Патент на изобретение №2315043
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) АМИДНОЕ ПРОИЗВОДНОЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА НА ЕГО ОСНОВЕ
(57) Реферат:
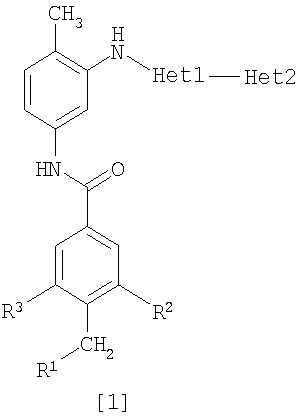
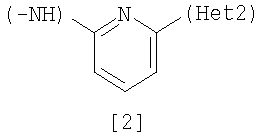
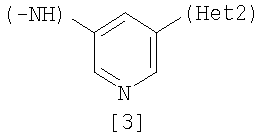
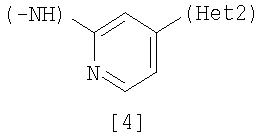
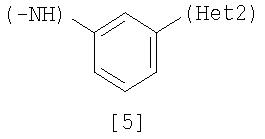
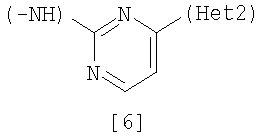
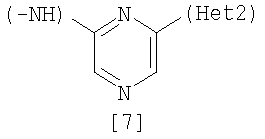
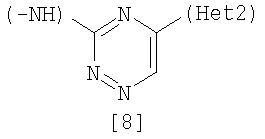
Изобретение относится к амидному производному, представленному следующей формулой [1] в любом из следующих случаев (А) и (В), или его соли. (А) R1 представляет 5-7 членную насыщенную циклическую группу, имеющую 1-2 атома азота в качестве атома, образующего цикл (насыщенная циклическая аминогруппа может быть замещена 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из С1-10алкила, C1-10алкоксикарбонила), моноС1-10алкиламино или диС1-10алкиламино; R2 представляет С1-10алкил, галоген, галогенС1-10алкил, С1-10алкокси, C1-10алкоксикарбонил, нитро, моноC1-10алкилкарбамоил, диС1-10алкилкарбамоил или циано; R3 представляет водород, галоген или C1-10алкокси; Het1 представляет любую из групп следующих формул [2]-[8]; Het2 представляет пиридил, пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил (Het2 может быть замещенным 1-3 одинаковыми или разными заместителями, выбранными из галогена); кроме соединения, в котором R1 является (i) пирролидинилом, пиперидинилом, пиперазинилом или морфолинилом, каждый из которых может быть замещен 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из алкила, алкоксикарбонила, галогена, галогеналкила, гидроксиалкила, амино, моноалкиламино, диалкиламино, карбамоила, моноалкилкарбамоила и диалкилкарбамоила, (ii) моноалкиламино или (iii) диалкиламино, Het1 является группой формулы [6], а Het2 является пиразинилом или пиридилом, который может быть замещенным алкилом; (В) R1 представляет 4-метилпиперазин-1-ил, 1-пирролидинил, пиперидино, 4-этилпиперазин-1-ил, 4-н-пропилпиперазин-1-ил, цис-3,5-диметилпиперазин-1-ил, морфолино, диметиламино или диэтиламино; R2 представляет метил, галоген, трифторметил, метокси, метоксикарбонил, нитро, диметилкарбамоил или циано; R3 представляет водород, бром или метокси; Het1 представляет группу формулы [6]; Het2 представляет 3-пиридил. Изобретение относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении BCR-ABL тирозинкиназы, содержащей в качестве активного ингредиента амидное производное формулы I или его соль и фармацевтически приемлемый нетоксичный и инертный носитель. Изобретение также относится к ингибитору BCR-ABL тирозинкиназы, терапевтическим средствам для лечения хронической миелогенной лейкемии, острой лимфобластной лейкемии, острой миелогенной лейкемии, содержащим в качестве активного ингредиента амидное производное формулы I или его соль и, необязательно, фармацевтически приемлемый нетоксичный и инертный носитель. Технический результат – амидное производное, ингибирующее активность BCR-ABL тирозинкиназы. 6 н. и 2 з.п. ф-лы, 2 табл.
