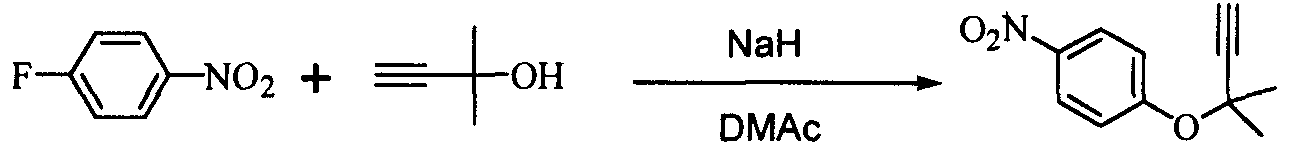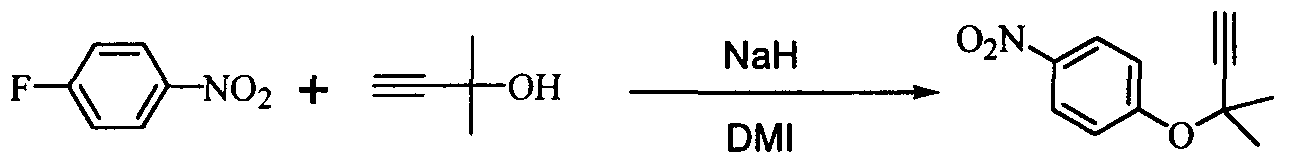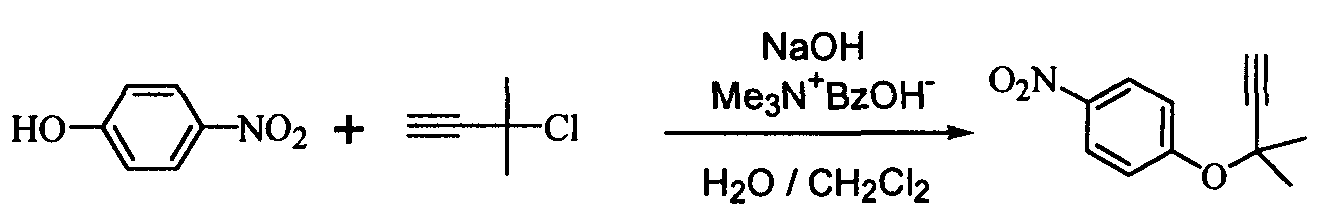Патент на изобретение №2315747
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ АЦЕТИЛЕНОВОГО СОЕДИНЕНИЯ
(57) Реферат:
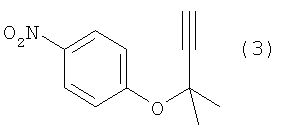
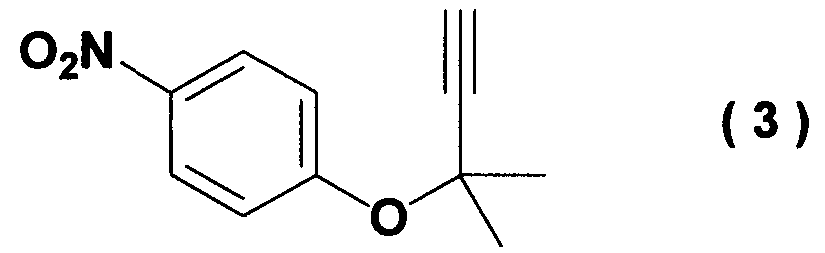
Изобретение относится к способу получения ацетиленового соединения, используемого в качестве промежуточного соединения при синтезе фармацевтических субстанций. Описывается способ получения ацетиленового соединения формулы (3)
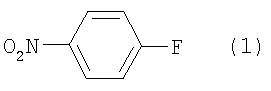
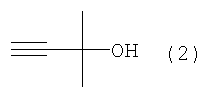
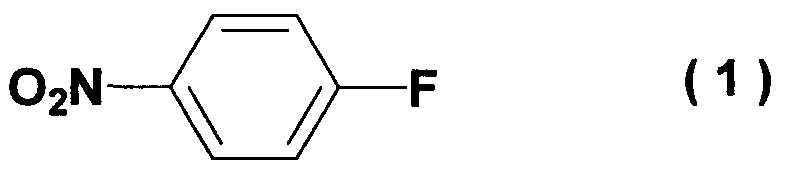
Область техники Настоящее изобретение относится к способу получения ацетиленового соединения из 4-нитрофторбензола и 2-метил-3-бутин-2-ола. Данное соединение используется в качестве промежуточного соединения для синтеза, например, антифибрилляторных агентов (см. JP-A-2001-151767) или гипотензивных агентов (см. J. Med. Chem., 1983, Vol.26, No.11, 1582-1589). Известный уровень техники В качестве известного способа получения ацетиленового соединения формулы (3) можно назвать способ, в котором 4-нитрофенол (соединение (5)) взаимодействует с 2-метил-3-бутин-2-хлоридом (соединение (6)) в присутствии основания (см., например, J. Med. Chem., 1983, Vol.26, No.11, p.1582 и JP-A-58-188880). Известен также способ, в котором 4-нитрофенол (соединение (5)) взаимодействует с 2-метил-3-бутин-2-хлоридом (соединение (6)) в присутствии катализатора йодида меди, йодида калия и карбоната калия (см., например, Synthesis, 1995, Vol.6, p.707). Кроме того, известен способ, в котором 4-нитрофенол (соединение (5)) взаимодействует с производным 2-метил-3-бутин-2-ола (соединение (7)) в присутствии медного катализатора и DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) (см., например, Tetrahedron Lett., 1994, Vol.35, p.6405). Кроме того, известен способ, в котором 4-нитрофенол (соединение (5)) взаимодействует с 2-метил-3-бутин-2-олом в присутствии трифенилфосфина и DEAD (диэтилазодикарбоксилата) (реакция Mitsunobu, см., например, Synth. Commun., 1989, vol.19, p.1255). Известен способ с использованием 4-нитрофторбензола в качестве исходного вещества, в котором 2-метил-3-бутин-2-ол используют вместо растворителя, и исходное вещество взаимодействует с калиевым алкоксидом 2-метил-3-бутин-2-ола (см., например, J. Org. Chem., 1972, vol.37, p.841).
(где Х означает Cl, -OCO2CH3 или -OCOCF3). При осуществлении способа, описанного в J. Med. Chem., 1983, vol.26, No.11, p.1582 и JP-A-58-188880, возникают некоторые проблемы, такие как низкий выход, и использование 2-метил-3-бутин-2-хлорида (соединение (6)), являющегося относительно нестабильным. Хотя способ, описанный в Synthesis, 1995, vol.6, p.707, дает увеличенный до 89% выход благодаря применению медного катализатора, он имеет некоторые недостатки, такие как удаление медного катализатора, являющегося тяжелым металлом, и применение большого количества йодида калия, о чем трудно сказать, что это недорого, в случае, когда применяют йодид калия, и т.п. Кроме того, остается проблема, связанная со стабильностью 2-метил-3-бутин-2-хлорида. Далее существует проблема с точки зрения стоимости и возможностей методики, такая как применение 2-метил-3-бутин-2-хлорида в количестве 2 моля на моль 4-нитрофторбензола (соединения (5)). Способ, описанный в Tetrahedron Lett., 1994, vol.35, p.6405, подобен вышеупомянутому способу, но дает выход 81% в большинстве случаев, когда используют 2-метил-3-бутин-2-хлорид; применение трифторацетата, которое