ундецил]-4-аза-1-азониабицикло[2.2.2]октана (19)
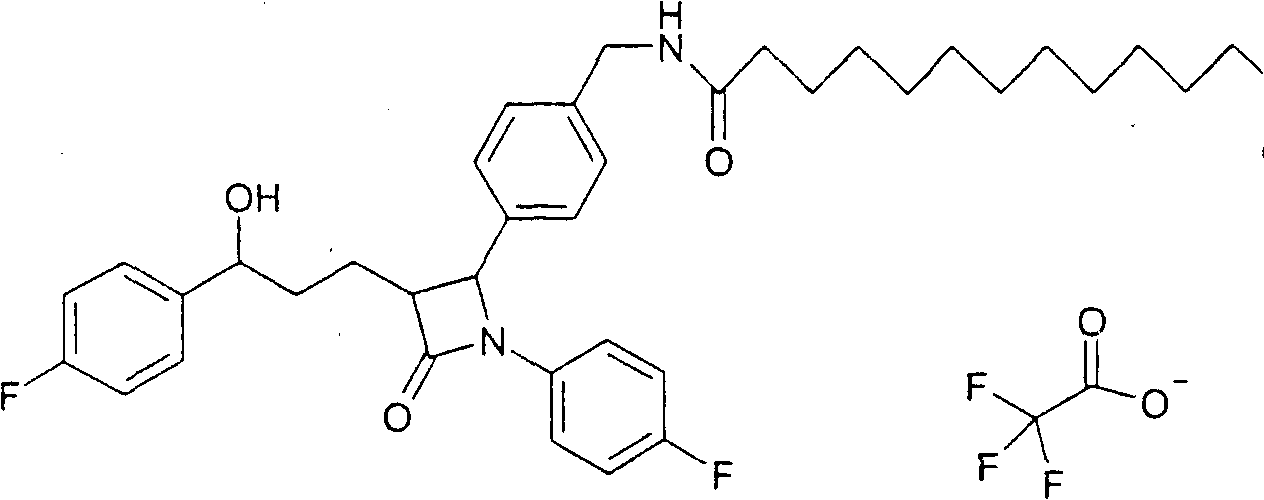
a) Бромид 1-(11-карбоксиундецил)-4-аза-1-азониабицикло[2.2.2]октана (18) Синтез осуществляли аналогично примеру VIIa, исходя из 495 мг 12-бромдодекановой кислоты. Получили продукт с молекулярным весом 311,49 (катион: C18H35N2O2 +); МС (ESI) 311,2 (M+). b) Трифторацетат 1-[11-(4-{1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензилкарбамоил)ундецил]-4-аза-1-азониабицикло[2.2.2]октана (19) Синтез осуществляли аналогично примеру VIIb. Получили продукт с молекулярным весом 715,96 (катион: C43H57F2N4O3); МС (ESI) 715,43 (M+). Пример X Трифторацетат 1-(11-{4-[3-(3-гидрокси-3-фенилпропил)-2-оксо-4-фенилазетидин-1-ил]бензилкарбамоил}ундецил)-4-аза-1-азониабицикло[2.2.2]октана (20)
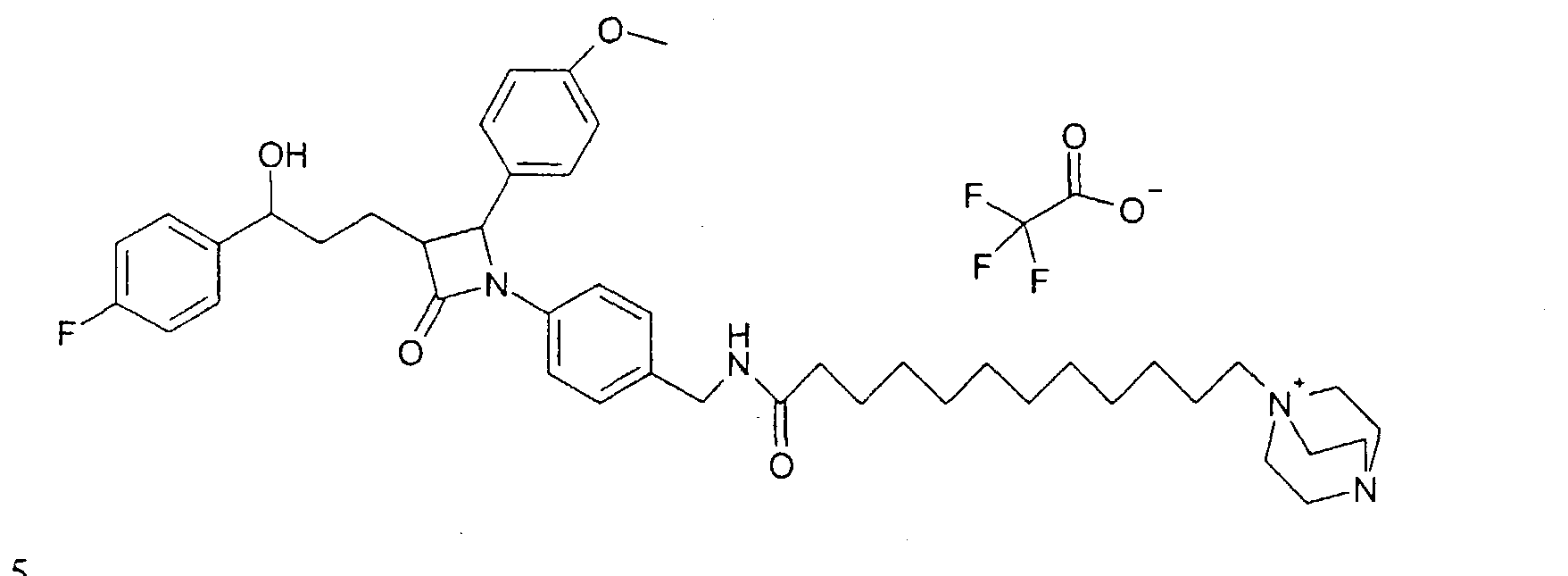
Синтез осуществляли аналогично примеру IXb, исходя из 74 мг 1-(4-аминометилфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-фенилазетидин-2-она. Получили продукт с молекулярным весом 679,97 (катион: C43H59N4O3); МС (ESI) 679,50 (M+). Пример XI Трифторацетат 1-(11-{4-[3-[3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензилкарбамоил}ундецил)-4-аза-1-азониабицикло[2.2.2]октана (21)
a) К раствору из 70 мг 12-бромдодекановой кислоты, 50 мг ЭДХ, 40 мг гидроксибензотриазола в 3 мл диметилформамида добавляли раствор из 50 мг 1-(4-аминометилфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-(4-метоксифенил)азетидин-2-она и 25 мл триэтиламина в 1 мл диметилформамида и перемешивали 1 час при комнатной температуре. Реакционный раствор разбавляли этилацетатом и трижды промывали водным раствором хлорида натрия. Органическую фазу фильтровали над силикагелем, концентрировали и разделяли путем флэш-хроматографии. Получили алкилбромид (72 мг) с молекулярным весом 695,72 (C38Н48BrFN2O4); МС (ESI) 695,4 (M+H+). b) 72 мг полученного ранее алкилбромида перемешивали вместе с 100 мг 1,4-диазабицикло{2.2.2]октана (DABCO) в 4 мл толуола в течение 20 часов при 100°C. Реакционный раствор концентрировали и разделяли путем ВЭЖХ (Knauer Eurospher-100-10-C18, вода (0,1% трифторуксусной кислоты)/ацетонитрил (0,1% трифторуксусной кислоты)=80/20 Пример XII N-{4-[3-(3-Гидрокси-3-фенилпропил)-2-оксо-4-фенилазетидин-1-ил]бензил}-N
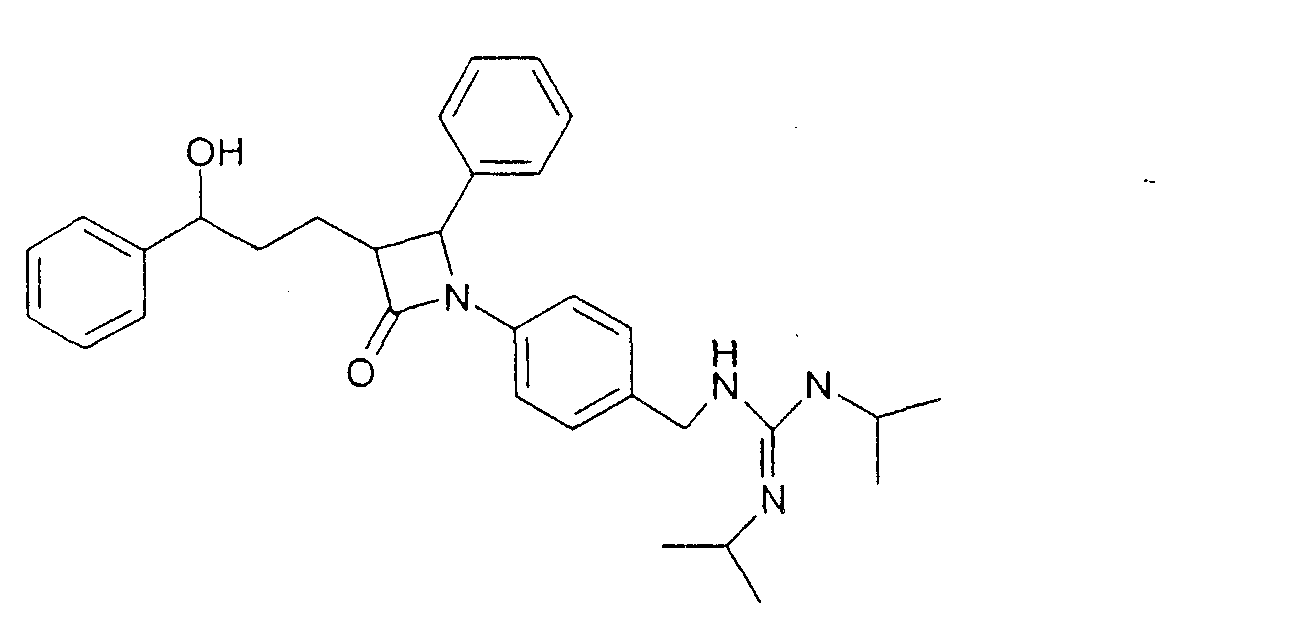
Раствор из 76 мг 1-(4-аминометилфенил)-3-[3-гидрокси-3-фенилпропил]-4-фенилазетидин-2-она, 64 мкл диизопропилкарбодиимида, 56 мг гидроксибензотриазола и 23 мкл триэтиламина в 2 мл диметилформамида перемешивали 22 часа при комнатной температуре. Реакционный раствор концентрировали и разделяли посредством ВЭЖХ (Knauer Eurospher-100-10-C18, вода (0,1% трифторуксусной кислоты)/ацетонитрил (0,1% трифторуксусной кислоты)=80/20 Пример XIII N-(3-{1-(4-Фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-4-оксоазетидин-2-ил}бензил)-N
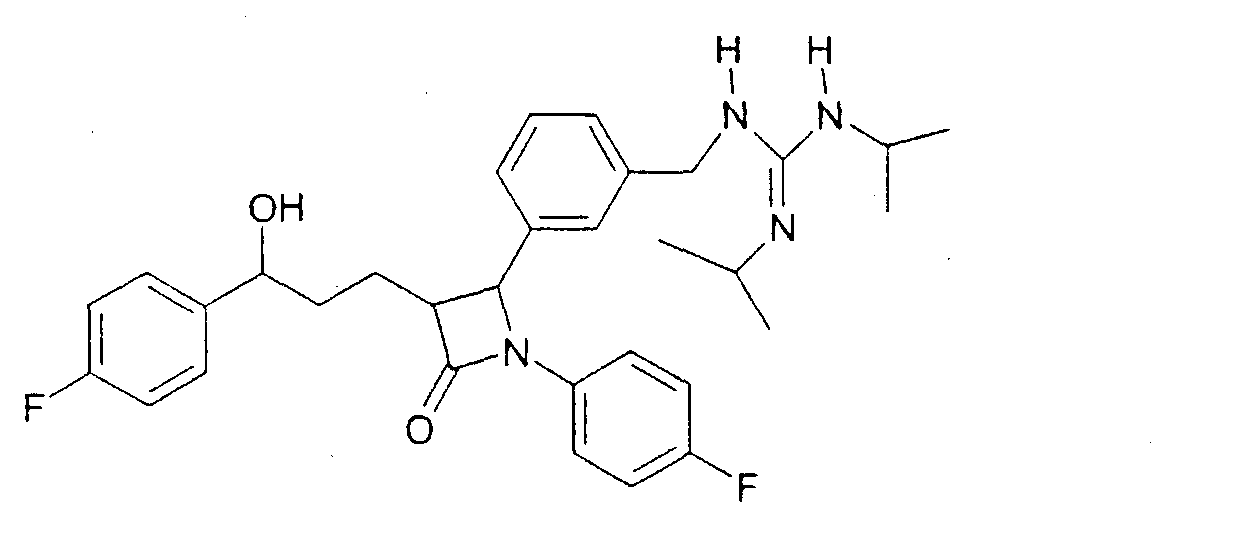
Синтез осуществляли аналогично примеру XII, исходя из 60 мг 4-(4-аминометилфенил)-1-(4-фторфенил)-3-[3-(4-фторфенил)-3-гидроксипропил]-азетидин-2-она. Получили продукт с молекулярным весом 548,68 (C32H38F2N4O2); МС (ESI) 549,4 (M+H+).
