Патент на изобретение №2315717
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ГЕКСАФТОРИДА УРАНА
(57) Реферат:
Изобретение относится к технологии получения и переработки гексафторида урана и направлено на исключение затрат тепла и потерь фтороводорода. Способ получения гексафторида урана осуществляют в замкнутом безотходном цикле, при котором закись-окись урана обрабатывают жидким фтороводородом, полученным гидролизом обедненного гексафторида урана, процесс обработки ведут во вращающемся реакторе при температуре 20-50°С и избыточном давлении до 150 КПа, а маточный раствор фтороводорода, полученный после фторирования закиси-окиси урана, используют при гидролизе обедненного гексафторида урана. Изобретение позволяет комплексно в замкнутом цикле решить две проблемы: осуществить фторирование U2O3 до UF4·2UO2F2 жидким HF, полученным гидролизом обедненного гексафторида урана, и, тем самым, сократить расход элементного фтора при последующем фторировании соли состава UF4·2UO2F2 вместо U2О3 на 44%, а также провести гидролиз обедненного гексафторида урана в среде маточного раствора HF со стадии фторирования U2O3.
Изобретение относится к технологии получения и переработки гексафторида урана. При получении гексафторида урана фторируют, как правило, закись-окись урана элементным фтором по реакции:
(Н.П.Галкин и др. Химия и технология фтористых соединений урана. М.: “Госатомиздат”, 1961. 173 с.). Известно предварительное фторирование U3O8 раствором плавиковой кислоты с получением UF4 и его последующим фторированием элементным фтором по реакции:
(Дж.Кац, Е.Рабинович. М.: “Иностранная литература”, 1954. 290 с.). Из-за сложности фильтрации UF4 из маточных растворов HF, содержащих растворенный UO2F2, и необходимости извлечения урана из фторидных маточных растворов данный способ не нашел применения. Наиболее близким по технической сути является способ фторирования U3O8 газообразным HF по реакции:
с последующим фторированием смеси фторсолей элементным фтором по реакции:
(Дж.Кац, Е.Рабинович. М.: “Иностранная литература”, 1954. 324 с., прототип). Реакция гидрофторирования протекает при высокой температуре и поддержании значительного избытка газообразного HF из-за его больших потерь, связанных с выделением воды и образованием паров разбавленной HF. Использование в рассмотренном способе 100% HF, являющегося товарным продуктом, также приводит к существенным материальным затратам, поэтому он не нашел промышленного применения. В то же время известна переработка обедненного (отвального) гексафторида урана гидролизом в среде 80-98% HF с образованием раствора фтороводорода, близкого к 100%
[Патент Великобритании №1075233. Усовершенствования, относящиеся к производству уранилфторида. МПК C01G 43/06. Опубликовано 12.07.67]. Фтористый водород, полученный в результате переработки обедненного UF6 путем его гидролиза в среде раствора HF, имеет ограниченное применение из-за необходимости его глубокой очистки от примеси урана, но, в тоже время, может быть использован для жидкофазного фторирования U3O8 без опасения загрязнения примесью урана:
При фторировании U3O8 жидким HF, в отношении масс Технической задачей, решаемой изобретением, является исключающее нагрев реакционной массы и потерю HF проведение жидкофазного фторирования закиси-окиси урана фтороводородом, полученным на стадии гидролиза отвального гексафторида урана, и использование раствора фтороводорода, образовавшегося на стадии жидкофазного фторирования закиси-окиси урана, для проведения гидролиза отвального гексафторида урана. Решение поставленной задачи достигается тем, что в способе получения гексафторида урана путем взаимодействия закиси-окиси урана с фтороводородом, последующего взаимодействия полученной смеси фторсолей с элементным фтором закись-окись урана обрабатывают жидким фтороводородом во вращающемся реакторе при температуре 20-50°С и избыточном давлении до 150 КПа. При этом для фторирования закиси-окиси урана используют фтороводород, полученный гидролизом обедненного гексафторида урана, а маточный раствор, полученный после фторирования закиси-окиси урана, используют при гидролизе обедненного гексафторида урана. В отличие от прототипа, при осуществлении которого для достижения полноты реакции гидрофторирования U3O8 используется нагрев до 500°С и значительный избыток газообразного HF, приводящий к образованию отходов в виде разбавленных растворов HF на стадии промывки печных газов с целью их обезвреживания, в предложенном способе, осуществляемом без нагрева, количество жидкого HF на стадию жидкофазного фторирования U3O8 строго не лимитируется, т.к. он выполняет кроме основного назначения транспортную роль жидкофазной среды при осуществлении процесса в замкнутом безотходном цикле и корректора концентрации раствора HF для осуществления гидролиза отвального UF6, что наглядно представлено реакциями (6); (5):