обеспечивает наивысший выход (88%), является явно невыгодным с точки зрения затрат, и используемый DBU также дорог. Поэтому данный способ не пригоден в качестве промышленного способа. Способ, описанный в Synth. Commun., 1989, vol.19, p.1255, также не пригоден в качестве промышленного способа с точки зрения низкого выхода (45%), стоимости дорогого DEAD и т.п. Способ, описанный в J. Org. Chem., 1972, vol.37, p.841, может быть назван способом, превосходным по затратам и возможностям методики, поскольку он использует в качестве исходных материалов 2-метил-3-бутин-2-ол и 4-нитрофторбензол, которые относительно недороги и стабильны, и не использует катализаторы, такие как тяжелые металлы. Однако данный способ имеет недостатки, такие как низкий выход (35%), длительное время реакции (3 суток при комнатной температуре) и т.п. Сущность изобретения Для того чтобы решить вышеупомянутые проблемы, авторы настоящего изобретения подробно исследовали условия реакции между 4-нитрофторбензолом и алкоксидом 2-метил-3-бутин-2-ола. В результате этого был найден способ, который является превосходным по возможностям методики и дает целевые соединения с хорошим выходом, и в результате было осуществлено настоящее изобретение. Настоящее изобретение относится к способу получения ацетиленового соединения формулы (3)
отличающемуся взаимодействием 4-нитрофторбензола формулы (1)
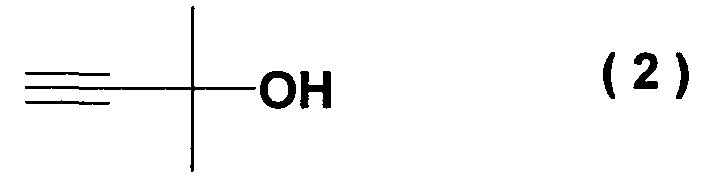
с алкоксидом 2-метил-3-бутин-2-ола формулы (2)
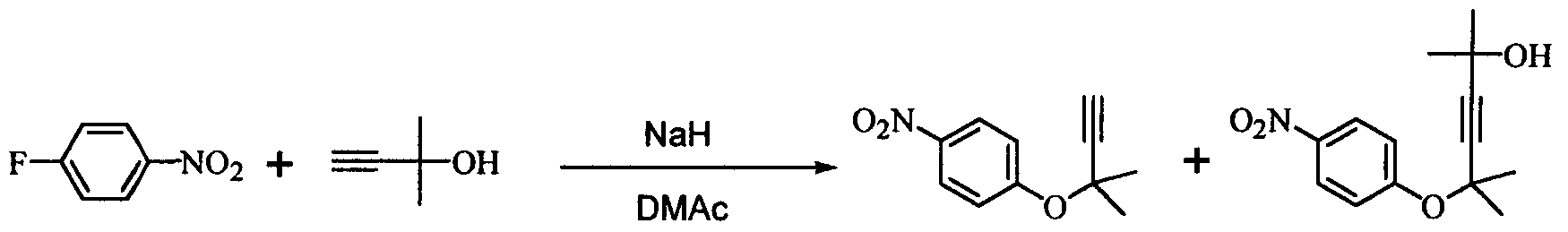
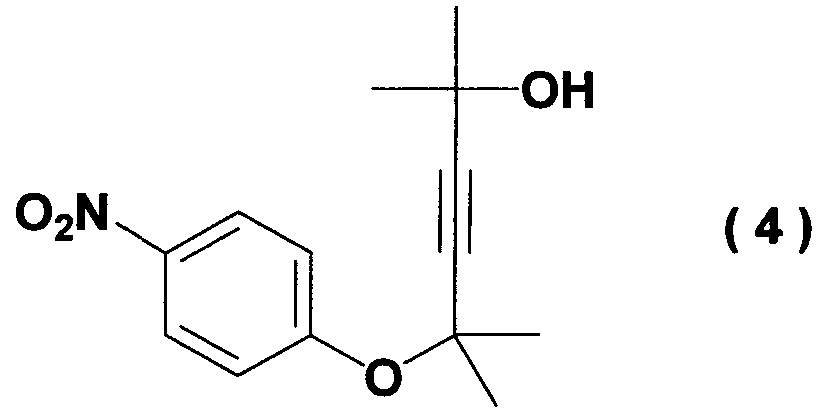
при температуре от -20 до 10°C. Кроме того, было найдено, что способ согласно настоящему изобретению может ингибировать образование побочного соединения (соединение (4)), которое не может быть легко удалено на последующих стадиях.
