Патент на изобретение №2315104
|
||||||||||||||||||||||||||
(54) СПОСОБ ОЧИСТКИ ДВУХЦЕПОЧЕЧНОЙ МОЛЕКУЛЫ ДНК, ПОДЛОЖКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СПОСОБ ИЗГОТОВЛЕНИЯ ПОДЛОЖКИ
(57) Реферат:
Изобретение относится к области биохимии и молекулярной биологии и может быть использовано в медико-биологической промышленности. Предложен способ очистки из смеси с другими компонентами двухцепочечной ДНК (дцДНК), включающей природную или искусственно введенную последовательность AGAAAAAAAGGA или AAGAAAAAAAAGAA. Способ по изобретению предусматривает а) пропускание раствора, содержащего указанную дцДНК через подложку, с которой ковалентно связан одноцепочечный олигонуклеотид с последовательностью (соответственно, ТСТТТТТТТССТ и ТТСТТТТТТТТСТТ), полностью комплементарной специфической последовательности, представленной в выделяемой дцДНК и образующей с ней стабильную триплексную структуру; б) элюирование целевой ДНК путем промывания подложки при щелочных значениях рН; и в) выделение дцДНК из элюата. Осуществление настоящего изобретения обеспечивает возможность получения значительных количеств фармацевтически чистой дцДНК, пригодной для использования, например, в генной терапии или в целях вакцинации. 3 с. и 17 з.п. ф-лы.
Родственные заявки Дата приоритета формулы изобретения настоящей заявки соответствует таковой заявки США S.N. 09/580923, поданной 26 мая 2000, которая является частичным продолжением заявки США S.N. 08/860038, поданной 9 июня 1997, которая, в свою очередь, соответствует поданной на национальной стадии в США 8 ноября 1995 заявки PCT FR 95/01468, содержание которой полностью включено в настоящее описание в виде ссылки. Основа изобретения Настоящее изобретение связано с новым способом очистки ДНК. Способ согласно изобретению дает возможность быстрой очистки фармакологически применяемой двухцепочечной ДНК. Более конкретно, способ очистки согласно изобретению включает в себя специфическую гибридизацию между последовательностью ДНК и олигонуклеотида. Технологии генной и клеточной терапии в настоящее время подвержены заметному развитию. Однако эти технологии сопряжены с необходимостью получения фармакологически чистой ДНК в больших количествах. И действительно, в этих новых способах терапии лекарственное средство зачастую состоит из ДНК как таковой, поэтому очень важно иметь возможность производить ее в соответствующих количествах, выделяя и очищая ее в соответствии с конкретной необходимостью ее терапевтического применения у человека. В настоящее время во многих публикациях была продемонстрирована возможность инъецирования плазмидной ДНК с целью генной терапии или вакцинации, что свидетельствует о том, что векторы экспрессии ДНК могут восприниматься различными типами клеток и впоследствии могут экпрессироваться гены, кодируемые этими плазмидами (Ledley, 1995 Hum. Gene Ther. 6, 1129). Гены, представляющие интерес для применения в генной терапии или при вакцинации, могут включать в себя, например, ген опухолевой супрессии, ген самоубийства или антисмысловые последовательности. Они могут кодировать также белки, такие как альфа-фетопротеин (AFP) (Morinaga, 1983, Proc. Natl. Acad. Sci. USA, 80, 4604), ферменты, гормоны, цитокины, факторы роста, такие как FGF (Jouanneau et al., 1991, Proc. Natl. Acad. Sci. USA, 88, 2893) или VEGFB (Olofsson B al., 1996, Proceedings 93, 576), факторы свертывания, такие как B-удаленный фактор VIII (Truett et al., 1985, DNA 4, 333), аполипопротеины, нейротрансмиттеры, нейротрофные факторы, природный или химерный иммуноглобулин. Также используются репортерные гены, такие как lacZ, кодирующий Основные требования при использовании плазмидной ДНК в качестве вектора для доставки генов у человека сводятся i) к производству и ii) к очистке данного лекарственного средства. В настоящее время развиты технологии получения плазмидных векторов с большим числом копий у Escherichia coli в качестве хозяев. Обычно используемыми плазмидами являются либо плазмиды, полученные из ColEl, такие как pBR322, pUC или pBluescript (Lahijani et al., 1996, Hum. Gene Ther., 7, 1971), либо плазмиды pCOR (Soubrier et al., 1999, Gene Therapy, 6, 1482). Вторым важным моментом, связанным с применением плазмидной ДНК в качестве вектора для генной терапии, является чистота плазмидного вектора как такового. Современные способы очистки, такие как ультрацентрифугирование в градиентах CsCI или хроматография, могут быть неэффективными в удалении примесей, таких как хозяйские геномные ДНК, или РНК, или белки. В частности, хозяйская геномная ДНК, химическая структура которой очень близка структуре плазмидной ДНК, исключительно трудно поддается удалению с помощью классической хроматографии. Обычно в препаратах плазмид, полученных с помощью методов классической хроматографии, обнаруживают хозяйскую геномную ДНК в концентрациях вплоть до 0,5-1%. Следовательно, для разработки препарата плазмидной ДНК в качестве безопасного вектора для применения в генной терапии человека необходимы технологии очистки, которые позволят снизить содержание хозяйской геномной ДНК до значительно более низких уровней, обычно до 0,1% или даже до 0,01% или ниже. В настоящем изобретении описывается простой и исключительно эффективный новый способ очистки ДНК. Благодаря этому способу становится возможным, в частности, получить особо высокую степень чистоты с высоким выходом. Способ согласно изобретению основан главным образом на специфическом взаимодействии между последовательностью, включенной в ДНК, которую подвергают очистке, и олигонуклеотидом, состоящим из природных или модифицированных оснований. В настоящее время показано, что олигонуклеотиды специфически взаимодействуют с широким желобком двойной спирали ДНК, образуя локально тройные спирали, что приводит к ингибированию транскрипции генов-мишеней (Helene et Toulme, Biochim. Biophys. Acta 1049 (1990) 99). Эти олигонуклеотиды выборочно распознают двойную спираль ДНК в олигопурин-олигопиримидиновых последовательностях, т.е. в областях, несущих олигопуриновые последовательности на одной цепи и олигопиримидиновые последовательности на комплементарной цепи, и там локально образуют тройную спираль. Основания тройной спирали (олигонуклеотид) образуют водородные связи (связи Hoogsteen, или обратимые связи Hoogsteen) с пуринами пар оснований Уотсона-Крика. На предшествующем уровне техники было описано использование взаимодействия такого типа для выделения плазмиды. Так, Ito et al. (PNAS 89 (1992) 495) описали применение биотинилированных олигонуклеотидов, распознающих конкретную последовательность плазмиды и образующих с ней локально тройную спираль. Затем образованные таким образом комплексы приводят в контакт с покрытыми стрептавидином магнитными бусинками. Затем взаимодействие между биотином и стрептавидином дает возможность выделить плазмиду путем магнитного отделения бусинок, сопровождаемого элюцией. Однако этот способ имеет некоторые недостатки. В частности, необходимо произвести два успешных этапа взаимодействий: первое – между олигонуклеотидом и плазмидой, а второе – между биотинилированным комплексом и стрептавидиновыми бусинками. Кроме того, конечный раствор может оказаться загрязненным биотинилированным олигонуклеотидом, а такой раствор не может быть использован в фармацевтической композиции. Краткое содержание изобретения В настоящем изобретении описан новый, улучшенный способ очистки ДНК с помощью такого типа взаимодействия. Более конкретно, в способе согласно изобретению используются олигонуклеотиды, ковалентно связанные с подложкой. Такой способ особенно быстр, и в результате получается особенно высокий выход продукции с повышенной степенью чистоты. Более того, он дает возможность очистить ДНК