Патент на изобретение №2315045
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ АМИНОПИРРОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ
(57) Реферат:
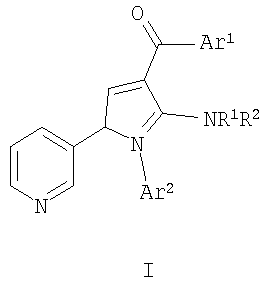
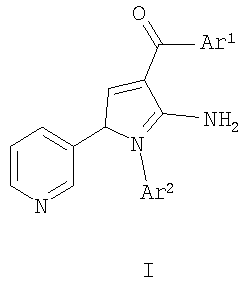
Изобретение относится к производным аминопиррола формулы (I)
где каждый из радикалов Ar1 и Ar2 независимо означает необязательно замещенный фенил, а каждый из R1 и R2 независимо означает водород, алкил или защитную группу при атоме азота, его изомер, смесь изомеров или его фармацевтически приемлемая соль; фармацевтическая композиция, обладающая противовоспалительной активностью, также изобретение включает в себя способ получения соединений формулы (I) и применение данных соединений для приготовления лекарственного средства для лечения воспалительного заболевания млекопитающего, в особенности такого, как артрит. Технический результат – получены новые соединения и фармацевтическая композиция на их основе. 3 н. и 8 з.п. ф-лы, 1 табл.
Настоящее изобретение относится к производным аминопиррола и к способам их получения и к их применению. Показано, что фактор некроза опухоли (TNF) и интерлейкин-1 (IL-1) играют центральную роль в патологических процессах, лежащих в основе многих хронических воспалительных и аутоиммунных заболеваний. IL-1 вовлечен в промежуточные заболевания или в обостряющиеся заболевания, как, например, ревматоидный артрит (см. Arend, W.P., Arthritis & Rheumatism 38(2): 151-160 (1995)), остеоартрит, резорбция костей, синдром токсического шока, туберкулез, атеросклероз, диабет, болезнь Ходжкина (см. Benharroch, D. и др., Euro. Cytokine Network 7(1): 51-57) и болезнь Альцгеймера. Избыточное или нерегулируемое продуцирование TNF вовлечено в промежуточные или обостряющиеся заболевания, как, например, ревматоидный артрит (см. Maini, R.N. и др., APMIS. 105 (4): 257-263 (1997); Feldmann, M., J. of the Royal College of Physicians of London 30 (6): 560-570 (1996); Lorenz, H.M. и др., J. of Immunology 156 (4): 1646-1853, (1996)), остеоартрит, спондилит, сепсис, септический шок (см. Abraham, Е. и др. JAMA. 277 (19): 1531-1538, (1997)), респираторный дистресс-синдром взрослых, астма (см. Shah, А. и др., Clin. & Exp.Allergy 1038-1044 (1995), и Lassale, Р. и др., Clin. & Exp.Immunol. 94 (I): 105-110 (1993)), заболевания, связанные с резорбцией костей, лихорадочное состояние (см. Cooper, A.L. и др., Am. J. of Phisiology 267 (6, часть 2): 1431-1436)), энцефаломиелит, демиелинизация (см. Klindert, W.E. и др., J. of Neuroimmunol. 72 (2): 163-168 (1997)) и периодонтальные заболевания. Клинические исследования с использованием антагонистов рецепторов IL-1 и TNF показали, что блокирование способности этих цитокинов подавать сигнал через их рецепторы приводит к значительному улучшению состояния людей с воспалительными заболеваниями. Поэтому модуляция этих воспалительных цитокинов рассматривается как одна из наиболее эффективных стратегий блокирования хронического воспаления и приводит к положительным терапевтическим последствиям. Было также показано, что р38 МАР-киназа (активированная митогеном протеинкиназа) играет важную роль в регуляции трансляции TNF и IL-1 и также вовлечена в биохимическую передачу сигнала этих молекул (см. Lee, J.C. и др., Nature, 372 (6508): 739-46 (1994)). Соединения, которые связываются с р38 МАР-киназой, эффективны при подавлении резорбции костей, воспаления и других иммунных патологий и патологий на основе воспаления. Характеристика р38 МАР-киназы и ее центральная роль в биосинтезе TNF АВ/23.07.2002 и IL-1 сделали эту киназу привлекательной целью для лечения заболеваний, опосредствованных этими цитокинами. Поэтому было бы желательно обеспечить ингибиторы р38 МАР-киназы и, таким образом, предоставить средства борьбы с заболеваниями, опосредствованными про-воспалительными цитокинами, как, например, TNF и IL-1. Один из аспектов настоящего изобретения (i) обеспечивает, таким образом, производное аминопиррола формулы
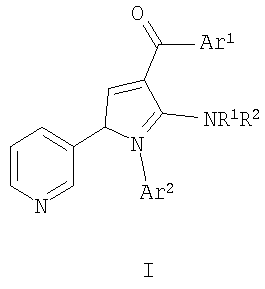
его пролекарство, отдельный изомер, смесь изомеров или его фармацевтически приемлемую соль и способы получения или применение таких соединений, где каждый из Ar1 и Ar2 означает независимо необязательно замещенный арил и каждый из R1 и R2 означает независимо водород, алкил или защитную группу при атоме азота. Более конкретно настоящее изобретение обеспечивает: (ii) соединение по (i), где R1 и R2 означают водород; (iii) соединения по (i) или (ii), где Ar2 означает галогензамещенный фенил; (iv) соединение по любому из (i) – (iii), где Ar2 означает 4-фторфенил; (v) соединение по любому из (i) – (iv), где Ar1 выбирается из группы, состоящей из фенила, замещенного алкоксигруппой фенила, замещенного гидроксильной группой фенила и замещенного гетероалкоксигруппой фенила; (vi) соединение по любому из (i) – (v), где Ar1 означает замещенный гетероалкоксигруппой фенил; (vii) соединение по любому из (i) – (vi), где Ar1 выбирается из группы, состоящей из фенила, 3-метоксифенила, 3-гидроксифенила и 3-(2,3-дигидроксипропокси)фенила. Другой аспект настоящего изобретения обеспечивает способ получения производного аминопиррола формулы
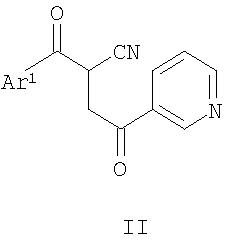
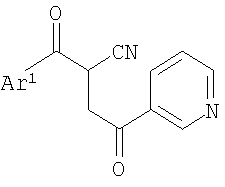
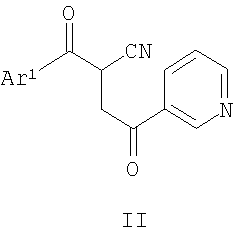
указанный способ включает образование кольцевой системы аминопиррола при взаимодействии содержащего циангруппу соединения формулы
с ариламином формулы Ar2-NH2 в условиях, подходящих для получения производного аминопиррола формулы I, где каждый из Ar1 и Ar2 является таким, как указано выше для (i) или для (iii) – (vii). Другой аспект настоящего изобретения обеспечивает композицию, включающую терапевтически эффективное количество соединения, указанного выше, и наполнитель. Еще один аспект настоящего изобретения обеспечивает способ ингибирования р38 МАР-киназы в клетке, включающий введение соединения формулы I в клетку, содержащую р38 МАР-киназу. Еще один аспект настоящего изобретения обеспечивает способ лечения заболевания у подвергаемого лечению млекопитающего путем введения ингибитора р38 МАР-киназы, включающий введение млекопитающему терапевтически эффективного количества соединения формулы I. Еще один аспект настоящего изобретения обеспечивает способ лечения заболевания у подвергаемого лечению млекопитающего путем введения ингибитора р38 МАР-киназы, а именно способ, включающий введение млекопитающему терапевтически эффективного количества одного или нескольких соединений, описанных выше, когда заболевание является воспалительным заболеванием и, более конкретно, когда заболевание является артритом. Согласно другому аспекту настоящее изобретение обеспечивает применение одного или нескольких соединений, как определено выше, для приготовления лекарственного средства для лечения заболевания у подвергаемого лечению млекопитающего путем введения ингибитора р38 МАР-киназы, а именно, когда заболевание является воспалительным заболеванием и, более конкретно, когда заболевание является