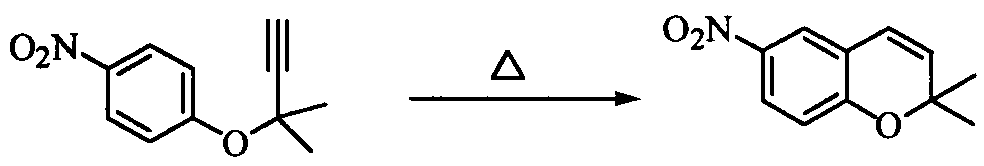
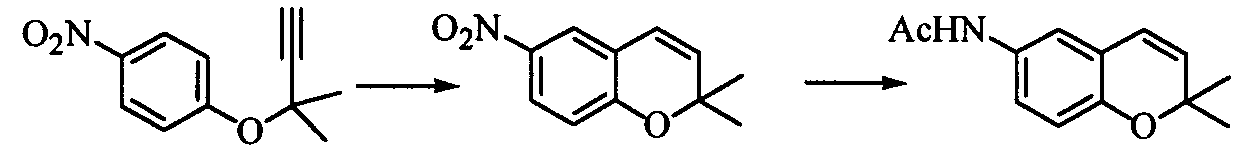
Наилучший способ осуществления изобретения Ниже поясняется способ получения ацетиленового соединения формулы (3). Ацетиленовое соединение формулы (3) может быть получено с хорошим выходом путем взаимодействия алкоксида 2-метил-3-бутин-2-ола формулы (2) с 4-нитрофторбензолом формулы (1) в растворителе при температуре от -20 до 10°C. В настоящем изобретении используют алкоксид 2-метил-3-бутин-2-ола формулы (2) так, как обычно используют алкоксиды металлов, и в качестве металла в алкоксиде металла предпочтительны щелочные металлы, такие как натрий, калий или литий, и т.п., и натрий является более предпочтительным с точки зрения легкости обращения и реакционной способности. Используемое количество алкоксида 2-метил-3-бутин-2-ола формулы (2) составляет от 0,5 до 20 молей на моль используемого количества 4-нитрофторбензола формулы (1). Между тем, поскольку выход снижается при используемом количестве 1 моль или менее, количество в 1 моль или более является предпочтительным, и количество от 1 до 3 молей является более предпочтительным с точки зрения затрат. Что касается методики реакции, предпочтительно добавлять по каплям 4-нитрофторбензол формулы (1) к раствору, состоящему из растворителя и алкоксида 2-метил-3-бутин-2-ола формулы (2). Время, требующееся для добавления по каплям, составляет предпочтительно от 0,5 до 5 часов, хотя оно не ограничено, до наступления быстрого роста температуры в реакционной системе и пока удерживается заданная температура. Растворители, используемые в настоящем изобретении, включают растворители типа амида, такие как N,N-диметилацетамид, N,N-диметилформамид, N-метилпирролидон, N,N’-диметилимидазилидинон или подобные, растворители типа ароматических углеводородов, таких как толуол, ксилол или подобные, растворители типа алифатических углеводородов, такие как гексан, гептан или подобные, растворители типа галогенсодержащих углеводородов, таких как дихлорметан, хлороформ или подобные, и смешанные или множественные растворители, упомянутые выше. Предпочтительные растворители включают растворители типа амида, более предпочтительно N,N-диметилацетамид и N,N’-диметилимидазилидинон, с точки зрения выхода ацетиленового соединения формулы (3). Используемое количество растворителя предпочтительно в 2 раза или более больше по массе количества 4-нитрофторбензола формулы (1) и, более предпочтительно, например, от 2 до 4 масс. раз или, например, от 2 до 3 масс. раз, с точки зрения затрат. Хотя температура реакции варьируется в интервале от -20 до 10°C, предпочтительным является интервал от -10 до 0°C с точки зрения удлинения времени реакции благодаря снижению температуры реакции и ингибированию образования побочного продукта формулы (4). И время реакции, и температура реакции не могут быть указаны в общем случае, так как они зависят от используемого количества алкоксида или подобного. Ацетиленовое соединение формулы (3) может быть получено в виде сырого продукта путем добавления воды с последующей экстракцией органическим растворителем, таким как толуол, промывки и затем отгонки растворителя. Сырой продукт может быть использован как таковой для получения бензопиранового промежуточного соединения, и он может быть очищен колоночной хроматографией или дистилляцией и т.