из сложных смесей, включающих в себя, в частности, другие нуклеиновые кислоты, белки, эндотоксины (такие как липополисахариды), нуклеазы и тому подобное. Используемые подложки могут, кроме того, быть легко циклизуемыми, и полученные ДНК могут обладать улучшенными свойствами фармацевтической безопасности. Наконец, этот способ, в отличие от способов, используемых в предшествующем уровне техники, выполняется всего лишь в одну стадию. Таким образом, первый объект изобретения связан со способом очистки двухцепочечной ДНК, в соответствии с которым раствор, содержащий указанную ДНК, смешанную с другими компонентами, пропускают через подложку, с которой ковалентно связан олигонуклеотид, образующий тройную спираль в результате гибридизации со специфической последовательностью, присутствующей в указанной ДНК. Специфической последовательностью может быть последовательность, естественным образом присутствующая в двухцепочечной ДНК, или же синтетическая последовательность, искусственно введенная в ДНК. Олигонуклеотиды, используемые в настоящем изобретении, являются олигонуклеотидами, которые напрямую гибридизуются с двухцепочечной ДНК. Эти олигонуклеотиды могут содержать следующие основания: – тимидин (T), который образует триплеты с дуплетами A-T двухцепочечной ДНК (Rajagopal et al., Biochem 28 (1989) 7859); – аденин (A), который образует триплеты с дуплетами A-T двухцепочечной ДНК; – гуанин (G), который образует триплеты с дуплетами G-C двухцепочечной ДНК; – протонированный цитозин (C+), который образует триплеты с дуплетами G-C двухцепочечной ДНК (Rajagopal et al., loc. cit.); – урацил (U), который образует триплеты с парами оснований A-U или A-T. Предпочтительно, чтобы используемый олигонуклеотид содержал богатую цитозином гомопиримидиновую последовательность, присутствующую в ДНК, и чтобы специфическая последовательность, присутствующая в ДНК, являлась гомопурин-гомопиримидиновой последовательностью. Присутствие цитозинов создает возможность существования тройной спирали, которая стабильна при кислых значениях рН, когда цитозины протонированы, и дестабилизируется при щелочных значениях pH, при которых цитозины нейтрализуются. Для того чтобы образование тройной спирали путем гибридизации стало возможным, важно, чтобы олигонуклеотид и специфическая последовательность, присутствующая в ДНК, были комплементарны друг другу. В этой связи, для получения наилучшего выхода и для наилучшей селективности в способе согласно изобретению используют олигонуклеотид и специфическую последовательность, которые полностью комплементарны друг другу. Это может быть, в частности, олигонуклеотид поли(CTT) и специфическая последовательность (GAA). В качестве примера можно привести олигонуклеотид последовательности 5′-GAGGCTTCTTCTTCTTCTTCTTCTT-3′ (GAGG(CTT)7; SEQ ID NO: 1), в котором основания GAGG не образуют тройную спираль, но дают возможность олигонуклеотиду разместиться на расстоянии от соединяющего плеча; может быть также указана последовательность (CTT)7(SEQ ID No: 26). Эти олигонуклеотиды образуют тройную спираль со специфической последовательностью, содержащей комплементарные единицы (GAA). Искомой последовательностью может быть, в частности, область, содержащая 7, 14 или 17 единиц GAA, как описано в Примерах. Другой последовательностью, представляющей особый интерес, является последовательность 5′-AAGGGAGGGAGGAGAGGAA-3′ (SEQ. ID NO: 5). Эта последовательность образует тройную спираль с олигонуклеотидами 5′-AAGGAGAGGAGGGAGGGAA-3′ (SEQ ID No: 6) или 5′-TTGGTGTGGTGGGTGGGTT-3′ (SEQ ID NO: 7). В этом случае олигонуклеотид связывается с полипуриновой цепью в антипараллельной ориентации. Эти тройные спирали являются стабильными только в присутствии Mg2+ (Vasquez et al., Biochemistry, 1995, 34, 7243-7251; Beal and Dervan, Science, 1991, 251, 1360-1363). Как указано выше, специфической последовательностью может являться последовательность, естественным образом присутствующая в двухцепочечной ДНК, или синтетическая последовательность, введенная в нее искусственным образом. Особенно предпочтительно использовать олигонуклеотид, образующий тройную спираль с последовательностью, естественным образом присутствующей в двухцепочечной ДНК, например, в сайте инициации репликации плазмиды или в маркерном гене. В этой связи, заявителем был предпринят анализ последовательностей, и было показано, что некоторые области этих ДНК, в частности, в сайте инициации репликации могут содержать гомопурин-гомопиримидиновые участки. Синтез олигонуклеотидов, образующих тройные спирали с этими природными гомопурин-гомопиримидиновыми участками, дают возможность применить способ согласно изобретению к немодифицированным плазмидам, в частности коммерческим плазмидам типа pUC, pBR322, pSV и т.п. Среди гомопурин-гомопиримидиновых последовательностей, естественным образом присутствующих в двухцепочечной ДНК, можно отметить последовательность, включающую в себя полностью или частично последовательность 5′-CTTCCCGAAGGGAGAAAGG-3′ (SEQ ID NO: 2), присутствующую в сайте инициации репликации плазмиды ColEl E.coli. В этом случае олигонуклеотид, образующий тройную спираль, содержит последовательность 5′-GAAGGGCTTCCCTCTTTCC-3′ (SEQ ID NO: 3) и альтернативно связывается с двумя цепями двойной цепи ДНК, как описано Beal и Dervan (J. Am. Chem. Soc. 1992, 114, 4976-4982) и Jayasena and Johnston (Nucleic Acids Res. 1992, 20, 5279-5288). Здесь можно также упомянуть последовательность 5′-GAAAAAGGAAGAG-3′ (SEQ ID NO: 4) гена Две дополнительные последовательности-мишени, которые могут образовывать структуры тройной спирали с отдельными олигонуклеотидами, были идентифицированы в сайтах инициации репликации в ColEl и в pCOR. Плазмиды, полученные из ColEl, содержат 12-членную гомопуриновую последовательность (5′- AGAAAAAAAGGA-3′) (SEQ ID NO: 27), картированную вверх по течению от транскрипта РНК-II, вовлеченного в репликацию плазмиды (Lacatena et al., 1981, Nature, 294, 623). Указанная последовательность образует стабильную триплексную структуру с 12-членным комплементарным олигонуклеотидом 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28). Остов pCOR содержит гомопуриновый участок (фрагмент секвенирования) из 14 неповторяющихся оснований (5′-AAGAAAAAAAAGAA-3′) (SEQ ID NO: 29), локализованных в A+T-обогащенном сегменте репликона Применение олигонуклеотида, образующего тройную спираль с последовательностью, присутствующей в сайте инициации репликации маркерного гена, является особенно благоприятным, поскольку создается возможность с помощью одного и того же олигонуклеотида очистить любую ДНК, содержащую указанный сайт инициации репликации указанного маркерного гена. Следовательно, нет необходимости модифицировать плазмиду или двухцепочечную ДНК, чтобы включить в нее искусственную специфическую последовательность. Несмотря на то что предпочтение отдается полностью комплементарным последовательностям, следует понимать, однако, что некоторое несоответствие между последовательностью олигонуклеотида и последовательностью, присутствующей в ДНК, может быть допустимым при условии, что оно не будет приводить к слишком большому снижению аффинности. Здесь может быть упомянута последовательность 5′- Согласно особому аспекту настоящего изобретения олигонуклеотиды согласно изобретению содержат последовательность (CCT)n, последовательность (CT)n или последовательность (CTT)n, где n представляет собой целое число между 1 и 15 включительно. Особенно предпочтительно применение последовательностей типа (CT)n или (CTT)n. В действительности заявителем было показано, что выход при очистке зависел от количества C в олигонуклеотиде. В частности, как показано в Примере 7, степень очистки на выходе увеличивается, когда олигонуклеотид