Из приведенных реакций следует, что для осуществления процесса фторирования U3O8 в замкнутом безотходном цикле требуется на 1 т U3О8 836 кг отвального гексафторида урана для связывания 85,5 кг воды, выделяющейся при осуществлении реакции фторирования U3O8 жидким фтороводородом. При этом концентрацию HF в маточном растворе после жидкофазного фторирования U3О8 можно варьировать в необходимых для последующего процесса гидролиза отвального UF6 пределах изменением оборотного количества HF при неизменном количестве воды, образующейся при протекании реакции взаимодействия U3O8 с HF. Пример исполнения: в цилиндрический реактор из нержавеющей спецстали объемом 300 мл загрузили 100 г U3O8 и прилили 200 мл 100% HF. Наблюдали незначительное вспенивание. Реактор загерметизировали. В течение 30 мин избыточное давление в реакторе возросло до 150 КПа, а температура до 50°С, после чего температура и давление стали снижаться и снизились в течение 30 мин до 22°С и 37 КПа соответственно. Реактор установили горизонтально на вращающиеся со скоростью 10 об/мин валки и вращали 30 мин, после чего вращение прекратили, установили реактор вертикально, подсоединили его к конденсатору. Нагрев верхней части реактора осуществляли рефлекторной лампой, а температуру в конденсаторе поддерживали минус 10°С. В начале температура в реакторе снизилась до 20°С, а абсолютное давление до 60 КПа (вакуум) за счет интенсивной конденсации в конденсаторе. Через 1 час температура в реакторе возросла до 50°С, после чего реактор разместили горизонтально на валки, установили в центральный патрубок реактора скользящий фторопластовый патрубок, соединенный с конденсатором, и, при вращении, продолжили нагрев рефлекторной лампой. В течение 1 часа температура стенки реактора возросла до 150°С. Нагрев и вращение прекратили, реактор вскрыли, выгрузили сыпучий негазящий продукт состава UF4·2UO2F2. Конденсат в объеме 180 мл с концентрацией 95,2% HF использовали для гидролиза обедненного гексафторида урана. После гидролиза обедненного UF6 концентрация HF в конденсате составила 99,8%, содержание примеси урана 0,15 г/л. Полученный HF был использован для фторирования U3O8 в последующем опыте. Данное изобретение позволяет комплексно, в замкнутом цикле, решить две проблемы, а именно осуществить фторирование U3O8 до UF4·2UO2F2 жидким HF, полученным гидролизом обедненного гексафторида урана, и, тем самым, сократить расход элементного фтора при последующем фторировании соли состава UF4·2UO2F2 вместо U3O8 на 44%, а также провести гидролиз обедненного гексафторида урана в среде маточного раствора HF со стадии фторирования U3O8. Из-за сложности утилизации содержащих примесь урана растворов HF как со стадии фторирования U3О8, так и со стадии гидролиза UF6 совместная реализация указанных способов экономически наиболее целесообразна.
Формула изобретения
Способ получения гексафторида урана путем взаимодействия закиси-окиси урана с фтористым водородом, последующего взаимодействия полученной смеси фторсолей с элементным фтором, отличающийся тем, что получение гексафторида урана осуществляют в замкнутом безотходном цикле, при котором закись-окись урана обрабатывают жидким фтороводородом, полученным гидролизом обедненного гексафторида урана, процесс обработки ведут во вращающемся реакторе при температуре 20-50°С и избыточном давлении до 150 кПа, а маточный раствор фтороводорода, полученный после фторирования закиси-окиси урана, используют при гидролизе обедненного гексафторида урана.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 21.02.2008
Извещение опубликовано: 27.12.2009 БИ: 36/2009
|
||||||||||||||||||||||||||
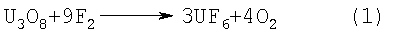
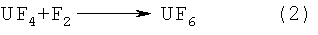
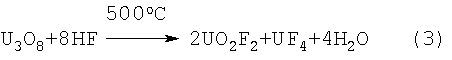
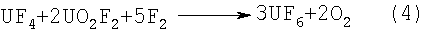


 1:2, в результате выделения воды образуется раствор 90-95% HF, который легко отделяется от осадка испарением. В сконденсированном 90-95% растворе HF содержание примеси урана находится в пределах 150÷200 мг/л. Полученную смесь фторсолей UF4·2UO2F2 после сушки направляют на фторирование элементным фтором, а конденсат 90-95% HF, незначительно загрязненный ураном, направляют на стадию гидролиза отвального гексафторида урана с целью извлечения
1:2, в результате выделения воды образуется раствор 90-95% HF, который легко отделяется от осадка испарением. В сконденсированном 90-95% растворе HF содержание примеси урана находится в пределах 150÷200 мг/л. Полученную смесь фторсолей UF4·2UO2F2 после сушки направляют на фторирование элементным фтором, а конденсат 90-95% HF, незначительно загрязненный ураном, направляют на стадию гидролиза отвального гексафторида урана с целью извлечения  фтора для дальнейшего использования его в сублиматно-разделительном цикле и перевода UF6 в безопасную форму хранения в виде соли UO2F2 или U3O8.
фтора для дальнейшего использования его в сублиматно-разделительном цикле и перевода UF6 в безопасную форму хранения в виде соли UO2F2 или U3O8.