п., если необходимо. Алкоксид 2-метил-3-бутин-2-ола формулы (2), являясь исходным веществом в настоящем изобретении, обычно может быть получен переработкой 2-метил-3-бутин-2-ола формулы (2) с гидридом металла, таким как гидрид натрия, гидрид калия или подобные, или с металлом, таким как металлический натрий, металлический калий, металлический литий или подобные. Ацетиленовое соединение формулы (3), полученное согласно настоящему изобретению, ведет к производным бензопирана, т.е. к промежуточному соединению для синтеза дефибриллирующего агента или гипотензивного агента препаративным методом, показанным в следующей схеме реакции:
Иначе говоря, ацетиленовое соединение формулы (3) может быть превращено в промежуточное соединение для синтеза вышеупомянутого дефибриллирующего агента или гипотензивного агента циклизацией его при нагревании для получения бензопиранового соединения и восстановлением, и ацетилированием полученного соединения. Побочный продукт формулы (4), образовавшийся при получении ацетиленового соединения формулы (3), не может быть полностью удален даже кристаллизацией вышеупомянутой ацетиламиновой формы. Поэтому для повышения эффективности последующего приготовления важно ингибировать образование побочного продукта формулы (4) при получении ацетиленового соединения формулы (3). Настоящее изобретение описано далее конкретно согласно примерам, которые не ограничивают изобретение. Во время опытов относительную площадь ВЭЖХ измеряли при следующих условиях анализа: Колонка: L-Column ODS Элюирующий раствор: от 0 до 45 мин CH3CN-0,01 M AcONH4 (45/55 об./об.); от 45 до 65 мин CH3CN-0,0 1 M AcONH4 (45/55 об./об от 65 до 85 мин CH3CN-0,01 M AcONH4 (95/5 об./об.). Детектирование: УФ (245 нм). Расход: 1 мл/мин. Температура колонки: 40°C. Пример 1
В 300 мл реакционную колбу, снабженную термометром, мешалкой и капельной воронкой, добавляли 96,0 г N,N-диметилацетамида (ДМА) и 11,6 г (290 ммоль) 60%-ного гидрида натрия (суспензия в минеральном масле) и добавляли по каплям при перемешивании и охлаждении льдом 25,2 г (300 ммоль) 2-метил-3-бутин-2-ола для получения алкоксида (время добавления по каплям: 2 часа). После перемешивания в течение 30 минут добавляли по каплям 33,8 г (240 ммоль) 4-нитрофторбензола (при перемешивании и охлаждении льдом; время добавления по каплям: 1,5 часа) и после завершения добавления по каплям полученную смесь перемешивали при той же температуре в течение 18 часов. К реакционной смеси добавляли 480 мл воды и 480 мл толуола и полученную смесь встряхивали. После отстаивания смесь разделяли на две фазы и толуольную фазу отбирали. Водную фазу вновь экстрагировали 240 мл толуола и полученную толуольную фазу объединяли с ранее полученной толуольной фазой, промывали 240 мл воды и затем отгоняли растворитель, получая сырой продукт (63,0 г) ацетиленового соединения формулы (3), являющегося целевым продуктом. Сырой продукт очищали колоночной хроматографией на силикагеле, получая 44,0 г (выход 90%) целевого продукта в виде желтого маслянистого продукта. 