содержит меньше цитозинов. Подразумевается, что олигонуклеотиды согласно изобретению могут содержать также комбинацию элементов (CCT), (CT) или (CTT). Используемые олигонуклеотиды могут быть природными (состоящими из немодифицированных природных оснований) или химически модифицированными. В частности, олигонуклеотид может предпочтительно содержать определенные химические модификации, придающие ему резистентность, или защищающие его от действия нуклеаз, или же повышающие его аффинность по отношению к специфической последовательности. Согласно настоящему изобретению под олигонуклеотидами подразумевается также любая родственная последовательность нуклеозидов, подвергнутая модификации остова с целью придания ей большей резистентности действию нуклеаз. К числу возможных модификаций можно отнести олигонуклеотидные фосфоротиоаты, которые могут образовывать тройные спирали с ДНК (Xodo et al., Nucleic Acids Res., 1994, 22, 3322-3330), а также олигонуклеотиды с формацетальным или метилфосфонатным остовами (Matteucci et al., J. Am. Chem. Soc., 1991, 113, 7767-7768). Возможно также использование олигонуклеотидов, синтезированных с Другой модификацией остова является фосфорамидатная связь. Например, среди таковых может быть упомянута межнуклеотидная фосфорамидатная связь N3′-P5′, описанная Gryaznov и Chen, которая дает олигонуклеотиды, которые образуют особенно стабильные тройные спирали с ДНК (J. Am. Chem. Soc., 1994, 116, 3143-3144). Среди других модификаций остова можно отметить также применение рибонуклеотидов 2′-O-метилрибозы, фосфодиэфира и т.д. (Sun and Hellene, Curr. Opinion Struct. Biol., 116, 3143-3144). Наконец, фосфорный остов может быть заменен на полиамидный остов, как в пептидно-нуклеиновых кислотах (PNA), которые также могут образовывать тройные спирали (Nielsen et al., Science, 1991, 254, 1497-1500; Kim et al., J. Am. Chem. Soc., 1993, 115, 6477-6481), или заменен на остов на основе гуанидина, как в дезоксирибонуклеиновом гуанидине (DNG, Proc. Natl. Acad. Sci. USA, 1995, 92, 6097-6101), или на поликатионные аналоги ДНК, которые также образуют тройные спирали. Тимин третьей цепочки может быть заменен на 5-бромурацил, который повышает аффинность олигонуклеотида к ДНК (Povsic and Dervan, J. Am. Chem. Soc., 1989, 111, 3059-3061). Третья цепочка может содержать также неприродные основания, среди которых могут быть упомянуты 7-деаза-2′-дезоксиксантозин (Milligan et al., Nucleic Acids Res., 1993, 21, 327-333), 1-(2-дезокси- Другой тип модификации олигонуклеотида связан с более конкретной задачей улучшить взаимодействие и/или аффинность между олигонуклеотидом и специфической последовательностью. В частности, наиболее преимущественная модификация согласно изобретению заключается в метилировании цитозинов олигонуклеотида (см. Пример 5). Метилированный таким образом олигонуклеотид приобретает примечательное свойство образовывать стабильную тройную спираль со специфической последовательностью в области значений pH, близкой к нейтральной ( Длина олигонуклеотида, используемого в способе согласно изобретению, составляет по меньшей мере 3 основания, а предпочтительно между 5 и 30. Предпочтительно использование олигонуклеотида с длиной более 10 оснований. Эта длина может быть адаптирована специалистом в данной области в каждом индивидуальном случае, в соответствии с необходимой селективностью и стабильностью взаимодействия. Олигонуклеотиды согласно изобретению могут быть синтезированы в соответствии с любой из известных технологий. В частности, они могут быть получены с использованием синтезаторов нуклеиновых кислот. Любой другой способ, известный специалистам в данной области, может быть использован с тем же успехом. Для того чтобы создать возможность ковалентного связывания с подложкой, олигонуклеотид обычно функционализируют. Таким образом, он может быть модифицирован тиолом, амином или концевой карбоксильной группой в 5′- или 3′-положении. В частности, добавление тиола, амина или карбоксильной группы создает возможность, например, соединить олигонуклеотид с подложкой, несущей дисульфидную, малеимидную, аминную, карбоксильную, сложно-эфирную, эпоксидную, цианогенбромидную или альдегидную функции. Эти соединения формируются за счет дисульфидных, тиоэфирных, сложно-эфирных, амидных или аминных связей между олигонуклеотидом и подложкой. Можно использовать любой другой способ, известный специалисту в данной области, такой, например, как бифункциональные сшивающие реагенты. Кроме того, для улучшения гибридизации со связанным олигонуклеотидом предпочтительно, чтобы олигонуклеотид содержал последовательность оснований “плеча” и “спейсера”. Использование “плеча” действительно создает возможность связывания олигонуклеотида на необходимом расстоянии от подложки, что, в свою очередь, позволяет улучшить условия взаимодействия с ДНК. Плечо предпочтительно состоит из линейной углеродной цепи, содержащей от 1 до 18, а предпочтительно от 6 до 12 (CH2)-групп, и амина, который способствует связыванию с колонкой. Плечо связано с фосфатом олигонуклеотида или “спейсера”, состоящего из оснований, которые не являются помехой для гибридизации. Таким образом, “спейсер” может включать в себя пуриновые основания. В качестве примера, “спейсер” может включать в себя последовательность GAGG. Плечо предпочтительно состоит из линейной углеродной цепи, содержащей 6 или 12 атомов углерода. Для реализации настоящего изобретения могут быть использованы различные виды подложек. Так, ими могут служить функционализированные хроматографические носители как в отдельной упаковке, так и предварительно набитые в колонку, функционализированные пластиковые поверхности или функционализированные латексные бусинки, магнитные или какие-либо иные. Предпочтительно используют хроматографические носители. В качестве примера, хроматографическими носителями, которые могут быть использованы, являются агароза, акриламид или декстран, а также их производные (такие как сефадекс, сефароза, супероза и т.д.), полимеры, такие как поли(стирол/дивинилбензол), или, например, привитой или непривитой диоксид кремния. Хроматографические колонки могут быть использованы в режиме диффузии или перфузии. Для получения лучшего выхода при очистке особенно предпочтительно использование на плазмиде последовательности, содержащей несколько положений для гибридизации с олигонуклеотидом. Наличие нескольких положений для гибридизации действительно стимулирует взаимодействия между указанной последовательностью и олигонуклеотидом, что дает лучший выход при очистке. Таким образом, для олигонуклеотида, содержащего n повторных мотивов (CCT), (CT) или (CTT), предпочтительно использовать последовательность ДНК, содержащую по меньшей мере n комплементарных мотивов, и предпочтительно n+1 комплементарный мотив. Последовательность, несущая n+1 комплементарный мотив, таким образом дает возможность для гибридизации с олигонуклеотидом по двум положениям. Предпочтительно, чтобы последовательность ДНК содержала вплоть до 11 положений для гибридизации, то есть n+10 комплементарных мотивов. Способ согласно изобретению может быть использован для очистки двухцепочечной ДНК любого типа. Примером последней может быть кольцевая ДНК, такая как плазмида, обычно несущая один или более генов, имеющих терапевтическое значение. Эта плазмида может также содержать сайт инициации репликации, маркерный ген и тому подобное. Способ согласно изобретению может быть приложен непосредственно к клеточному лизату. В таком воплощении, плазмиду, амплифицированную путем трансформации и дальнейшего культивирования клеток, очищали непосредственно после лизиса клеток. Способ согласно изобретению может быть применен также к прозрачному лизату, то есть к