артритом. Если не указано иначе, следующие термины, используемые в спецификации и пунктах формулы изобретения, имеют приведенные ниже значения. “Алкил” означает насыщенный одновалентный углеводородный остаток с прямой цепью, содержащий от одного до шести углеродных атомов, или насыщенный одновалентный углеводородный радикал с разветвленной цепью, содержащий от трех до шести атомов углерода, например метил, этил, пропил, 2-пропил, пентил и им подобные. “Алкоксигруппа” означает остаток -OR, где R означает алкил, как определено выше, например метокси-, этокси-, пропокси-, 2-пропоксигруппа и им подобные. “Ацил” означает остаток -C(O)R, где R означает водород, алкил, галоидалкил или гетероалкил, например ацетил, трифторацетил и им подобные. “Арил” означает одновалентный моноциклический или бициклический ароматический углеводородный радикал с 6-10 кольцевыми атомами, например фенил, 1-нафтил, 2-нафтил и им подобные. “Галогенид” означает фторид, хлорид, бромид или иодид. “Галоидалкил” означает алкил, замещенный одним или несколькими одинаковыми или различными атомами галоида, например -CH2Cl, -CF3, -СН2CF3, -СН2CCl3 и им подобные. “Гетероалкил” означает алкильный остаток, как определено выше, имеющий один или несколько, предпочтительно один, два или три заместителя, выбранные из -NRaRb, -ORc, где Ra, Rb и Rc независимо друг от друга означают водород, алкил или соответствующую защитную группу. Типичные примеры включают, но не ограничиваются этим, гидроксиметил, 3-гидроксипропил, 1,2-дигидроксиэтил, 2-метоксиэтил, 2-аминоэтил, 2-диметиламиноэтил и им подобные группы. “Гетероалкоксигруппа” означает остаток -OR, где R означает гетероалкильную группу, как определено выше, например 2-гидроксиэтокси-, 3-гидроксипропокси-, 2,3-дигидроксипропокси-, 2,3-дигидрокси-1-метилпропокси-, 2-аминоэтоксигруппу и им подобные группы. “Необязательно замещенный арил” означает арильное кольцо, как определено выше, которое необязательно замещено независимо одним или несколькими, предпочтительно одним или двумя, заместителями, выбранными из алкила, алкоксигруппы, гетероалкила, галогенида, циангруппы, ацила, -NRR’ (где R и R’ независимо выбираются из водорода, алкила или ацила), -NHCOR (где R означает алкил), -NRS(O)nR’ (где R означает водород или алкил, n означает целое число от 0 до 2 и R’ означает водород, алкил или гетероалкил), -NRS(O)nNR’R” (где R означает водород или алкил, n означает целое число от 0 до 2 и R’ и R” означают независимо водород, алкил или гетероалкил), -S(O)nR (где n является целым числом от 0 до 2 и R означает водород, алкил или гетероалкил), -S(O)nNRR’ (где n является целым числом от 0 до 2 и R и R’ означают независимо водород, алкил или гетероалкил), -COOR, -(алкилен)COOR (где R означает водород или алкил), -CONR’R” или -(алкилен)-CONR’R” (где R’ и R” означают независимо водород или алкил). “Необязательно замещенный фенил” означает фенил, который необязательно замещен независимо одним или несколькими, предпочтительно одним или двумя, заместителями, выбранными из алкила, алкоксигруппы, гетероалкила, галогенида, циангруппы, ацила, -NRR’ (где R и R’ независимо выбираются из водорода, алкила или ацила), -NHCOR (где R означает алкил), -NRS(O)nR’ (где R означает водород или алкил, n означает целое число от 0 до 2 и R’ означает водород, алкил или гетероалкил), -NRS(O)nNR’R’ (где R означает водород или алкил, n означает целое число от 0 до 2 и R’ и R” означают независимо водород, алкил или гетероалкил), -S(O)nR (где n является целым числом от 0 до 2 и R означает водород, алкил или гетероалкил), -S(O)nNRR'(где n является целым числом от 0 до 2 и R и R’ означают независимо водород, алкил или гетероалкил), -COOR, -(алкилен)COOR (где R означает водород или алкил), -CONR’R” или -(алкилен)CONR’R” (где R’ и R” означают независимо водород или алкил). “Необязательный” или “необязательно” означает, что описанное затем явление или обстоятельство может необязательно иметь место и что описание включает примеры, когда такое явление или обстоятельство имеет место, и примеры, когда это не имеет места. Например, “арильная группа, необязательно моно- или дизамещенная алкильной группой” означает, что алкил может присутствовать или может отсутствовать и описание подразумевает ситуации, когда арильная группа моно- или дизамещена алкильной группой, и ситуации, когда гетероциклическая группа не замещена алкильной группой. “Фармацевтически приемлемый наполнитель” означает наполнитель, который полезен для приготовления фармацевтической композиции, который обычно безопасен, нетоксичен и не является нежелательным ни биологически, ни по другим соображениям, и включает наполнитель, который приемлем для использования как в ветеринарии, так и при фармацевтическом применении для людей. “Фармацевтически приемлемый наполнитель”, как он используется в спецификации и пунктах формулы изобретения, включает как один, так и более чем один, из таких наполнителей. “Пролекарства” означает любое соединение, которое высвобождает in vivo активное исходное лекарственное средство согласно формуле (I), когда такое пролекарство вводится млекопитающему. Пролекарства соединения формулы (I) получают при модифицировании присутствующих в соединении формулы (I) функциональных групп таким образом, чтобы модификации могли расщепляться in vivo для высвобождения исходного соединения. Пролекарства включают соединения формулы (I), где гидроксил, аминогруппа или сульфгидрильная группа в соединении (I) связана с любой группой, которая может быть расщеплена in vivo с целью регенерации свободного гидроксила, свободной аминогруппы или сульфгидрильной группы соответственно. Примеры пролекарств включают, но не ограничивают этим, сложные эфиры (например, ацетаты, формиаты и бензоаты), карбаматы (например, N,N-диметиламинокарбонильное производное) по гидроксильным функциональным группам в соединениях формулы (I) и им подобные. “Обработка” или “лечение” заболевания включает (1) предупреждение заболевания, то есть способствование тому, чтобы у млекопитающего не развились клинические симптомы заболевания, которые могут подвергать опасности заболевания или провоцировать заболевание, хотя млекопитающее еще не испытывает или не обнаруживает симптомы заболевания, (2) подавление заболевания, то есть задержка или уменьшение развития заболевания или его клинических симптомов или (3) облегчение заболевания, то есть способствование регрессии заболевания или его клинических симптомов. Когда имеется в виду химическая реакция, термины “обработка”, “связывание” и “взаимодействие” используются здесь взаимозаменяемо и относятся они к прибавлению или смешиванию двух или нескольких реагентов при определенных условиях для получения указанного и/или желаемого продукта. Следует принять во внимание, что реакция, которая приводит к указанному и/или желаемому продукту, может не обязательно непосредственно быть результатом соединения двух реагентов, которые прибавляли сначала, то есть может наблюдаться одно или несколько промежуточных соединений, которые образуются в смеси, что в конце концов приводит к образованию указанного и/или желаемого продукта. “Терапевтически эффективное количество” означает количество соединения, которое, будучи введенным млекопитающему для лечения заболевания, достаточно для достижения эффективного лечения заболевания. “Терапевтически эффективное количество” варьируется в зависимости от соединения, заболевания и его тяжести, возраста, массы и т.д. подвергающегося лечению млекопитающего. Один аспект настоящего изобретения обеспечивает