1Н-ЯМР (CDCl3) ч/млн: 8,18 (2Н, д, J=9,2 Гц), 7,30 (2Н, д, J=9,2 Гц), 2,68 (1Н, с), 1,73 (6Н, с). Пример 2 В 2 л реакционную колбу, снабженную термометром, мешалкой и капельной воронкой, добавляли 283 г N,N-диметилацетамида (ДМА), полученную смесь охлаждали до (-13)-(-12°C) и к ней добавляли 34,3 г (856 ммоль) 60%-ного гидрида натрия (суспензия в минеральном масле). Затем добавляли по каплям 74,5 г (886 ммоль) 2-метил-3-бутин-2-ола для получения алкоксида (внутренняя температура: (-10)-(-8°C), время добавления по каплям: 3,5 часа. После перемешивания в течение 1,5 часов добавляли по каплям 100 г (709 ммоль) 4-нитрофторбензола (внутренняя температура: (-10)-(-5°C), время добавления по каплям: 1,5 часа и после завершения добавления по каплям полученную смесь перемешивали при той же температуре в течение 38 часов. При условии ниже 10°C к реакционной смеси добавляли 1420 мл воды, после перемешивания в течение 1 часа к ней добавляли 1420 мл этилацетата и встряхивали; полученной смеси давали отстояться, разделяли на две фазы и этилацетатную фазу отбирали. Водную фазу экстрагировали 709 мл этилацетата и полученную этилацетатную фазу объединяли с ранее полученной этилацетатной фазой, промывали 709 мл воды и затем отгоняли растворитель, получая сырой продукт (177 г) ацетиленового соединения формулы (3), являющегося целевым продуктом. Относительные проценты площади при ВЭЖХ ацетиленового соединения формулы (3) и побочного продукта формулы (4) после реакции в течение 38 часов и в сыром продукте приведены в таблице 1.
Ссылочный пример 1
В 1 л реакционную колбу, снабженную термометром, мешалкой, конденсатором Димрота и капельной воронкой, добавляли 162 г о-дихлорбензола, нагретого до 170°C, и на протяжении 3 ч 40 мин туда добавляли по каплям все количество сырого продукта ацетиленового соединения формулы (3), полученного в примере 2, растворенного в 186 г о-дихлорбензола (внутренняя температура: 168-176°C). После завершения добавления по каплям полученную смесь перемешивали при той же температуре в течение 1 часа и отгоняли растворитель, получая 169 г сырого продукта 2,2-диметил-6-нитро-2Н-1-бензопирана, являющегося целевым продуктом. Сырой продукт растворяли при нагревании в смешанном растворителе из 317 г метанола и 56 г воды и постепенно охлаждали до 2°C, а затем подвергали кристаллизации в течение 2 ч 30 мин при температуре от 0 до 5°C. Полученные кристаллы отбирали фильтрованием и промывали, затем сушили при пониженном давлении при 50°C, получая 137 г (выход: 94%) 2,2-диметил-6-нитро-2Н-1-бензопирана. Поскольку кристаллы были загрязнены минеральным маслом гидрида натрия и тому подобное, они были подвергнуты определению с внутренним стандартом и имели чистоту 89,4%. Следовательно, выход в совокупности двух стадий составил 84%. Значения физических свойств образца, полученного очисткой колоночной хроматографией с силикагелем, были следующими: Т.пл.: 74,6-74,7°C 1Н-ЯМР (CDCl3) ч/млн: 8,02 (1Н, дд, J=8,9, 2,8 Гц), 7,89 (1Н, д, J=2,8 Гц), 6,81 (1Н, д, J=8,9 Гц), 6,36 (1Н, д, J=9,9 Гц), 5,75 (1Н, д, J=9,9 Гц), 1,48 (6Н, с). Примеры 3-5 (Влияние температуры реакции и количества растворителя) Реакцию проводили в тех же условиях, как в примере 2, за исключением температуры реакции и количества растворителя, которые были изменены. Относительные проценты площади при ВЭЖХ ацетиленового соединения формулы (3) и побочного продукта формулы (4) после завершения реакции и в сыром продукте приведены в таблице 2. Во время опытов время реакции изменяли в зависимости от условий реакции. Кроме того, количество растворителя проведено в массовых частях на часть использованного количества 4-нитрофторбензола формулы (1) Для сравнения приведены также результаты примера 2.