супернатанту, полученному после нейтрализации и цетрифугирования клеточного лизата. Он со всей очевидностью может быть применен также и к раствору, предварительно очищенному с помощью известных способов. Этот способ также создает возможность очистки линейной или кольцевой ДНК, несущей значимую последовательность, из смеси, содержащей молекул ДНК с различными последовательностями. Способ согласно изобретению может быть использован также для очистки двухцепочечной ДНК. Клеточным лизатом может быть лизат прокариотических или эукариотических клеток. Что касается прокариотических клеток, в качестве примеров можно привести бактерии E. coli, B. subtilis, S. typhimurium или Strepomyces. В качестве примеров эукариотических клеток могут быть упомянуты клетки животных, дрожжи, грибки и тому подобное, в особенности дрожжи Kluyveromyces или Saccharomyces, или клетки COS, CHO, C127, NIH3T3 и тому подобное. Способ согласно изобретению полезен, в частности, тем, что он дает возможность быстро и просто получить плазмидную ДНК очень высокой степени чистоты. В частности, как показано в примерах, этот способ дает возможность эффективного отделения заданной ДНК от загрязняющих компонентов, таких как фрагментированной хромосомной ДНК, эндотоксинов, протеинов, нуклеаз и тому подобного. Более конкретно, способ согласно изобретению дает возможность получить двухцепочечную ДНК, в частности, плазмидной природы, имеющую содержание хромосомной ДНК, равное 0,5% или ниже. Еще более предпочтительно, чтобы полученные препараты ДНК содержали хромосомную ДНК в количестве, меньшем или равном 0,2%. Таким образом, в настоящем изобретении описаны композиции, содержащие плазмидную ДНК, которая может быть использована фармацевтически, в частности, в генной или клеточной терапии. В этой связи, объектом изобретения является также и фармацевтическая композиция, содержащая двухцепочечную ДНК линейной или плазмидной природы, полученную в соответствии с описанным выше способом. Изобретение связано также с препаратами плазмидной ДНК, имеющими содержание хромосомной ДНК, меньшее или равное 0,5%, предпочтительно меньшее или равное 0,2% и более предпочтительно меньшее или равное 0,1%, еще более предпочтительно меньшее или равное 0,01%. Как показано ниже в примерах, стадия аффинного взаимодействия триплета была включена в процесс очистки после классических хроматографических стадий. Эта аффинная стадия значительно повысила чистоту плазмидного препарата, какой бы ни была его первоначальная чистота. Образование триплексной структуры между олигонуклеотидом (ковалентно связанным с хроматографическим субстратом (подложкой)) и плазмидой, подлежащей очистке, зависит от присутствия на плазмиде последовательности, которая может образовать триплексную структуру с олигонуклеотидом. Эта триплексная структура стабильна только при кислых значениях pH, когда цитозины олигонуклеотида протонированы. После этого плазмидную ДНК элюируют с колонки просто путем повышения pH до нейтральных значений. Композиция может содержать плазмидную ДНК, либо “обнаженную”, либо комбинированную с носителями-переносчиками, такими как липосомы, наночастицы, катионные липиды, полимеры, рекомбинантные вирусы или белки и тому подобное. В одном аспекте способ согласно изобретению может быть использован для очистки двухцепочечной ДНК одного типа из смеси, содержащей две или более двухцепочечные ДНК различных типов и последовательностей. Этот способ может быть применен непосредственно к клеточному лизату, в котором двухцепочечные ДНК, амплифицированные в клеточной культуре, очищаются после лизиса и культивирования клеток. Этот способ может быть применен также и к прозрачному лизату, т.е. к супернатанту, полученному после нейтрализации и цетрифугирования клеточного лизата. Он может быть также применен также и к предварительно очищенному раствору. Более конкретно, способ очистки первой двухцепочечной ДНК из раствора, содержащего первую и вторую двухцепочечные ДНК, включает в себя (i) пропускание раствора через первую подложку, содержащую ковалентно связанный олигонуклеотид, образующий тройную спираль со второй двухцепочечной ДНК в результате гибридизации с ее специфической последовательностью, (ii) восстановление раствора, который пропускается через первую подложку, который будет обогащен несвязанной первой двухцепочечной ДНК, и (iii) пропускание восстановленного раствора через вторую подложку, содержащую ковалентно связанный олигонуклеотид, образующий тройную спираль в результате гибридизации со специфической последовательностью второй двухцепочечной ДНК. После необязательной стадии промывания первую двухцепочечную ДНК можно элюировать со второй подложки. С помощью такого способа двойной очистки первая двухцепочечная ДНК может быть восстановлена со второй подложки без каких-либо значительных примесей второй двухцепочечной ДНК. В особом аспекте данного изобретения молекула первой двухцепочечной ДНК представляет собой плазмиду pCOR, имеющую специфическую последовательность 5’AAGAAAAAAAAGAA-3′ (SEQ ID NO: 29), которая образует стабильную триплексную структуру с олигонуклеотидом, имеющим последовательность 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30). Молекула второй двухцепочечной ДНК представляет собой полученную из ColEl плазмиду, имеющую специфическую последовательность 5′-AGAAAAAAAGGA-3′ (SEQ ID NO: 27), которая образует триплекс с олигонуклеотидом, имеющим последовательность 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28). Соответственно, плазмида pCOR успешно выделяется из раствора, содержащего другие плазмиды, такие как плазмиды, полученные из ColEl, с использованием способа двойной очистки согласно изобретению. Данное изобретение будет описано подробно с помощью следующих далее примеров, которые следует рассматривать исключительно как иллюстративные, но не как ограничительные. ПОДРОБНОЕ ОПИСАНИЕ Основные технологии клонирования и молекулярной биологии Общепринятые методы молекулярной биологии, такие как переваривание под действием ферментов рестрикции, гель-электрофорез, трансформация в E. coli, преципитация нуклеиновых кислот и тому подобное, описаны в литературе (Maniatis et al., T., E.F.Fritsch, and J. Sambrook, 1989. Molecular cloning: a laboratory manual, second edition. Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, New York; Ausubel F.M., R. Brent, R.E.Kinston, D.D.Moore, J.A.Smith, J.G.Seidman and K.Struhl. 1987. Current protocols in molecular biology 1987-1988. John Willey and Sons, New York.). Нуклеотидные последовательности определяли с помощью метода терминации цепей, согласно опубликованной методике (Ausubel et al., 1987). Ферменты рестрикции были получены из New England Biolabs, Beverly, MA (Biolabs). Для проведения лигирования фрагменты ДНК инкубировали в буфере, содержащем 50 мМ Tris-HCl pH 7,4, 10 мМ MgCl2,10 мМ DTT, 2 мМ АТФ в присутствии ДНК-лигазы фага T4 (Biolabs). Олигонуклеотиды синтезировали, используя химию фосфорамидитов с помощью фосфорамидитов, защищенных в Лигированные ДНК или ДНК, подлежащие тестированию в отношении эффективности их трансформации, использовали для трансформации следующих признанных компетентными штаммов: E. coli DH5 Минипрепараты плазмидной ДНК получали в соответствии с методикой Klein et al., 1980. Для роста штаммов E. coli использовали культуральную среду LB (Maniatis et al., 1982). Штаммы инкубировали при 37оC. Бактерии высевали на чашках со средой LB medium, обогащенной соответствующими антибиотиками. Пример 1 1.1. Подготовка колонки Оборудование Используемой колонкой являлась колонка HiTrap объемом в 1 мл, активированная NHS (N-гидроксисукцинимид, Pharmacia), соединенная с перистальтическим насосом (выход < 1 мл/мин). Используемый специфический