его пролекарство, отдельный изомер, смесь изомеров или его фармацевтически приемлемую соль и способы их получения или их применение, где Ar1, Ar2, R1 и R2 являются такими, как указано выше. Предпочтительно R1 и R2 означают водород. Предпочтительно Ar2 означает необязательно замещенный фенил, более предпочтительно означает галогензамещенный фенил и наиболее предпочтительно означает 4-галоидфенил, в особенности 4-фторфенил. Предпочтительно Ar1 выбирают из группы, состоящей из фенила, замещенного алкоксигруппой фенила, замещенного гидроксильной группой фенила и замещенного гетероалкоксигруппой фенила. Более предпочтительно Ar1 выбирается из группы, состоящей из фенила, 3-метоксифенила, 3-гидроксифенила и 3-(2,3-дигидроксипропокси)фенила. В одном варианте осуществления настоящего изобретения R1 и R2 означают водород и Ar2 означает необязательно замещенный фенил. В другом варианте осуществления изобретения R1 и R2 означают водород и Ar2 означает галогензамещенный фенил, предпочтительно 4-фторфенил. Еще в одном варианте осуществления изобретения R1 и R2 означают водород, Ar2 означает 4-фторфенил и Ar1 выбирается из группы, состоящей из фенила, замещенного алкоксигруппой фенила, замещенного гидроксильной группой фенила и замещенного гетероалкоксигруппой фенила. Предпочтительно Ar1 означает замещенный гетероалкоксигруппой фенил. Более предпочтительно Ar1 означает 3-(2,3-дигидроксипропокси)фенил. Однако еще в одном варианте осуществления изобретения R1 и R2 означают водород, Ar2 означает 4-фторфенил и Ar1 означает фенил, 3-метоксифенил, 3-гидроксифенил или 3-(2,3-дигидроксипропокси)фенил. В другом варианте осуществления изобретения R1 и R2 означают водород и Ar1 означает необязательно замещенный фенил. Предпочтительно Ar1 означает фенил, замещенный алкоксигруппой фенил, замещенный гидроксильной группой фенил или замещенный гетероалкоксигруппой фенил. Предпочтительно Ar1 означает замещенный гетероалкоксигруппой фенил. Более предпочтительно Ar1 означает 3-(2,3-дигидроксипропокси)фенил. В рамках данного варианта осуществления изобретения Ar2 предпочтительно означает галогензамещенный фенил, предпочтительно 4-фторфенил. Комбинации предпочтительных групп, описанных выше, также составляют другие предпочтительные варианты осуществления изобретения. Так, например, предпочтительные заместители R1 и R2 являются также предпочтительными заместителями соединении, содержащих предпочтительные заместители Ar1 и/или Ar2. Соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. Обычно сольватированные формы, включая гидратированные формы, эквивалентны несольватированным формам и подразумевается, что они охватываются объемом настоящего изобретения. Кроме того, как установлено выше, настоящее изобретение также включает все фармацевтически приемлемые соли таких соединений вместе с пролекарственными формами соединений и все стереоизомеры или в чистой хиральной форме, или в виде рацемической смеси, или в виде смеси другого вида. Соединения формулы I способны также образовывать фармацевтически приемлемые кислотно-аддитивные соли. Все из этих форм входят в объем настоящего изобретения. Фармацевтически приемлемые кислотно-аддитивные соли соединений формулы I включают соли, производные неорганических кислот, как, например, соляная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, иодистоводородная кислота, фосфористая кислота и им подобные кислоты, а также соли, производные органических кислот, как, например, алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые кислоты, алкандиовые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты и т.д. Такие соли, таким образом, включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, нитраты, фосфаты, вторичные фосфаты, первичные фосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, соли каприловой кислоты, соли изомасляной кислоты, оксалаты, малонаты, соли янтарной кислоты, соли пробковой кислоты, соли себациновой кислоты, соли фумаровой кислоты, малеиновой кислоты, миндальной кислоты, бензоаты, соли хлорбензойной кислоты, соли толуиловой кислоты, соли динитробензойной кислоты, фталаты, бензолсульфонаты, толуолсульфонаты, соли фенилуксусной кислоты, цитраты, соли молочной кислоты, соли малеиновой кислоты, соли винной кислоты, соли метансульфокислоты и им подобные соли. Также рассматриваются соли аминокислот, как, например, соли аргинина и им подобные, и соли глюконовой кислоты и галактуроновой кислоты (см., например, Berge и др., “Фармацевтические соли,” J. of Pharmaceutical Science, 1977, 66, 1-19). Кислотно-аддитивные соли основных соединений могут быть синтезированы при взаимодействии соединения в виде свободного основания с достаточным количеством желаемой кислоты для получения соли общепринятым способом. Форма в виде свободного основания может быть регенерирована при взаимодействии соли с основанием и выделении свободного основания общепринятым способом. Формы в виде свободных оснований могут несколько отличаться от соответствующих форм в виде солей некоторыми физическими свойствами, как, например, растворимость в полярных растворителях, но, с другой стороны, соли эквивалентны соответствующим им свободным основаниям для целей настоящего изобретения. Фармацевтически приемлемые основно-аддитивные соли могут быть образованы с помощью ионов металлов или аминов, как, например, ионы щелочных и щелочноземельных металлов или органические амины. Примеры ионов металлов, которые применяются в качестве катионов, включают натрий, калий, магний, кальций и им подобные. Примерами подходящих аминов являются N,N’-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метилглюкамин и прокаин (см., например, Berge и др., “Фармацевтические соли,” J. of Pharmaceutical Science, 1977, 66, 1-19). Основно-аддитивные соли кислотных соединений могут быть синтезированы при взаимодействии формы в виде свободной кислоты с достаточным количеством желаемого основания для получения соли обычным способом. Форма в виде свободной кислоты может быть регенерирована при взаимодействии соли с кислотой и выделении свободной кислоты общепринятым способом. Формы в виде свободных кислот могут несколько отличаться от соответствующих форм в виде солей некоторыми физическими свойствами, как, например, растворимость в полярных растворителях, но, с другой стороны, соли эквивалентны соответствующим им свободным кислотам для целей настоящего изобретения. Примеры соединений по настоящему изобретению представлены в таблице.