Данное изобретение относится к амидному производному или его соли и фармацевтической композиции, содержащей амидное производное или его соль в качестве активного ингредиента. Поскольку BCR-ABL тирозинкиназа (см., например, непатентный документ 1) вызывает аберрантный рост клеток, соединение, которое подавляет ее активность, пригодно для профилактики или лечения заболеваний, вызванных активностью BCR-ABL тирозинкиназы, например, хронической миелогенной лейкемии, острой лимфобластной лейкемии и острой миелогенной лейкемии (см., например, непатентный документ 2). BCR является геном, который находится в человеческой двадцать второй хромосоме, а ABL является геном, который находится в человеческой девятой хромосоме, и хромосома Philadelphia образуется транслокацией человеческой двадцать второй и девятой хромосом. Известно, что генный продукт хромосомы, BCR-ABL, является протеином, обладающим тирозинкиназной активностью, и постоянно генерирует рост сигнала, который вызывает аберрантный рост клеток (см., например, непатентный документ 2). Поэтому подавление активности BCR-ABL тирозинкиназы делает возможным подавление роста клеток, вызванного киназой, и соединение, которое подавляет данную активность, пригодно для использования в качестве терапевтического средства таких заболеваний, как хроническая миелогенная лейкемия, острая лимфобластная лейкемия и острая миелогенная лейкемия. Известен препарат Гливек® (Glyvek®) (см., например, патентный документ 1) в качестве лекарственного средства, обладающего аналогичным действием, однако другие лекарственные средства, имеющие такой же механизм действия, никогда не поступали на рынок, и, таким образом, было необходимо разработать лучшие лекарственные средства. Известно, что у пациентов, у которых достигнута ремиссия в результате применения Гливека® при BCR-ABL-положительной острой лимфобластной лейкемии, рецидивы часто регистрируют в дополнение к примерам бластного криза хронической миелогенной лейкемии (см., например, непатентный документ 3). В результате изучения лейкемических клеток пациентов, страдающих от рецидива заболевания, определено появление такого варианта, как Е255К (см., например, непатентные документы с 4 по 7). Также при применении Гливека® у пациентов с BCR-ABL-положительной острой лимфобластной лейкемией зарегистрировано появление устойчивых клеток, которые в основном представляют вариант Е255К (см., например, непатентный документ 8). С увеличением использования Гливека® число резистентных пациентов дополнительно увеличивается, и, таким образом, необходимо разрабатывать терапию. Патентный документ 1: Japanese Unexamined Patent No. 6-87834 Патентный документ 2: Pamphlet of International Publication WO 02/22597 Непатентный документ 1: Shtivelman E., et al.: Nature, 1985, 315, 550-554 Непатентный документ 2: Daley G. Q., et al.: Science, 1990, 247, 824-830 Непатентный документ 3: Druker B. J., et al.: N. Engi. J. Med., 2001, 344, 1038-1042 Непатентный документ 4: Weisberg E., et al.: Drug Resist Updat, 2001, 4, 22-28 Непатентный документ 5: Gorre M. E., et al.: Science, 2001, 293, 876-880 Непатентный документ 6: Blagosklonny M. V.: Leukemia, 2002, 16, 570-572 Непатентный документ 7: Hochhaus A., et al.: Leukemia, 2002, 16, 2190-2196 Непатентный документ 8: Hofmann W.-K., et al.: blood, 2002, 99, 1860-1862 Непатентный документ 9: Deninger W. N., et al.: blood, 2000, 96, 3343-3356 Непатентный документ 10: J.Org.Chem., 1996, 61, 1133-1135 Непатентный документ 11: J.Org.Chem., 2000, 65, 1144-1157 Непатентный документ 12: Recl.Trav.Chim.Pays-Bas., 1950, 69, 673-699 Непатентный документ 13: J.Med.Chem., 2000, 43, 1508-1518 Непатентный документ 14: J.Med.Chem., 1975, 18, 1077-1088 Непатентный документ 15: Bioorg.Med.Chem.Lett., 2001, 11, 2235-2239 Непатентный документ 16: J.Heterocyclic Chem., 2000, 37, 1457-1462 Непатентный документ 17: J.Med.Chem., 2000, 43(8), 1508-1518 Непатентный документ 18: Khim.Geterotsikl.Soedim., 1981, (7), 958-962 Непатентный документ 19: J.Heterocyclic Chem., 1990, 27, 579-582 Непатентный документ 20: Arzneim.