олигонуклеотид на своем 5′-конце содержал NH2-группу и имел следующую последовательность: 5′-GAGGCTTCTTCTTCTTCTTCTTCTT-3′ (SEQ ID NO: 1) В этом примере были использованы следующие буферы: Буфер для присоединения: 0,2 M NaHCO3, 0,5 M NaCl, pH 8,3. Буфер A: 0,5 M этаноламин, 0,5 M NaCl, pH 8,3. Буфер B: 0,1 M ацетат, 0,5 M NaCl, pH 4. Методика: Колонку промывали 6 мл 1 мМ HCl и затем олигонуклеотид, разведенный в буфере для присоединения (50 нмоль в 1 мл), наносили на колонку и выдерживали в течение 30 минут при комнатной температуре. Затем колонку трижды промывали 6 мл буфера A, а затем 6 мл буфера B. Таким образом олигонуклеотид ковалентно связывался с колонкой посредством CONH-связи. Колонку хранили при 4°C в PBS, 0,1% NaN3, и ее можно было использовать по меньшей мере четыре раза. 1.2. Конструирование плазмид Было синтезировано два следующих олигонуклеотида: олигонуклеотид 4817: 5′-GATCCGAAGAAGAAGAAGAAGAAGAAGAAGAAGAAGAAGAAGAAGA AGAA GAAGAAGG-3′ (SEQ ID NO: 9) олигонуклеотид 4818: 5′-AATTCCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCTTCG-3′ (SEQ ID NO: 10) Эти олигонуклеотиды при гибридизации и клонировании в плазмиду интродуцируют гомопурин-гомопиримидиновую последовательность (GAA) 17 (SEQ ID NO: 33) в соответствующую плазмиду, как описано выше. Последовательность, соответствующая этим двум гибридизованным олигонуклеотидам, была клонирована в сайт множественного клонирования плазмиды pBKS+ (Stratagene Cloning System, La Jolla CA), которая является носителем ампициллин-резистентного гена. С этой целью олигонуклеотиды были гибридизованы следующим образом: один мкг этих двух олигонуклеотидов был добавлен к 40 мл конечного буфера, содержащего 50 мМ Tris-HCl, pH 7,4, 10 мМ MgCl2. Эту смесь нагревали до 95°C, а затем переносили в атмосферу комнатной температуры, давая смеси медленно остыть до комнатной температуры. Десять нг смеси гибридизованных олигонуклеотидов лигировали с 200 нг плазмиды pBKS+ (Stratagene Cloning System, La Jolla CA), переваренной под действием BamHI и EcoRI в конечной [концентрации] 30 мкл. После лигирования аликвоту трансформировали в DH5a. Смеси для трансформации помещали в среду L, обогащенную ампициллином (50 мг/л) и X-gal (20 мг/л). В рекомбинантных клонах должна отсутствовать голубая окраска среды в отличие от родительской плазмиды (pBKS+), которая допускает 1.3. Очистка плазмид Оборудование: Плазмиду pXL2563 (описанную в разделе 1.2) очищали на колонке HiTrap, связанной с нуклеотидом, описанным в разделе 1.1., из раствора, содержащего также плазмиду pBKS+. В этих стадиях очистки использовали следующие буферы: Буфер F: 2 M NaCl, 0,2 M ацетат, pH 4,5-5. Буфер E: 1 M Tris-HCl, pH 9, 0,5 мМ EDTA. Методика: Колонку промывали 6 мл буфера F, плазмиды (20 мкг pXL2563 и 20 мкг pBKS+ в 400 мкл буфера F) наносили на колонку и инкубировали в течение 2 часов при комнатной температуре. Колонку промывали 10 мл буфера F, а затем элюировали буфером E. Плазмиды определяли после электрофореза в 1% агарозном геле и окрашивания бромидом этидия. Соотношение плазмид в растворе определяли путем измерения их трансформирующей активности на E. coli. Результат: Начиная со смеси, содержащей 30% pXL2563 и 70% pBKS+, на выходе с колонки получали раствор, содержащий 100% pXL2563. Степень чистоты, определяемая по отношению показателей дисперсии оптического вращения (OD) при 260 и 280 нм, возрастала от 1,9 до 2,5, что указывает на то, что примесные белки при таком способе очистки удаляются. Пример 2 2.1. – В этом примере описан опыт очистки плазмидной ДНК. Присоединение олигонуклеотида (5′-GAGGCTTCTTCTTCTTCTTCTTCTT-3′ (SEQ ID NO: 1)) к колонке производили так, как описано в примере 1. Для присоединения олигонуклеотид модифицировали на 5′-конце с помощью аминогруппы, связанной с фосфатом спейсера плечом, содержащим 6 атомов углерода (Modified oligonucleotide Eurogentec SA, Belgium). Плазмиду pXL2563 очищали с помощью набора Wizard Megaprep (Promega Corp., Madison, WI) в соответствии с инструкциями изготовителя. В этом примере использовались следующие буферы: Буфер F: 0-2 M NaCl, 0,2 M ацетат, pH 4,5-5. Буфер E: 1 M Tris-HCl pH 9, 0,5 мМ EDTA. Колонку промывали 6 мл буфера F, 100 мкл плазмиды pXL2563 в 400 мкл буфера F наносили затем на колонку и инкубировали в течение 2 часов при комнатной температуре. Колонку промывали 10 мл буфера F, а затем элюировали буфером E. Количественное определение плазмиды проводили, измеряя оптическую плотность при 260 нм. В этом примере связывание проводили в буфере, в котором молярность NaCl варьировали от 0 до 2 M (буфер F). Выход продукта при такой очистке снижался, когда молярность раствора NaCl падала. Величина pH связывающего буфера может варьировать от 4,5 до 5, выход при очистке улучшался при 4,5. Можно использовать также и другой элюирующий буфер с основным pH: в этом случае элюирование проводили с помощью 50 мМ боратного буфера, pH 9, 0,5 мМ EDTA. 2.2. – Связывание олигонуклеотида (5′-GAGGCTTCTTCTTCTTCTTCTTCTT-3′ (SEQ ID NO: 1) с колонкой производили, как описано в Примере 1. Плазмиду pXL2563 очищали с помощью набора Wizard Megaprep (Promega Corp., Madison, WI), руководствуясь рекомендациями изготовителя. В этом примере были использованы следующие буферы: Буфер F: 0,1 M NaCl, 0,2 M ацетат, pH 5. Буфер E: 1 M Tris-HCl pH 9, 0,5 мМ EDTA. Колонку промывали 6 мл буфера F, 100 мкл плазмиды pXL2563, разбавленной 400 мкл буфера F, наносили затем на колонку и инкубировали в течение 1 часа при комнатной температуре. Колонку промывали 10 мл буфера F, а затем элюировали буфером E. Измеряли содержание геномной или хромосомной ДНК E. Coli, присутствующей в плазмидных образцах, до и после пропускания через олигонуклеотидную колонку. Эту геномную ДНК количественно определяли методом ПЦР, используя праймеры в гене galK E. coli, согласно следующей методике: Последовательность этих праймеров описана у Debouck et al. (Nucleic Acids Res. 1985, 13, 1841-1853): 5′-CCGAATTCTGGGGACCAAAGCAGTTTC-3′ (SEQ ID NO: 24) and 5′-CCAAGCTTCACTGTTCACGACGGGTGT-3′ (SEQ ID NO: 25). Реакционная среда содержит в 25 мкл буфера ПЦР (Promega France, Charbonnières) 1,5 мМ MgCl2; 0,2 мМ dXTP (Pharmacia, Orsay); 0,5 мкМ праймера; 20 ед./мл Taq-полимеразы (Promega). Реакцию проводили в следующей последовательности: – 5 мин при 95°C – 30 циклов по 10 сек при 95°C – 30 сек при 60°C – 1 мин при 78°C – 10 мин при 78°C. Фрагмент амплифицированной ДНК длиной в 124 пары нуклеотидов отделяли с помощью электрофореза в 3% агарозном геле в присутствии SybrGreen I (Molecular Probes, Eugene, USA), а затем определяли количественно, со ссылкой на серии геномной ДНК Ultrapur из штамма B E. coli (Sigma, ref D4889). В образце, наносимом на колонку, содержался 1% хромосомной ДНК, а в образце, очищенном на колонке с олигонуклеотидом, содержалось 0.2% хромосомной ДНК. Пример 3. Эксперимент на прозрачном лизате В данном примере описывается очистка плазмидной ДНК из прозрачного лизата бактериальной культуры, на так называемой шкале “miniprep”: центрифугировали 1,5 мл культивируемых в течение ночи клеток штаммов DH5 Пример 4 4.1: В этом примере описан эксперимент по очистке плазмидной ДНК, проведенный в таких же условиях, что и описанные в Примере 3, начиная с 20 мл бактериальной культуры штаммов DH5 4.2: Использованная плазмида содержит кассету, включающую в себя промотор цитомегаловируса, ген, кодирующий люциферазу, и гомопурин-гомопиримидиновую последовательность (GAA) 17 (SEQ ID NO: 33), происходящую из плазмиды pXL2563. Штамм DH1 (Maniatis et al., 1989), содержащий эту плазмиду, культивировали в 7-литровом ферментере. Прозрачный лизат получали из 200 граммов клеток: клеточный осадок растворяли в 2 литрах 25 мМ Tris, pH 6,8, содержащих 50 мМ глюкозы, 10 мМ EDTA, и к этому раствору добавляли 2 литра 0,2 M NaOH, содержащего 1% SDS. Нейтрализацию лизата проводили путем добавления к нему одного литра 3 M ацетата калия. После диафильтрации 4 мл этого лизата наносили на 5 мл колонку HiTrap-NHS со связанным с ней олигонуклеотидом, имеющим последовательнсть 5′-GAGGCTTCTTCTTCTTCTTCTTCTT-3′ (SEQ ID NO: 1), в соответствии со способом, описанным в Примере 1.1. Процессы промывки и элюции проводили согласно способам, описанным в Примере 1. Получали приблизительно 400 микрограммов плазмиды. Уровень геномной ДНК в этом образце, определяемый в соответствии с технологией, описанной в Примере 2.2, составлял 0,1%. Пример 5: Применение модифицированного олигонуклеотида В этом примере описано применение олигонуклеотида, несущего метилированные цитозины. Последовательность использованного олигонуклеотида была следующей: 5′-GAGGMeCTTMeCTTMeCTTMeCTTMeCCTMeCTTMeCTT-3′ (SEQ ID NO: 12). Этот олигонуклеотид содержит на 5′-конце NH2-группу. MeC = 5-метилцитозин. Этот олигонуклеотид позволяет очистить плазмиду pXL2563 в условиях, описанных в Примере 1, с помощью связывающего буфера, имеющего pH 5 (таким образом снижается риск возможной деградации плазмиды). Пример 6 Использованные в приведенных выше примерах олигонуклеотиды модифицированы на 5′-конце аминогруппой, связанной с фосфатом посредством плеча, содержащего 6 атомов углерода: NH2-(CH2)6. В данном примере аминогруппа связана с 5′-концевым фосфатом посредством плеча, содержащего 12 атомов углерода: NH2-(CH2)12. Связывание олигонуклеотида и пропускание через колонку производили так же, как и в Примере 2, с использованием буфера F: 2 M NaCl, 0,2 M ацетат, pH 4,5. Применение такого олигонуклеотида создает в результате возможность увеличения выхода при очистке: был получен 53% выход, тогда как при применении олигонуклеотида, содержащего 6 атомов углерода, этот выход в таких же условиях составляет 45%. Пример 7 Следуя методике клонирования, описанной в Примере 1.2, сконструировали еще две плазмиды, несущие гомопурин-гомопиридиновые последовательности: плазмида pXL2725, которая содержит последовательность (GGA)16, (SEQ ID NO: 34), и плазмида pXL2726, которая содержит последовательность (GA)25 (SEQ ID NO: 35). Пример 7.1: Конструирование плазмид Плазмиды pXL2725 и pXL2726, аналогичные плазмиде pXL2563, были сконструированы согласно методике клонирования, описанной в Примере 1.2, с использованием следующих олигонуклеотидных пар: 5986: 5′-GATCC(GA)25GGG-3′ (SEQ ID NO: 13) 5987: 5′-AATTCCC(TC)25G-3′(SEQ ID NO: 14) 5981: 5′-GATCC(GGA)17GG-3′(SEQ ID NO: 15) 5982: 5′-AATT(CCT)17CCG-3′(SEQ ID NO: 16) Пару олигонуклеотидов 5986 и 5987 использовали для конструирования плазмиды pXL2726 путем клонирования олигонуклеотидов в сайтах BamHI и EcoRI pBKS+ (Stratagene Cloning System, La Jolla CA), тогда как олигонуклеотиды 5981 и 5982 использовали для конструирования плазмиды pXL2725. Были использованы те же самые условия, что и в случае конструирования плазмиды pXL2563, но только с заменой пар олигонуклеотидов. Сходным образом, клонированные последовательности выверяли путем секвенирования плазмид. Это давало возможность воочию убедиться в том, что плазмида pXL2725 содержит модификацию ожидаемой последовательности: вместо повторенной 17 раз последовательности GGA она содержала последовательность GGAGA(GGA)15 (SEQ ID NO: 17). Пример 7.2: Приготовление колонок и очистка Олигонуклеотиды, образующие тройные спирали с этими гомопуриновыми последовательностями, были связаны с колонками HiTrap в соответствии со способом, описанным в Примере 1.1. Олигонуклеотид с последовательностью 5′-AATGCCTCCTCCTCCTCCTCCTCCT-3′ (SEQ ID NO: 18) использовали для очистки плазмиды pXL2725, а олигонуклеотид с последовательностью 5′-AGTGCTCTCTCTCTCTCTCTCTCTCT-3′ (SEQ ID NO: 19) использовали для очистки плазмиды pXL2726. Полученные таким образом две колонки позволяли очищать соответствующие плазмиды согласно способу, описанному в Примере 2, с помощью следующих буферных растворов: Буфер F: 2 M NaCl, 0,2 M ацетат, pH 4,5. Буфер E: 1 M Tris-HCl, pH 9, 0,5 мМ EDTA. Выход плазмид pXL2725 и pXL2726 составлял соответственно 23% и 31%. Пример 8 В этом примере показано влияние длины специфической последовательности, присутствующей в плазмиде, на количественный выход при очистке. Пример 8.1: Конструирование плазмид Репортерным геном, используемым в этих экспериментах для демонстрации активности композиций согласно изобретению, является ген, кодирующий люциферазу (Luc). Плазмида pXL2621 содержит кассету, включающую в себя промотор цитомегаловируса (CMV), насчитывающий 661 пар нуклеотидов, экстрагированный из pcDNA3 (Invitrogen Corp., San Diego, CA) путем расщепления ферментами рестрикции MluI и HindIII, клонировали вверх по течению от гена, кодирующего люциферазу, в сайтах MluI и HindIII, в вектор pGL basic Vector (Promega Corp., Madison, WI). Эта плазмида была сконструирована с использованием стандартных технологий молекулярной биологии. Плазмиды pXL2727-1 и pXL2727-2 были сконструированы следующим способом. Два микрограмма плазмиды pXL2621 были линеаризованы с помощью BamHI; фермент был инактивирован путем 10-минутной обработки при 65°C; в то же самое время олигонуклеотиды 6006 и 6008 были гибридизованы, как описано для случая конструирования плазмиды pXL2563. 6006: 5′-GATCT(GAA)17CTGCAGATCT-3′ (SEQ ID NO: 20) 6008: 5′-GATCAGATCTGCAG(TTC)17A-3′ (SEQ ID NO: 21). Эту смесь для гибридизации клонировали на BamHI-концах плазмиды pXL2621 и после трансформации в DH5 Первый клон (pXL2727-1) содержит последовательность GAA, повторенную 10 раз. Второй клон (pXL2727-2) содержит последовательность 5′-GAAGAAGAG(GAA)7GGAAGAGAA-3′ (SEQ ID NO: 23). Пример 8.2: Подготовка колонок и очистка Была использована колонка наподобие описанной в Примере 1, которая была связана с олигонуклеотидом 5′- GAGGCTTCTTCTTCTTCTTCTTCTT-3′ (SEQ ID NO: 1). Плазмида pXL2727-1 содержит 14 повторов последовательности GAA. Описанный выше олигонуклеотид, который содержит только 7 повторов соответствующей гибридизующейся последовательности CTT, может, следовательно, гибридизоваться с плазмидой в 8 различных положениях. Плазмида pXL2727-2, наоборот, содержит гибридизующуюся последовательность (GAA)7 (SEQ ID NO: 36) такой же самой длины, что и олигонуклеотид, связанный с колонкой. Этот олигонуклеотид может, следовательно, гибридизоваться только с одним положением на pXL2727-2. Эксперимент идентичен описанному в Примере 2, при этом используются следующие буферы: Буфер F: 2 M NaCl, 0,2 M acetate, pH 4,5. Буфер E: I M Tris-HCl, pH 9, 0,5 мМ EDTA. Выход после очистки составляет 29% в случае плазмиды pXL2727-1 и 19% в случае плазмиды pXL2727-2. Пример 8.3: трансфекция клеток млекопитающих in vitro Используемыми клетками являлись клетки NIH 3T3, инокулированные в предшествующий эксперименту день в 24-луночные культуральные планшеты из расчета 50000 клеток/лунку. Плазмиду разбавляли 150 мМ NaCl и смешивали с липофектантом RPR115335. Использовали отношение положительных зарядов липофектанта к отрицательным зарядам ДНК, равное 6. Смесь перемешивали на vortex’e, выдерживали в течение десяти минут при комнатной температуре, разбавляли средой, не содержащей эмбриональной телячьей сыворотки, и затем добавляли к клеткам в соотношении 1 мкг ДНК на культуральную лунку. После двух часов инкубации при 37оC добавляли 10% объем/объем эмбриональной тельячьей сыворотки и клетки инкубировали в течение 48 часов при 37°C в присутствии 5% CO2. Клетки дважды промывали буфером PBS и измеряли люциферазную активность согласно описанной методике (Promega kit, Promega Corp.Madison, WI), на люминометре Lumat LB9501 (EG and G Berthold, Evry). Плазмида pXL2727-1, очищенная, как