Соединения формулы I согласно изобретению проверялись на их действенность описываемыми далее методами. Влияние на поглощение холестерина + выделение 3H-таурохолевой кислоты посредством фекальных выделений мышей, крыс или хомяков Мыши линии NMRI, крысы линии Wistar или золотые сирийские хомячки (в группах из n=4-6) содержались при стандартной диете (альтромин, производства Lage (Липпе)) в клетках для изучения обмена веществ. Во второй половине дня перед получением радиоактивной метки (14C-холестерина) животных не кормили и выдерживали в клетке. Дополнительно за 24 часа перед пероральным приемом пробной пищи (14C-холестерин в Интралипиде® 20, Pharmacia-Upjohn) животных метили подкожно 3H-TCA (таурохолевая кислота) (например, от 1 мкКи на мышь до 5 мкКи на крысу). Тест на поглощение холестерина: 0,25 мл/мышь Интралипида® 20 (Pharmacia-Upjohn) (добавляя 0,25 мкКи 14C-холестерина в 0,1 мг холестерина) вводили перорально с помощью желудочного зонда. Испытываемые вещества раздельно вмешивали в смесь 0,5% метилцеллюлоза (Sigma)/5% солютол (BASF, Людвигсхафен) или в подходящее растворяющее вещество. Объем нанесения испытуемого вещества составлял 0,5 мл на мышь. Испытываемое вещество (интралипид с меткой 14C-холестерин) давалось непосредственно перед пробным приемом пищи (тест на поглощение холестерина). Экскременты собирали через 24 часа: определяли удаление 14C-холестерина и 3H-таурохолевой кислоты (TCA) через фекалии через 24 часа. Печень удаляли, гомогенизировали и аликвоту сжигали в Oximaten (модель 307, Паккард) для определения поглощенного/усвоенного количества 14C-холестерина. Результаты Пробы экскрементов Определяли полный вес, разбавляли водой до определенного объема, затем гомогенизировали, аликвоту высушивали и сжигали в Oximat (модель 307, Паккард для сжигания радиоактивно меченных проб): количество радиоактивных 3H-H2O и 14C-CO2 подсчитывалось приблизительно по осажденному количеству 3H-таурохолевой кислоты или 14C-холестерина (техника двойных изотопов). Значения ED200 интерполировались из кривой действия дозы как такая доза, которая удваивает выделение TCA или холестерина, по сравнению с одновременно обрабатываемой контрольной группой. Пробы печени Поглощенное количество 14C-холестерина в печени пересчитывалось на полученную дозу. Значения ED50 интерполировали из кривой действия дозы как такая доза, которая наполовину (50%) уменьшает поглощение 14C-холестерина в печени по сравнению с контрольной группой. Следующие значения ED50 подтверждают активность соединений формулы I.
Из вышеприведенного следует, что соединения формулы I обладают очень высокой активностью в отношении снижения холестерина. Всасываемость Всасываемость соединений формулы I проверялась на модели Caco-ячейки (A.R.Hilgers et al., Caco-2 cell monolayers as a model for drug transport across the intestinal mucosa, Pharm. Res. 1990, 7, 902). Из данных измерений следует, что соединения формулы I согласно изобретению по сравнению с соединениями, описанными в уровне техники (стандартная структура), имеют заметно меньшую всасываемость.