Пример 6 (пример, в котором в качестве растворителя использован N,N’-диметилимидазолидинон (ДМИ))
В 300 мл реакционную колбу, снабженную термометром, мешалкой и капельной воронкой, добавляли 96,0 г N,N’-диметилимидазолидинон (ДМИ) и 11,6 г (290 ммоль) 60%-ного гидрида натрия (суспензия в минеральном масле) и добавляли по каплям при перемешивании и охлаждении льдом 25,2 г (300 ммоль) 2-метил-3-бутин-2-ола для получения алкоксида (внутренняя температура: 10°C или менее, время добавления по каплям: 1,5 часа, перемешивание в течение 30 мин после добавления по каплям). Затем добавляли по каплям 33,8 г (240 ммоль) 4-нитрофторбензола, и полученная смесь реагировала при перемешивании в течение 18 часов при охлаждении льдом. К реакционной смеси добавляли 480 мл толуола и 480 мл воды и полученную смесь встряхивали. После отстаивания смесь разделяли на две фазы и толуольную фазу отбирали. Водную фазу вновь экстрагировали 240 мл толуола и полученную толуольную фазу объединяли с ранее полученной толуольной фазой. Объединенную толуольную фазу сушили над безводным сульфатом натрия и фильтровали, затем отгоняли растворитель, получая сырой продукт ацетиленового соединения формулы (3), являющегося целевым продуктом. Сырой продукт подвергали колоночной хроматографии на силикагеле (500 г силикагеля, элюирующий раствор: этилацетат-гексан =1/20), получая 43,5 г (выход 89%) очищенного продукта ацетиленового соединения формулы (3) в виде желтого маслянистого продукта. Анализ ВЭЖХ показал, что относительный процент площади целевого продукта составил 91,2% и относительный процент площади 4-нитрофторбензола составил 7,9%. Поскольку 4-нитрофторбензол показал отношение чувствительности в 4 раза, корректированная ВЭЖХ чистота составила 97,9%. Сравнительный пример 1
В 1 л реакционную колбу, оборудованную термометром, мешалкой и капельной воронкой, добавляли 96 г N,N-диметилацетамида (ДМА), полученную смесь охлаждали до 3°C и добавляли к ней 11,6 г (289 ммоль) 60% гидрида натрия (суспензия в минеральном масле). Затем по каплям добавляли 25,2 г (299 ммоль) 2-метил-3-бутин-2-ола для получения алкоксида (внутренняя температура: от 3 до 10°C, время добавления по каплям: 1 час). Затем по каплям добавляли 33,8 г (239 ммоль) 4-нитрофторбензола (внутренняя температура: от 2 до 5°C, время добавления по каплям: 1,5 часа) и после завершения добавления по каплям полученную смесь перемешивали при 10-15°C в течение 20 часов. После этого, подобно методике примера 2, был получен сырой продукт ацетиленового соединения формулы (3) Относительные проценты площади ВЭЖХ ацетиленового соединения формулы (3) и побочного продукта формулы (4) после реакции в течение 18 часов и в сыром продукте приведены в таблице 3.