описано в Примере 8.2, дает на выходе трансфекцию, вдвое превосходящую таковую, полученную при очистке этой же плазмиды с помощью набора Wizard Megaprep (Promega Corp.Madison, WI). Пример 9: Очистка плазмид, полученных на основе pCOR-plasmids В следующем примере показана очистка плазмид, полученных на основе pCOR, с использованием трехспиральной аффинной хроматографии. Было показано, что такая технология позволяет удалять примеси нуклеиновых кислот (в частности, геномные ДНК и РНК хозяина), доводя их до таких уровней, каких не удавалось достичь с помощью традиционно применяемых методов хроматографической очистки. Аффинный к триплексу гель был синтезирован на основе сефакрила S-1000 SF (Amersham-Pharmacia Biotech) как хроматографического матрикса. Сефакрил S-1000 был впервые активирован m-перйодатом натрия (3 мМ, комнатная температура, 1 час) в 0,2 M ацетате натрия (pH 4,7). Затем олигонуклеотид соединяли через его 5′-NH2-концевой фрагмент с альдегидными группами активированного матрикса путем редуктивного аминирования в присутствии аскорбиновой кислоты (5 мМ), как описано ранее для соединения белков (Hornsey et al., J. Immunol. Methods, 1986, 93, 83-88). Использованный в этих экспериментах гомопиридиновый олигонуклеотид (из Eurogentec, HPLC-очищенный) имел последовательность, комплементарную короткой 14-членной гомопуриновой последовательности (5′-AAGAAAAAAAAGAA-3′) (SEQ ID NO: 29), присутствующей в сайте начала репликации (ori Хроматографии были подвергнуты следующие плазмиды: pXL3296 (pCOR без трансгена, 2.0 т.п.н), pXL3179 (pCOR-FGF, 2.4 т.п.н.), pXL3579 (pCOR-VEGFB, 2.5 т.п.н.), pXL3678 (pCOR-AFP, 3.7 т.п.н.), pXL3227 (pCOR-lacZ, 5.4 т.п.н.) и pXL3397 (pCOR-B-удаленный FVIII, 6.6 т.п.н.). Все эти плазмиды были очищшены из прозрачных лизатов, полученных, как было описано в Примере 4, с помощью двух стадий анионообменной хроматографии. Была изучена также плазмида pBKS+ (pBluescript II KS + from Stratagene), плазмида, полученная из ColEl, очищенная путем ультрацентрифугирования в CsCl. Все использованные плазмиды топологически находились в суперскрученном (>95%) состоянии. В каждом эксперименте по очистке плазмидной ДНК 300 мкг плазмидной ДНК в 6 мл 2 M NaCl, 0,2 M ацетата калия (pH 5,0) наносили со скоростью тока 30 см/час на аффинную колонку, содержащую указанный выше олигонуклеотид 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30). После промывки колонки 5 объемами того же самого буфера плазмиду элюировали 1 M Tris/HCl, 0,5 мMEDTA (pH 9,0) и количественно определяли с помощью УФ- (260 нм) и ионообменной хроматографии на колонке Millipore Gen-Pak (Marquet et al., BioPharm, 1995, 8, 26-37). Восстановленные плазмиды в собранных фракциях составляли 207 мкг плазмиды pXL3296, 196 мкг плазмиды pXL3179, 192 мкг плазмиды pXL3579, 139 мкг плазмиды pXL3678, 97 мкг плазмиды pXL 3227 и 79 мкг плазмиды pXL 3397. Никакого связывания плазмиды (<3 мкг) не было обнаружено при хроматографии на этой колонке pBKS. Это указывает на то, что олигонуклеотид 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30) придает стабильность триплексным структурам с комплементарной 14-членной последовательностью 5′-AAGAAAAAAAAGAA-3′ (SEQ ID NO: 29), присутствующей в pCOR (ori В качестве контроля, никакого связывания плазмиды (<1 мкг) не было обнаружено при хроматографии pXL3179 на контрольной колонке, полученной точно в таких же условиях, но без олигонуклеотида. Манипулируя указанной аффинной очистительной колонкой в условиях, описанных выше, удавалось снизить уровень примесей хозяйской геномной ДНК от 2,6% до 0,07% в препарате плазмиды pXL3296. Сходным образом, когда образец хроматографировали через ту же самую аффинную колонку, уровень примесей хозяйской геномной ДНК в препарате плазмиды pXL3179 был снижен от 0,5% до 0,008%. Кроме того, с помощью этой очистительной аффинной колонки удалось при получении плазмиды pXL3179 значительно снизить также уровень примесей РНК, от 43% РНК до 0,2%. Кроме того, восстановление плазмиды PXL3579 составляло менее 8%, когда олигонуклеотид 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30) был заменен на аффинной колонке олигонуклеотидом 5′-TTTTTTTTCTT-3′ (SEQ ID NO: 31). Несмотря на то что олигонуклеотид, соответствующий последовательности SEQ ID NO: 31, комплементарен части последовательности VEGFB внутри pXL3579 (т.e. нуклеотиды 379-389 относительно ATG), никакой значительной триплексной аффинности не возникало. Это указывает на то, что такая аффинная очистка требует не случайной гомопурин-гомопиримидиновой последовательности ДНК. Пример 10: Очистка плазмиды, полученной на основе ColEl В следующем примере показана очистка плазмид, полученных на основе ColEl, с помощью хроматографии на основе аффинности к тройной спирали. Было показано, что с помощью этой технологии можно удалить примеси нуклеиновых кислот (в частности, хозяйскую геномную ДНК и РНК), снизив их до таких уровней, каких не удавалось достичь с помощью общеизвестных хроматографических методик. Аффинный к триплексу гель был синтезирован путем связывания олигонуклеотида, имеющего последовательность 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28), на окисленном перйодатом сефакриле S-1000 SF, как было описано в Примере 9. Плазмиды pXL3296 (pCOR без трансгена) и pBKS, плазмида, полученная на основе ColEl, хроматографировали на 1-миллилитровой колонке, содержащей олигонуклеотид 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28), в условиях, описанных в Примере 9. Восстановление плазмид в собранных фракциях составляло 175 мкг плазмиды pBKS и < 1 мкг плазмиды pXL3296. Это указывает на то, что олигонуклеотид 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28) придает стабильность структурам тройной спирали с комплементарной 12-членной последовательностью (5′-AGAAAAAAAGGA-3′) (SEQ ID NO: 27), присутствующей в pBKS, чего, однако, не происходит в случае очень схожей 12-членной последовательности (5′-AGAAAAAAAAGA-3′) (SEQ ID NO: 32), присутствующей в pCOR. Это указывает на то, что введение единственной неканонической триады (в данном случае C*AT) может приводить к полной дестабилизации структуры триплекса. Пример 11: Способ двойной очистки В следующем примере показана очистка суперскрученной двухцепочечной молекулы ДНК, такой как pXL3296, в смеси, содержащей другую суперскрученную двухцепочечную молекулу ДНК, такую как pBSK, с помощью хроматографии на основе аффинности к тройной спирали. Обе двухцепочечные молекулы ДНК могут иметь одинаковый размер, но каждая молекула ДНК содержит уникальную последовательность, которая образует тройную спираль с различными последовательностями-мишенями. Как обсуждалось ранее, молекулы, такие как pXL3296, содержат последовательность 5′-AAGAAAAAAAAGAA-3′ (SEQ ID NO: 29), но не содержат последовательность 5′-AGAAAAAAAGGA-3′ (SEQ ID NO: 27). И наоборот, молекулы, такие как pBSK, содержат последовательность SEQ ID NO: 27, но не содержат последовательность SEQID NO: 29. На первой стадии смесь, содержащую pXL3296 и pBSK, наносили на первую аффинную колонку, содержащую олигонуклеотид 5′-TCTTTTTTTCCTT-3′ (SEQ ID NO: 28), такую как колонка, описанная в Примере 10. Раствор пропускали через первую колонку, которая содержала несвязанные молекулы ДНК. На второй стадии несвязанные молекулы ДНК из первой стадии наносили на вторую аффинную колонку, содержащую олигонуклеотид 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30), такую как колонка, описанная в Примере 9. Затем вторую колонку промывали и связанные молекулы элюировали, как описано в Примере 9. Со второй колонки элюировались только молекулы pXL3296. В элюате со второй колонки (т.e. растворе, который элюируется с колонки) не было обнаружено молекул pBSK.