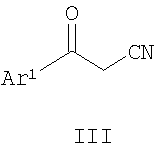
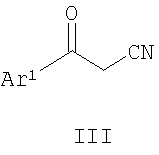
Соединения по настоящему изобретению могут быть получены с помощью описанных ниже способов. Исходные вещества и реагенты, используемые для получения этих соединений, либо доступны от коммерческих поставщиков, как, например, Aldrich Chemical Co. (Милуоки, штат Висконсин, США), Bachem (Торранс, штат Калифорния, США), Emka-Chemie или Sigma (Сент-Луис, штат Миссури, США), или их получают способами, известными специалистам в данной области, следуя методикам, представленным в ссылках, таких как Fieser and Fieser’s Reagents for Organic Synthesis, тома 1-17 (изд-во John Wiley and Sons, 1991); Rodd’s Chemistry of Carbon Compounds, тома 1-5 и Приложения (изд-во Elsevier Science Publishers, 1989), Organic Reactions, тома 1-40 (изд-во John Wiley and Sons, 1991), March’s Advanced Organic Chemistry (изд-во John Wiley and Sons, 4-ое издание) и Larock’s Comprehensive Organic Transformations (изд-во VCH Publishers Inc., 1989). Эти схемы являются только иллюстрацией некоторых способов, с помощью которых могут быть синтезированы соединения по изобретению, и могут быть осуществлены различные модификации этих схем, очевидные для специалиста в данной области, относящиеся к данному раскрытию. Исходные вещества и промежуточные соединения реакции могут быть выделены и очищены, если желательно, с применением обычных методик, включая, но не ограничивая этим, фильтрацию, перегонку, кристаллизацию, хроматографию и им подобные методики. Такие вещества могут быть охарактеризованы с применением обычных способов, включая физические константы и спектральные данные. В одном варианте осуществления изобретения Ar2 означает 4-фторфенил. В другом варианте осуществления изобретения Ar2 означает 4-фторфенил и Ar1 означает замещенный алкоксигруппой фенил. Еще в одном варианте осуществления изобретения Ar2 означает 4-фторфенил и Ar1 означает замещенный алкоксигруппой фенил и способ дополнительно включает превращение алкоксильного заместителя в гидроксильный заместитель при взаимодействии производного аминопиррола формулы I с кислотой Льюиса в условиях, подходящих для получения производного аминопиррола формулы I, где Ar1 означает замещенный гидроксильной группой фенил. Согласно еще одному варианту осуществления изобретения способ дополнительно включает алкилирование гидроксильной группы в Ar1 путем взаимодействия производного аминопиррола формулы I, где Ar1 означает замещенный гидроксильной группой фенил, с гетероалкильным производным, включающим уходящую группу, в условиях, требующихся для получения производного аминопиррола формулы I, где Ar1 является фенилом, замещенным гетероалкоксигруппой. Однако еще в одном варианте осуществления изобретения содержащее циангруппу соединение формулы II получают при реакции ароильного производного ацетонитрила формулы
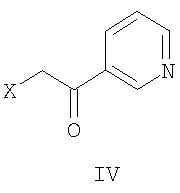
с 3-галоидацетилпиридином формулы
в присутствии основания в условиях, подходящих для получения упомянутого содержащего циангруппу соединения формулы II, где Ar1 означает необязательно замещенный арил и Х является уходящей группой. Один характерный способ получения соединений формулы I включает образование кольцевой системы аминопиррола при реакции содержащего циангруппу соединения формулы
с ариламином формулы Ar2-NH2 в условиях, подходящих для получения производного аминопиррола формулы I, где Ar1 и Ar2 являются такими, как установлено выше. Реакция образования аминопиррольного кольца является типичной катализируемой кислотой реакцией циклизации. Предпочтительно кислота является сильной кислотой с рН около 2 или меньше. Подходящие кислотные катализаторы включают неорганические кислоты, как, например, серная кислота, фосфорная кислота, соляная кислота, бромистоводородная кислота, иодистоводородная кислота, а также кислоты Льюиса, как, например, AlCl3, BBr3, BCl3 и им подобные соединения. Следует учесть, что когда источник протонов доступен, кислоты Льюиса могут также генерировать неорганическую протонную кислоту, которая также может катализировать реакцию циклизации. Циклизацию обычно проводят в полярном растворителе, как, например, этанол, изопропанол и им подобные. Температура реакции циклизации зависит от ряда факторов, включая применяемый специфический катализатор, растворитель для реакции, реакционную способность исходного вещества и т.д. Обычно температура реакции циклизации составляет, как минимум, около 80°С. Практически реакцию циклизации проводят в условиях кипения растворителя реакции с обратным холодильником. Время реакции циклизации также зависит от ряда факторов, например таких, которые описаны выше, включая температуру реакции. Обычно, однако, время реакции циклизации составляет, как минимум, 8 часов в условиях кипячения с обратным холодильником. Как правило, время реакции циклизации составляет примерно от 6 часов до 16 часов. Содержащее циангруппу соединение формулы II может быть легко получено при реакции ароильного производного ацетонитрила формулы
с 3-галоидацетилпиридином формулы
в присутствии основания в условиях, необходимых для получения содержащего циангруппу соединения формулы II, где Ar1 является таким, как установлено выше, и Х является уходящей группой, как, например, галогенид, предпочтительно бромид или хлорид. Подходящими основаниями для реакции замещения являются обычно совсем не нуклеофильные основания. Предпочтительно основание является достаточно сильным, чтобы депротонировать ароильное производное ацетонитрила формулы III. Подходящие основания включают гидриды металлов, трет-бутоксиды металлов и им подобные соединения. Так как обычно применяют сильное основание, первоначальная реакция депротонирования, протекающая между основанием и ароильным производным ацетонитрила формулы III, является экзотермической реакцией. По существу температура реакции обычно поддерживается около 0°С или ниже. Типичным растворителем для реакции является апротонный растворитель, как, например, тетрагидрофуран и диэтиловый эфир. Способы получения соединений формулы I могут также включать модификацию арильной группы Ar1 или Ar2. Например, когда арильная группа Ar1 содержит заместитель, способы по настоящему изобретению могут включать замещение или модификацию заместителя при арильной группе. Это, в частности, применимо, где Ar1 имеет один или несколько заместителей, представляющих аминогруппу, карбонил, гидроксильную или алкоксигруппу. Когда Ar1 замещен алкоксигруппой, последняя может быть превращена в гидроксильную группу при взаимодействии соединения формулы I с кислотой