Стандартная структура: эзетимиб
Формула изобретения
1. Соединения формулы I
где означают R1, R2, R3, R4, R5, R6 независимо друг от друга (С0-С30)-алкилен-(LAG)n, где n равно 1, и причем один или несколько С-атомов алкиленового остатка могут быть заменены -О-, -(С=0)-, -СН=СН- или -NH-; Н, F, Cl, Br, I, (С1-С6)-алкил, O-(С1-С6)-алкил, (LAG)n означает моно- или трициклический триалкиламмонийалкильный остаток, выбранный из
или
где Alk1 означает линейный или разветвленный алкильный остаток с 1-20 атомами углерода, причем n, m и р независимо друг от друга могут быть 0-10, и одна или несколько СН2-групп алкиленового остатка независимо друг от друга могут быть заменены -О- или -NH-, и LAG может означать -(CH2)0-10-C(=NH)(NH2), -(CH2)0-10-C(=NH)(NHOH) или -NR7-C(=NR8)(NR9R10), причем всегда, по меньшей мере, один из остатков R1-R6 должен означать (С0-С30)-алкилен-(LAG)n, где n равно 1, причем один или несколько С-атомов алкиленового остатка могут быть заменены -О-, -(С=0)-, -СН=СН- или -NH-; R7, R8, R9 и R10 независимо друг от друга означают Н или (C1-С6)-алкил, а также их фармацевтически приемлемые соли. 2. Соединения формулы I по п.1, отличающиеся тем, что в них означают R2, R4, R5, R6 независимо друг от друга Н, F, Cl, Br, I, (С1-С6)-алкил, O-(С1-С6)-алкил, R1, R3 независимо друг от друга (С0-С30)-алкилен-(LAG), и причем один или несколько С-атомов алкиленового остатка могут быть заменены -O-, -(С=0)- или -NH-; Н, F, Cl, Br, I, (С1-С6)-алкил, (O-(С1-С6)-алкил, причем всегда, по меньшей мере, один из остатков R1 или R3 должен означать (С0-С30)-алкилен-(LAG), и причем один или несколько С-атомов алкиленового остатка могут быть заменены -О-, -(С=0)- или -NH-, а также их фармацевтически приемлемые соли. 3. Соединения формулы I по п.1 или 2, отличающиеся тем, что в них означают R2, R4, R5, R6 независимо друг от друга Н, F, Cl, Br, I, (С1-С6)-алкил, O-(С1-С6)-алкил, R1, R3 независимо друг от друга – (СН2)0-1-Y-W-(С0-С25)-алкилен-Y Н, F, Cl, Br, I, (С1-С6)-алкил, O-(С1-С6)-алкил, Y, W, Y или Y-W или Y причем всегда, по меньшей мере, один из остатков R1 или R3 должен означать -(CH2)0-1-Y-W-(C0-C25)-алкилен-Y а также их фармацевтически приемлемые соли. 4. Соединения формулы I по одному или нескольким пп.1-3, отличающиеся тем, что LAG означает трициклический триалкиламмонийалкильный остаток, определенный в п.1, а также его фармацевтически приемлемые соли. 5. Лекарственное средство для снижения уровня холестерина в сыворотке, содержащее одно или несколько соединений по любому из пп.1-4. 6. Способ получения лекарственного средства для снижения уровня холестерина в сыворотке, содержащего одно или несколько соединений по одному или нескольким пп.1-4, отличающийся тем, что активное вещество смешивают с фармацевтически приемлемым носителем и эту смесь переводят в подходящую для введения форму.
|
||||||||||||||||||||||||||||||||||
 С-, -N((C1-С6)-алкил)-, -N(фенил)-, -N((C1-С6)-алкилфенил)-, -N(CO-(CH2)1-10-COOH)- или -NH-,
С-, -N((C1-С6)-алкил)-, -N(фенил)-, -N((C1-С6)-алкилфенил)-, -N(CO-(CH2)1-10-COOH)- или -NH-, -W
-W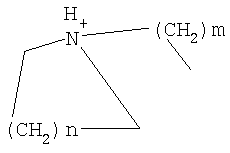 или
или 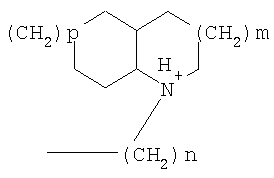 или
или 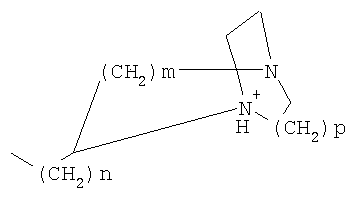
 или
или 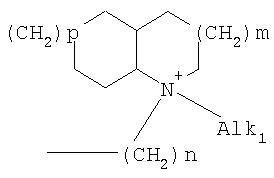 или
или 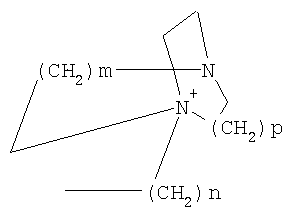
 или
или 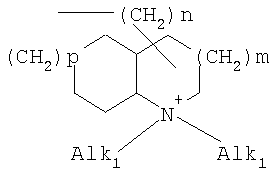
 или
или 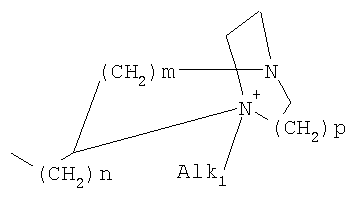
 -глюкозидазы, таким как, например, миглитол или акарбоза.