Сравнительный пример 2 (реакция между 4-нитрофенолом и 2-метил-3-бутин-2-хлоридом)
В 3 л реакционную колбу, снабженную термометром и мешалкой, добавляли 148 г (1,06 моль) нитрофенола и 1000 мл воды, добавляли и растворяли в этом водный раствор гидроксида натрия [NaOH 64,7 г (1,62 моль)/100 мл Н2О] (температура поднималась до 35°C благодаря экзотермической реакции). Затем последовательно добавляли 1100 мл дихлорметана, 166 г (1,62 моль) 2-метил-3-бутин-2-хлорида и 92,0 г гидроксида триметилбензиламмония (40% метанольный раствор), полученную смесь перемешивали при комнатной температуре в течение 5 суток. Перемешанному раствору давали отстояться, делили на 2 фазы и отбирали дихлорметановую фазу. Водную фазу экстрагировали 500 мл хлороформа и полученную хлороформную фазу объединяли с ранее полученной дихлорметановой фазой. Объединенную фазу промывали последовательно 1000 г 10% водного раствора гидроксида натрия, 700 мл воды и 500 мл воды, затем сушили над безводным сульфатом натрия. Полученный раствор подвергали фильтрованию и затем отгоняли растворитель, получая 71,3 г (выход 33%) ацетиленового соединения формулы (3), являющегося целевым продуктом, в виде темно-коричневого маслянистого продукта. Настоящее изобретение может обеспечить процесс получения ацетиленового соединения формулы (3), используемого как промежуточное соединение фармацевтических субстанций, из 4-нитрофторбензола, который является доступным и недорогим, промышленным и экономически выгодным путем. Применимость в промышленности Ацетиленовое соединение формулы (3), полученное согласно способу по настоящему изобретению, используется, например, в качестве промежуточного соединения для синтеза дефибриллирующего агента или гипотензивного агента и поэтому является полезным для фармацевтической промышленности и т.п.
Формула изобретения
1. Способ получения ацетиленового соединения формулы (3)
отличающийся взаимодействием 4-нитрофторбензола формулы (1)
с алкоксидом 2-метил-3-бутин-2-ола формулы (2)
при температуре от -20 до 10°С в растворителе. 2. Способ получения ацетиленового соединения по п.1, в котором в качестве растворителя используют растворитель типа амида. 3. Способ получения ацетиленового соединения по п.1 или 2, в котором количество растворителя по массе в 2 раза или более больше количества 4-нитрофторбензола.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
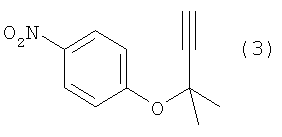 , отличающийся взаимодействием 4-нитрофторбензола с алкоксидом 2-метил-3-бутин-2-ола при температуре от -20 до 10°С в растворителе. Технический результат – способ приводит к получению целевого продукта с высоким выходом, промышленным и экономически выгодным путем. 2 з.п. ф-лы, 3 табл.
, отличающийся взаимодействием 4-нитрофторбензола с алкоксидом 2-метил-3-бутин-2-ола при температуре от -20 до 10°С в растворителе. Технический результат – способ приводит к получению целевого продукта с высоким выходом, промышленным и экономически выгодным путем. 2 з.п. ф-лы, 3 табл.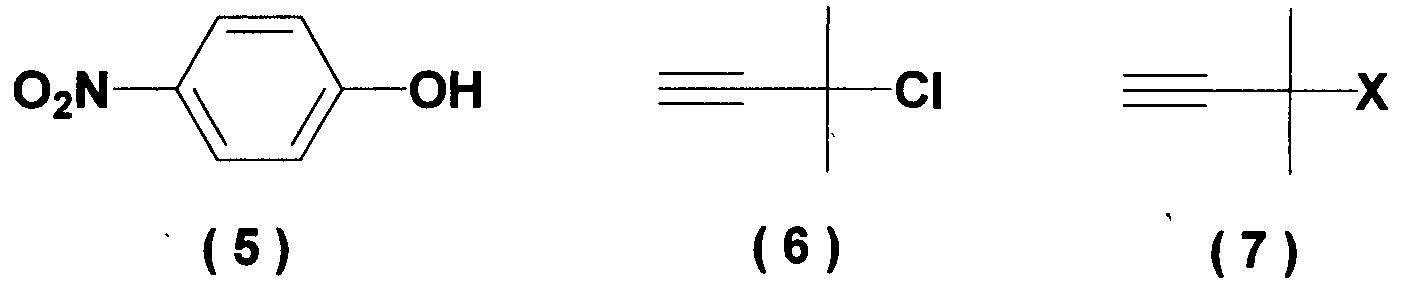
 4,6 (250 мм (произведена в Chemical Evaluation Research Institute, Japan);
4,6 (250 мм (произведена в Chemical Evaluation Research Institute, Japan); 95/5 об./об.);
95/5 об./об.);