Льюиса. Подходящие кислоты Льюиса включают такие, которые описаны в Protective Groups in Organic Synthesis, 3-е издание, T.W.Greene и P.G.M.Wuts, изд-во John Wiley & Sons, Нью-Йорк, 1999, это во всей полноте включено сюда в виде ссылок. Свободная гидроксильная группа может быть замещена (например, проалкилирована) желаемым заместителем. Например, взаимодействие гидроксильной группы с гетероалкильным производным, включающим уходящую группу, приводит к производному аминопиррола формулы I, где Ar1 является фенилом, замещенным гетероалкоксигруппой. Соединения по настоящему изобретению характеризуются широким разнообразием фармацевтической активности. Например, согласно настоящему изобретению было обнаружено, что соединения по изобретению являются ингибиторами р38 МАР-киназы. Таким образом, такие соединения пригодны для лечения воспалительных заболеваний, в частности артрита. Следовательно, соединения по настоящему изобретению полезны при лечении заболевания, опосредствованного р38 МАР-киназой, включая ревматоидный артрит, остеоартрит, спондилит, заболевания, связанные с резорбцией костей, сепсис, септический шок, синдром токсического шока, эндотоксический шок, туберкулез, атеросклероз, диабет, респираторный дистресс-синдром взрослых, хроническое воспалительное легочное заболевание, лихорадочное состояние, периодонтальные заболевания, язвенный колит, изжога, болезни Альцгеймера и Паркинсона. Способность соединений по настоящему изобретению ингибировать р38 МАР-киназу была продемонстрирована при испытании in vitro, описанном в примере 4. Способность соединений по настоящему изобретению ингибировать высвобождение TNF- Обычно соединения по настоящему изобретению вводят в терапевтически эффективном количестве с помощью любого из принятых способов введения средств, которые служат таким же целям. Фактическое количество соединения по настоящему изобретению, то есть активного ингредиента, обычно зависит от многих факторов, как, например, тяжесть подвергающегося лечению заболевания, возраст и относительное здоровье субъекта, эффективность используемого соединения, способ и форма введения и другие факторы. Терапевтически эффективные количества соединений по настоящему изобретению могут варьироваться примерно от 0,1 до 50 мг на килограмм массы тела реципиента в день, предпочтительно примерно 1-30 мг/кг/день. Так, для введения больному с массой тела 70 кг дозы наиболее предпочтительно составляют примерно от 70 мг до 2,1 г в день. Обычно соединения по настоящему изобретению вводят в виде фармацевтических композиций любым из следующих способов: путем перорального, системного введения (например, чрескожного, интраназального или с помощью суппозитория) или путем парентерального введения (например, внутримышечно, внутривенно или подкожно). Предпочтительным способом введения является пероральный, использующий подходящую ежедневную схему приема лекарственного средства, которая может быть подобрана в соответствии со степенью поражения болезнью. Композиции могут иметь форму таблеток, пилюль, капсул, полутвердых веществ, порошков, лекарственных форм с поддерживаемым высвобождением, растворов, суспензий, эликсиров, аэрозолей или любых других подходящих композиций. Выбор лекарственной формы зависит от различных факторов, как, например, способ введения лекарственного средства (например, для перорального введения предпочтительны формы в виде таблеток, пилюль или капсул) и биологическая доступность лекарственного вещества. Недавно были разработаны фармацевтические композиции, особенно для лекарственных средств с низкой биологической доступностью, основанные на том принципе, что биологическая доступность может быть повышена за счет увеличения площади поверхности, то есть уменьшения размера частиц. Например, в патенте US №4107288 описывается фармацевтическая композиция с размером частиц в диапазоне от 10 до 1,000 нм, в которой активное вещество поддерживается на матрице из макромолекул с поперечной сшивкой. Патент US №5145684 описывает получение фармацевтической композиции, в которой лекарственное вещество измельчается до наночастиц (средний размер частиц 400 нм) в присутствии модификатора поверхности и затем диспергируется в жидкой среде для получения фармацевтической композиции, которая характеризуется необыкновенно высокой биологической доступностью. Композиции состоят обычно из соединения формулы (I) в комбинации с, как минимум, одним фармацевтически приемлемым наполнителем. Приемлемые наполнители являются нетоксичными, помогают введению и не оказывают неблагоприятного воздействия на полезный эффект соединения формулы (I). Таким наполнителем может быть любой твердый, жидкий, полутвердый или, в случае аэрозольной композиции, газообразный наполнитель, который обычно доступен специалисту в данной области. Твердые фармацевтические наполнители включают крахмал, целлюлозу, тальк, глюкозу, лактозу, сахарозу, желатину, солод, рис, муку, мел, силикагель, стеарат магния, стеарат натрия, моностеарат глицерина, хлористый натрий, высушенные снятые сливки с молока и им подобные вещества. Жидкие и полутвердые наполнители могут быть выбраны из глицерина, пропиленгликоля, воды, этанола и различных масел, включая таковые нефтяного, животного, растительного или синтетического происхождения, например масло земляного ореха, масло сои культурной, минеральное масло, кунжутное масло и т.д. Предпочтительные жидкие носители, особенно для инъецируемых растворов, включают воду, физиологический раствор, водную декстрозу и гликоли. Сжатые газы могут быть применены для диспергирования вещества по данному изобретению в аэрозольной форме. Инертными газами, подходящими для этой цели, являются азот, углекислый газ и т.д. Другие подходящие фармацевтические наполнители и технологии приготовления лекарственных форм с их участием описаны в Remington’s Pharmaceutical Sciences, под редакцией Е.W.Martin (изд-во Mack Publishing Company, 18-oe издание, 1990). Количество соединения в готовой лекарственной форме может варьироваться в широком диапазоне при применении специалистами в данной области. Обычно лекарственная форма содержит, исходя из массовых процентов (мас.%), примерно от 0,01-99,99 мас.% соединения формулы (I) от общей массы формы, уравновешенных одним или несколькими подходящими фармацевтическими наполнителями. Предпочтительно содержание соединения составляет примерно 1-80 мас.%. Образцы лекарственных форм, содержащих соединение формулы (I), описаны в примере 3. ПРИМЕРЫ Пример 1 Данный пример иллюстрирует способ получения [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]фенилметанона.