-глюкозидазы, таким как, например, миглитол или акарбоза. 3-агонистами, антагонистами MCH (меланоцитконцентрирующих гормонов), агонистами CCK, ингибиторами восстановления серотонина, смешанными серотонинергическими и норадренергическими соединениями, агонистами 5HT, агонистами бомбезина, антагонистами галанина, гормоном роста, соединениями, выделяющими гормоны роста, агонистами TRH, 2- и 3-модуляторами, расцепляющими белок, агонистами лептина, агонистами DA (бромокриптин, допрексин), ингибиторами липазы/амилазы, модуляторами PPAR, модуляторами RXR или агонистами TR-
3-агонистами, антагонистами MCH (меланоцитконцентрирующих гормонов), агонистами CCK, ингибиторами восстановления серотонина, смешанными серотонинергическими и норадренергическими соединениями, агонистами 5HT, агонистами бомбезина, антагонистами галанина, гормоном роста, соединениями, выделяющими гормоны роста, агонистами TRH, 2- и 3-модуляторами, расцепляющими белок, агонистами лептина, агонистами DA (бромокриптин, допрексин), ингибиторами липазы/амилазы, модуляторами PPAR, модуляторами RXR или агонистами TR-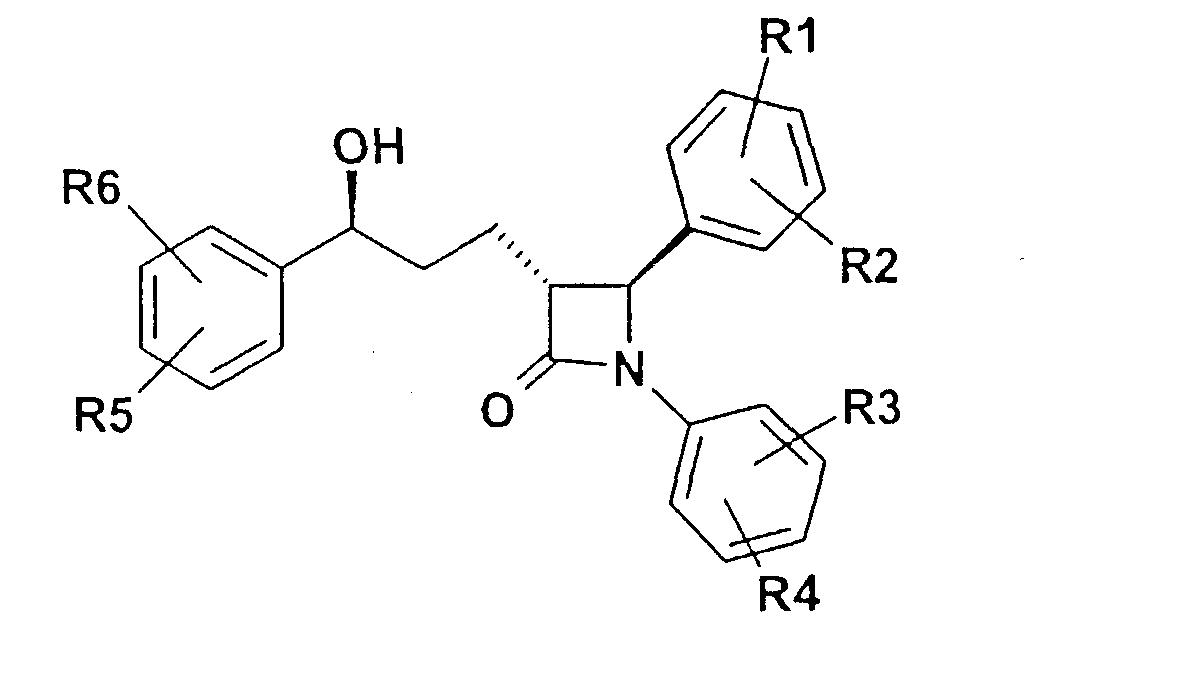
 10/90). Получили продукт с молекулярным весом 435,48 (C25H23F2N3О2); МС (ESI) 436,18 (M + H+)
10/90). Получили продукт с молекулярным весом 435,48 (C25H23F2N3О2); МС (ESI) 436,18 (M + H+)