Формула изобретения
1. Способ очистки двухцепочечной молекулы ДНК из раствора, содержащего указанную двухцепочечную молекулу ДНК, смешанную с другими компонентами, предусматривающий (i) пропускание раствора через подложку, содержащую ковалентно связанный олигонуклеотид, способный к образованию тройной спирали с двухцепочечной ДНК путем гибридизации со специфической последовательностью, присутствующей в двухцепочечной молекуле ДНК, где ковалентно связанный олигонуклеотид содержит последовательность ТСТТТТТТТССТ (SEQ ID NO: 28) или ТТСТТТТТТТТСТТ (SEQ ID NO: 30), которая способна к гибридизации с образованием тройной спирали, соответственно, с последовательностями AGAAAAAAAGGA (SEQ ID NO: 27) или AAGAAAAAAAAGAA (SEQ ID NO: 29) указанной ДНК, но которая не гибридизуется с последовательностями, приводящими к образованию, по меньшей мере, одной неканонической триады (T*GC или С*АТ), (ii) элюирование указанной ДНК путем промывания подложки в щелочной области рН и (iii) получение указанной молекулы ДНК. 2. Способ по п.1, где раствор представляет собой клеточный лизат. 3. Способ по п.2, где клеточный лизат является прозрачным клеточным лизатом. 4. Способ по любому из пп.1-3, где двухцепочечная ДНК является предварительно очищенной. 5. Способ по любому из пп.1-4, где специфическую последовательность искусственно вводят в двухцепочечную ДНК или она естественным образом присутствует в двухцепочечной ДНК. 6. Способ по любому из пп.1-5, где олигонуклеотид соединен с подложкой посредством дисульфидной, тиоэфирной, сложноэфирной, амидной или аминной связи. 7. Способ по п.6, где олигонуклеотид соединен с колонкой через плечо, содержащее углеродную цепь (СН2)n, где n представляет собой целое число от 1 до 18 включительно и где плечо связано с олигонуклеотидом посредством фосфатной связи, а с колонкой – посредством амидной связи. 8. Способ по любому из пп.1-7, где олигонуклеотид содержит, по меньшей мере, одну химическую модификацию, которая способствует его резистентности к нуклеазам или защищает его от действия нуклеаз, или же способствует увеличению его аффинности в отношении специфической последовательности. 9. Способ по п.8, где, по меньшей мере, один из цитозинов олигонуклеотида является метилированным. 10. Способ по любому из пп.1-9, где двухцепочечная ДНК является кольцевой ДНК. 11. Способ по п.10, где кольцевая ДНК представляет собой плазмиду. 12. Способ по любому из пп.1-11, где специфическая последовательность, присутствующая в двухцепочечной ДНК, содержит несколько положений для гибридизации с олигонуклеотидом. 13. Способ по любому из пп.1-12, где подложка представляет собой функционализированную хроматографическую подложку, функционализированную пластическую поверхность или функционализированные латексные бусы. 14. Способ по п.13, где подложка представляет собой функционализированную хроматографическую подложку. 15. Способ по п.14, где очищенная двухцепочечная ДНК содержит хромосомную ДНК в количестве, меньшем или равном 0,5%. 16. Способ по п.15, где очищенная двухцепочечная ДНК содержит хромосомную ДНК в количестве, меньшем или равном 0,01%. 17. Способ изготовления подложки для очистки двухцепочечной молекулы нуклеиновой кислоты, предусматривающий ковалентное связывание олигонуклеотида с подложкой, где олигонуклеотид содержит обогащенную пиримидином последовательность, выбранную из последовательностей SEQ ID NO: 28 или 30, и способен к образованию тройной спирали с двухцепочечной ДНК путем гибридизации с нуклеотидной последовательностью в двухцепочечной ДНК. 18. Способ по п.17, где подложка содержит смолу, содержащую гидроксильные остатки, где гидроксильные остатки активированы путем эстерификации N-гидроксисукцинимидом и где контактирование активированных остатков с олигонуклеотидом создает ковалентное связывание с образованием амидной связи между олигонуклеотидом и остатками подложки. 19. Способ по любому из пп.17-18, где смола представляет собой агарозу, декстран, сефадекс или привитой кремний. 20. Подложка для очистки двухцепочечной ДНК, полученная способом по любому из пп.17-19.
РИСУНКИ
|
||||||||||||||||||||||||||
 -галактозидазу Escherichia coli.
-галактозидазу Escherichia coli. -ориджин pCOR (Levchenko et al., 1996, Nucleic Acids Res., 24, 1936). Эта последовательность образует стабильную триплексную структуру с 14-членным комплементарным олигонуклеотидом 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30). В соответствующих олигонуклеотидах 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28) и 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30) эффективно и специфически нацелены их соответствующие комплементарные последовательности, локализованные в пределах сайта инициации репликации либо ColEl ori, либо pCOR (
-ориджин pCOR (Levchenko et al., 1996, Nucleic Acids Res., 24, 1936). Эта последовательность образует стабильную триплексную структуру с 14-членным комплементарным олигонуклеотидом 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30). В соответствующих олигонуклеотидах 5′-TCTTTTTTTCCT-3′ (SEQ ID NO: 28) и 5′-TTCTTTTTTTTCTT-3′ (SEQ ID NO: 30) эффективно и специфически нацелены их соответствующие комплементарные последовательности, локализованные в пределах сайта инициации репликации либо ColEl ori, либо pCOR (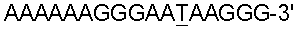 (SEQ ID NO: 8), присутствующая в гене
(SEQ ID NO: 8), присутствующая в гене  -аномерами нуклеотидов, которые также образуют тройные спирали с ДНК (Le Doan et al., Nucleic Acids Res., 1987,15, 7749-7760).
-аномерами нуклеотидов, которые также образуют тройные спирали с ДНК (Le Doan et al., Nucleic Acids Res., 1987,15, 7749-7760). 5). Это, следовательно, дает возможность работать с олигонуклеотидами при более высоких значениях pH, чем на предшествующем уровне техники, то есть при значениях pH, при которых риск деградации плазмидной ДНК значительно снижается.
5). Это, следовательно, дает возможность работать с олигонуклеотидами при более высоких значениях pH, чем на предшествующем уровне техники, то есть при значениях pH, при которых риск деградации плазмидной ДНК значительно снижается. (lacZYA-arqF) U169, deoR,
(lacZYA-arqF) U169, deoR,  80dlac (lacZ
80dlac (lacZ E. coli
E. coli 