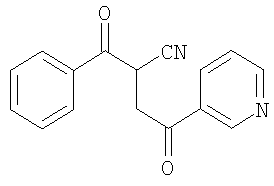
Стадия а: получение 2-бензоил-4-оксо-4-пиридин-3-илбутиронитрила
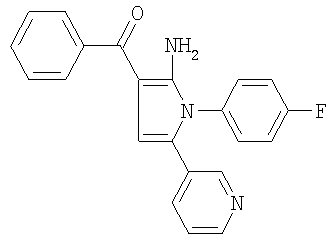
К 3,0 г (21 ммоль) бензоилацетонитрила в 50 мл тетрагидрофурана, охлажденного в бане с тающим льдом, прибавляли 0,84 г (21 ммоль) гидрида натрия (60% дисперсия в масле). Через 1 час прибавляли 2,8 г (10 ммолей) гидробромида 3-бромацетилпиридина. Через 3 часа реакционную смесь выливали в соляной раствор, экстрагировали этилацетатом, сушили над сульфатом натрия, концентрировали в вакууме и очищали с помощью флэш-хроматографии (градиентное элюирование: 40-80% этилацетат/гексан), получали 2,6 г (93%) 2-бензоил-4-оксо-4-пиридин-3-илбутиронитрила (МН+=265). Стадия б: получение [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]фенилметанона
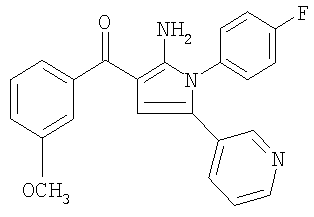
К раствору 2,6 г (9,8 ммоля) 2-бензоил-4-оксо-4-пиридин-3-илбутиронитрила и 0,93 мл (9,8 ммоля) 4-фторанилина в 30 мл этилового спирта прибавляли 6 капель концентрированной соляной кислоты и смесь кипятили с обратным холодильником. Через 16 часов реакционную смесь охлаждали до комнатной температуры и твердое вещество желтого цвета выделяли с помощью фильтрации. Твердое вещество перекристаллизовывали из метилового спирта/этилацетата, получали 1,5 г (42%) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]фенилметанона (tпл=231,4-231,8°С). Обработка раствора полученного свободного основания в этилацетате с помощью HCl/диэтилового эфира приводила к хлористоводородной соли [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]фенилметанона (tпл 218-222°С). Пример 2 Данный пример иллюстрирует способ получения [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-{[2(S),3-дигидроксипропокси]фенил)метанона.
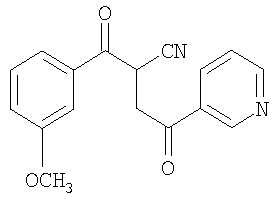
Стадия а: получение 2-(3-метоксибензоил)-4-оксо-4-пиридин-3-илбутиронитрила
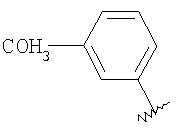
К 1,3 г (7,5 ммоля) 3-метоксибензоилацетонитрила в 30 мл тетрагидрофурана, охлажденного в бане с тающим льдом, прибавляли 0,3 г (7,5 ммоля) гидрида натрия (60% дисперсия в масле). Через 1 час прибавляли 1,0 г (3,6 ммоля) гидробромида 3-бромацетилпиридина. Через 3 часа реакционную смесь выливали в соляной раствор, экстрагировали этилацетатом, сушили над сульфатом натрия, концентрировали в вакууме и очищали с помощью флэш-хроматографии (градиентное элюирование: 40-100% этилацетат/гексан), получали 0,95 г (43%) 2-(3-метоксибензоил)-4-оксо-4-пиридин-3-илбутиронитрила (МН+=295). Стадия б: получение [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-(метоксифенил)метанона
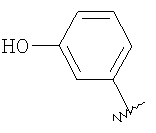
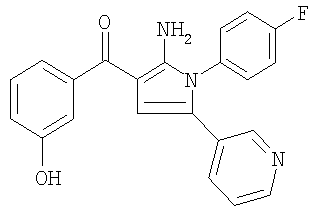
Смесь 3,0 г (10,2 ммоля) 2-(3-метоксибензоил)-4-оксо-4-пиридин-3-илбутиронитрила, 0,97 мл (10,2 ммоля) 4-фторанилина и 6 капель концентрированной соляной кислоты в 30 мл этилового спирта кипятили с обратным холодильником. Через 16 часов реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, разбавляли водным раствором бикарбоната натрия и экстрагировали этилацетатом. Экстракты промывали соляным раствором, сушили над сульфатом натрия, концентрировали в вакууме и очищали с помощью флэш-хроматографии (градиентное элюирование: 20-40% этилацетат/гексан), получали 1,0 г (25%) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-(метоксифенил)метанона (МН+=388). Стадия в: получение [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-(гидроксифенил)метанона
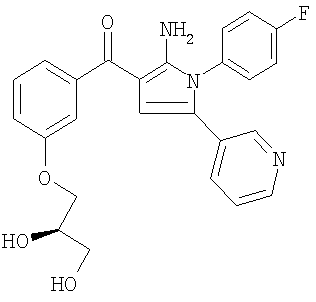
К раствору 1,0 г (2,6 ммоля) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-(метоксифенил)метанона в 25 мл дихлорметана, охлажденному в бане с тающим льдом, прибавляли 15,5 мл (155 ммолей) трехбромистого брома (1,0 М в дихлорметане). Реакционной смеси давали нагреться до комнатной температуры. Через 16 часов реакционную смесь повторно охлаждали в бане с тающим льдом и прибавляли по каплям воду. Устанавливали в реакционной смеси рН 10 с помощью концентрированной гидроокиси аммония и затем экстрагировали дихлорметаном. Органические экстракты промывали соляным раствором, сушили над сульфатом натрия, концентрировали в вакууме и очищали с помощью флэш-хроматографии (градиентное элюирование: 40-80% этилацетат/гексан), получали 0,6 г (62%) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-(гидроксифенил)метанона (t пл 240,3-242,5°С). Стадия г: получение [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-[3-(2,2-диметил[1,3]диоксолан-4(S)-илметокси)фенил]метанона
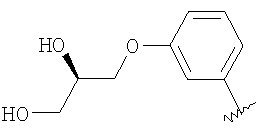
Смесь 0,6 г (1,6 ммоля) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-(гидроксифенил)метанона, 1,06 г (3,7 ммоля) L- Стадия д: получение [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-{[2(S),3-дигидроксипропокси]фенил}метанона
Смесь 0,7 г (1,44 ммоля) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-[3-(2,2-диметил[1,3]диоксолан-4(S)-илметокси)фенил]метанона и 0,35 г п-толуолсульфокислоты в 20 мл метилового спирта и 5 мл воды нагревали до 50°С. Через 18 часов реакционную смесь охлаждали до комнатной температуры и растворитель выпаривали при пониженном давлении. Остаток распределяли между водным бикарбонатом натрия и этилацетатом. Органические экстракты промывали соляным раствором, сушили над сульфатом натрия и концентрировали в вакууме. Продукт очищали с помощью флэш-хроматографии (градиентное элюирование: 100% этилацетат-10% метиловый спирт/этилацетат/0,4% гидроокись аммония). Очищенный продукт превращали в хлористоводородную соль при обработке раствора в этилацетате с помощью HCl/диэтилового эфира. Соль выделяли при фильтрации и сушили, получали 0,4 г (56%) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-3-{[2(S),3-дигидрокси-пропокси]фенил}метанона (МН+=448). Пример 3 Далее приведены образцы приготовленных лекарственных форм, содержащих соединение формулы (I). Лекарственная форма в виде таблеток Следующие ингредиенты тщательно смешивают и прессуют в таблетки с одной бороздкой.
Лекарственная форма в виде капсул Следующие ингредиенты тщательно смешивают и помещают в желатиновую капсулу с твердой оболочкой.
Лекарственная форма в виде суспензии Смешивают следующие ингредиенты для получения суспезии для перорального введения
Лекарственная форма для инъекций Смешивают следующие ингредиенты для получения лекарственной формы для инъекций.
Все из указанных выше ингредиентов, кроме воды, смешивают и нагревают до 60-70°С при перемешивании. Затем при 60°С прибавляют при интенсивном перемешивании достаточное количество воды для эмульгирования ингредиентов и затем прибавляют достаточное количество воды до 100 г. Лекарственная форма в виде суппозиториев Суппозиторий общей массой 2,5 г изготавливают при смешивании соединения по изобретению с Вайтпсолом® Н-15 (триглицериды насыщенной растительной жирной кислоты; фирма Riches-Nelson, Inc., Нью-Йорк), суппозиторий имеет следующий состав:
Пример 4 Ингибирование р38 МАР-киназы при анализе in vitro Ингибирующую активность соединений по данному изобретению в отношении р38 МАР-киназы in vitro определяли путем измерения переноса Фосфорилированную форму рекомбинантной р38 МАР-киназы экспрессировали с киназами SEK-1 и МЕКК в E.Coli и затем очищали с помощью аффинной хроматографии, используя никелевую колонку. Фосфорилированную р38 МАР-киназу разбавляли в буфере для киназы (20 мМ 3-(N-морфолин)пропансульфокислота, рН 7,2, 25 мМ Соединения по изобретению проявляли активность в данном анализе. Ингибирующая активность в отношении р-38 (выраженная как IC50, концентрация, вызывающая 50%-ное ингибирование исследуемого фермента р-38) некоторых соединений по изобретению составляет:
Пример 5 Ингибирование индуцированного липополисахаридами продуцирования TNF- Способность соединений по настоящему изобретению ингибировать высвобождение TNF- (а) Индуцирование биосинтеза TNF Клетки ТНР-1 суспендировали в культуральной среде [RPMI (Gibco-BRL, Gailthersburg, MD), содержащая 15%-ную сыворотку плода коровы, 0,02 мМ 2-меркаптоэтанол] в концентрации 2,5×10 клеток/мл и затем помещали в 96-ячеечный планшет (аликвоты в 0,2 мл в каждую ячейку). Исследуемые соединения растворяли в диметилсульфоксиде и затем разбавляли культуральной средой таким образом, чтобы конечная концентрация диметилсульфоксида составляла 5%. В каждую ячейку прибавляли аликвоты в 20 мкл исследуемого раствора или только среду с ДМСО (контроль). Клетки инкубировали 30 минут при 37°С. В ячейки прибавляли липополисахарид (фирма Sigma, Сент-Луис, штат Миссури, США) до конечной концентрации 0,5 мкг/мл и клетки инкубировали еще 2 часа. В конце периода инкубирования собирали надосадочные культуральные жидкости и определяли количество присутствующего TNF-a, применяя ферментный иммуносорбентный анализ (ELISA), как описано ниже. (б) ELISA-анализ Количество присутствующего человеческого TNF- В ячейки полистирольных 96-ячеечных планшетов помещали по 50 мкл в ячейку антитела 2TNF-H22 в забуференном фосфатом физиологическом растворе (ЗФР) (10 мкг/мл) и инкубировали в увлажненной камере при 4°С в течение ночи. Планшеты промывали забуференным фосфатом физиологическим раствором, затем блокировали 5%-ным нежирным сухим молоком в ЗФР в течение 1 часа при комнатной температуре и промывали 0,1% бычьим сывороточным альбумином (БСА) в ЗФР. Стандарты TNF получали из исходного раствора человеческого рекомбинантного TNF- Аликвоты в 25 мкл указанных выше культуральных надосадочных жидкостей или стандартов TNF или только среды (контроль) смешивали с аликвотами в 25 мкл биотинилированного моноклонального антитела 2TNF-H34 (2 мкг/мл в ЗФР, содержащем 0,1% БСА) и затем прибавляли в каждую ячейку. Образцы инкубировали 2 часа при комнатной температуре при осторожном встряхивании и затем трижды промывали 0,1% БСА в ЗФР. В каждую ячейку прибавляли 50 мкл раствора пероксидазы-стрептавидина (Zymed, S., Сан-Франциско, Калифорния), содержащего 0,416 мкг/мл пероксидазы-стрептавидина и 0,1% БСА в ЗФР. Образцы инкубировали дополнительно 1 час при комнатной температуре и затем промывали 4 раза 0,1% БСА в ЗФР. В каждую ячейку прибавляли 50 мкл раствора О-фенилендиамина (1 мкг/мл О-фенилендиамина и 0,03% перекиси водорода в 0,2 М цитратном буфере, рН 4,5) и образцы инкубировали в темноте 30 минут при комнатной температуре. Оптическую плотность образца и стандарта определяли при 450 и 650 нм соответственно. Содержание TNF- Величину IC50 определяют как концентрацию исследуемого соединения, соответствующую уменьшению максимального поглощения при 450 нм на 50%. Соединения по изобретению проявляли активность в этом анализе. Пример 6 Ингибирование индуцированного липополисахаридами продуцирования TNF- Способность соединений по изобретению ингибировать высвобождение TNF- Самок крыс линии Sprague-Dawley массой 110-140 г (Charles River, Холлистер, Калифорния, США) подвергают акклиматизации в течение недели. Группам, состоящим из 8 мышей каждая, вводят перорально или исследуемые соединения, растворенные в водном носителе, содержащем 0,9% хлорида натрия, 0,5% натрийкарбоксиметилцеллюлозы, 0,4% полисорбата 80, 0,9% бензилового спирта (карбоксиметилцеллюлозный носитель), или только носитель (контрольная группа). Через 30 минут мышам инъецируют внутрибрюшинно (50 мкг/кг) липополисахариды (фирма Sigma, Сент-Луис, штат Миссури, США). Через 1,5 часа мышей умерщвляют путем ингаляции CO2 и кровь собирают с помощью пункции сердца. Кровь осветляют путем центрифугирования при 15,600 × g в течение 5 минут и сыворотки переносят в чистые пробирки и замораживают при -20°С до проведения ELISA-анализа на TNF- Пример 7 Анализ адъювантного артрита у крыс: анализ in vivo Противовоспалительная активность соединений по настоящему изобретению может быть определена путем использования вызванного адъювантом артрита у крыс. Если кратко, самки крыс линии Sprague-Dawley массой 120-155 г (Charles River, Холлистер, штат Калифорния, США) подвергаются акклиматизации в помещении в течение примерно 1 недели до испытания. В день 1 животным впрыскивают внутрикожно в 1/4 проксимального участка хвоста 0,1 мл суспензии погибших в результате термообработки и высушенных Mycobacterium Butyricum (Difco, Bacto., Des., серия 115979JA/EXP9/99) в минеральном масле (фирма Sigma, Сент-Луис, штат Миссури, США) в концентрации 1 мг/0,1 мл. В день 7 вводят исследуемые соединения в карбоксиметилцеллюлозном носителе в течение периода до дня 18. В день 18 после введения соединения животных взвешивают. Получают клинические показатели для оценки интенсивности отека на четырех лапах и хвосте. Балльный показатель от 0 до 4 устанавливают для каждой лапы и от 0 до 3 для хвоста, так чтобы максимальный балл составлял 19. Животным с полиартритом устанавливают балл 0, когда не наблюдается воспалительных признаков (припухлость и покраснение) в любом из малых суставов (интрафаланговых, пястно-фаланговых, плюснефаланговых) и крупных суставов (лучезапястный сустав/запястье, голеностопный сустав/предплюсна). Животных оценивают баллом 1, когда наблюдается слабое воспаление, баллом 2 при умеренном отеке, баллом 3 при серьезном отеке и баллом 4, когда имеет место очень серьезный отек. Хвосту присваивают балл 0, когда не наблюдается никаких признаков отека или омертвевшей ткани, балл 1, когда в местах впрыскивания и в непосредственно окружающих их тканях наблюдается легкий отек, балл 2, когда примерно 1/4 хвоста либо воспалена, либо там обнаруживается некротическая ткань, и балл 3, когда на площади более 1/4 хвоста обнаруживается тяжелый некроз или отеки. После клинических оценок на задних лапах делают поперечные разрезы по периферической большеберцовой кости, ближе к предплюсневому суставу. Левую и правую задние лапы взвешивают отдельно и показатели регистрируют.
Формула изобретения
1. Соединение формулы
его изомер, смесь изомеров или его фармацевтически приемлемая соль, где каждый из радикалов Ar1 и Ar2 независимо означает необязательно замещенный фенил, а каждый из R1 и R2 независимо означает водород, алкил или защитную группу при атоме азота. 2. Соединение по п.1, где R1 и R2 означают водород. 3. Соединение по п.1, где Ar2 означает галогензамещенный фенил. 4. Соединение по п.1, где Ar2 означает 4-фторфенил. 5. Соединение по п.1, где Ar1 выбран из группы, включающей фенил, замещенный алкоксигруппой фенил, замещенный гидроксильной группой фенил и замещенный гетероалкоксигруппой фенил. 6. Соединение по п.1, где Ar1 означает замещенный гетероалкоксигруппой фенил. 7. Соединение по п.1, где Ar1 выбран из группы, включающей фенил, 3-метоксифенил, 3-гидроксифенил и 3-(2,3-дигидроксипропокси)фенил. 8. Фармацевтическая композиция, обладающая противовоспалительной активностью и включающая терапевтически эффективное количество одного или нескольких соединений по пп.1-7 и наполнитель. 9. Способ получения производного аминопиррола формулы I
где радикалы Ar1 и Ar2 определены в одном из пп.1, 3-7, включающий взаимодействие содержащего циангруппу соединения формулы II
где Ar1 определен в одном из пп.1, 5-7, с ариламином формулы Ar2-NH2 в условиях, подходящих для образования кольцевой системы аминопиррола, с получением производного аминопиррола формулы I. 10. Соединение по одному из пп.1, 3-7, полученное способом по п.9. 11. Применение соединения по одному из пп.1-7 для приготовления лекарственного средства, предназначенного для лечения воспалительного заболевания млекопитающего, в особенности такого, как артрит.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 27.08.2009
Извещение опубликовано: 20.07.2010 БИ: 20/2010
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
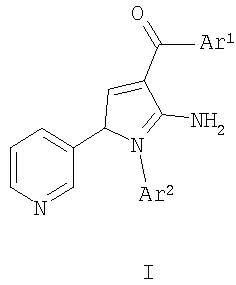
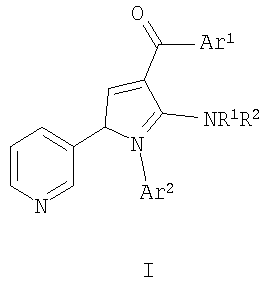
 была продемонстрирована при испытаниях in vitro и in vivo, описанных подробно в примерах 5 и 6 соответственно. Противовоспалительная активность соединений по настоящему изобретению может быть определена, используя анализ индуцированного адъювантом артрита у крыс, описанный в примере 7.
была продемонстрирована при испытаниях in vitro и in vivo, описанных подробно в примерах 5 и 6 соответственно. Противовоспалительная активность соединений по настоящему изобретению может быть определена, используя анализ индуцированного адъювантом артрита у крыс, описанный в примере 7.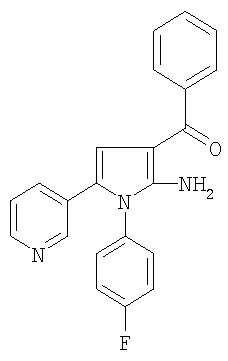
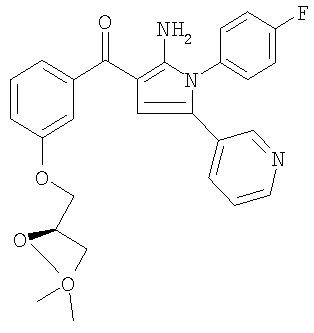
 -изопропилиденглицерин-
-изопропилиденглицерин- -тозилата и 1,2 г (8,7 ммоля) карбоната калия в 10 мл диметилформамида нагревали при 80°С. Через 16 часов реакционную смесь охлаждали до комнатной температуры, выливали в соляной раствор и экстрагировали этилацетатом. Экстракты сушили над сульфатом натрия, концентрировали в вакууме и очищали с помощью флэш-хроматографии (градиентное элюирование: 15-50% этилацетат/гексан), получали 0,7 г (90%) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-[3-(2,2-диметил[1,3]диоксолан-4(S)-илметокси)фенил]метанона.
-тозилата и 1,2 г (8,7 ммоля) карбоната калия в 10 мл диметилформамида нагревали при 80°С. Через 16 часов реакционную смесь охлаждали до комнатной температуры, выливали в соляной раствор и экстрагировали этилацетатом. Экстракты сушили над сульфатом натрия, концентрировали в вакууме и очищали с помощью флэш-хроматографии (градиентное элюирование: 15-50% этилацетат/гексан), получали 0,7 г (90%) [2-амино-1-(4-фторфенил)-5-пиридин-3-ил-1Н-пиррол-3-ил]-[3-(2,2-диметил[1,3]диоксолан-4(S)-илметокси)фенил]метанона.