-Forsch./Drug Res., 1989, 39(2), 1196-1201 Непатентный документ 21: J.Org.Chem., 1996, 61, 7240-7241 ОПИСАНИЕ ИЗОБРЕТЕНИЯ Задачей данного изобретения является создание амидного производного, обладающего превосходной подавляющей BCR-ABL тирозинкиназу активностью, или его соли. Были проведены интенсивные исследования в отношении разных соединений, и было обнаружено, что поставленная задача достигается с помощью амидного производного в соответствии с данным изобретением, и, таким образом, было выполнено данное изобретение. Данное изобретение относится к амидному производному общей формулы [1] в любом из следующих случаев [А] и [В] или его соли (здесь далее называемым «соединением данного изобретения»).
(А) R1 представляет насыщенную циклическую аминогруппу (насыщенная циклическая аминогруппа может быть замещена 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из алкила, алкоксикарбонила, галогена, галогеналкила, гидроксиалкила, амино, моноалкиламино, диалкиламино, карбамоила, моноалкилкарбамоила и диалкилкарбамоила), моноалкиламино или диалкиламино, R2 представляет алкил, галоген, галогеналкил, гидроксиалкил, алкокси, алкоксиалкил, алкоксикарбонил, ацил, амино, моноалкиламино, диалкиламино, нитро, карбамоил, моноалкилкарбамоил, диалкилкарбамоил или циано, R3 представляет водород, галоген или алкокси, Неt1 представляет любую из групп следующих формул с [2] по [8]:
Неt2 представляет пиридил, пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил (Неt2 может быть замещенным 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из алкила, галогена и амино). Кроме соединения, в котором R1 является (i) пирролидинилом, пиперидинилом, пиперазинилом или морфолинилом, каждый из которых может быть замещен 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из алкила, алкоксикарбонила, галогена, галогеналкила, гидроксиалкила, амино, моноалкиламино, диалкиламино, карбамоила, моноалкилкарбамоила и диалкилкарбамоила, (ii) моноалкиламино или (iii) диалкиламино, Неt1 является группой формулы [6], а Неt2 является пиразинилом или пиридилом, который может быть замещенным алкилом; (В) R1 представляет 4-метилпиперазин-1-ил, 1-пирролидинил, пиперидино, 4-этилпиперазин-1-ил, 4-н-пропилпиперазин-1-ил, цис-3,5-диметилпиперазин-1-ил, морфолино, диметиламино или диэтиламино; R2 представляет метил, галоген, трифторметил, метокси, метоксикарбонил, нитро, диметилкарбамоил или циано; R3 представляет водород, бром или метокси; Неt1 представляет группу формулы [6]; Неt2 представляет 3-пиридил. Данное изобретение относится также к фармацевтической композиции, содержащей представленное выше амидное производное или его соль в качестве активного ингредиента, и более конкретно, к ингибитору BCR-ABL тирозинкиназы, содержащему вышеуказанное амидное производное или его соль в качестве активного ингредиента. Специфическое терапевтическое средство для лечения болезней включает терапевтическое средство для лечения хронической миелогенной лейкемии, терапевтическое средство для лечения острой лимфобластной лейкемии и терапевтическое средство для лечения острой миелогенной лейкемии. Примеры предпочтительных амидных производных и их солей включают следующие амидные производные или их соли. Амидное производное общей формулы [1], где R1 является насыщенной циклической аминогруппой (насыщенная циклическая аминогруппа может быть замещенной 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из алкила и алкоксикарбонила), моноалкиламино или диалкиламино, R2 представляет алкил, галоген, галогеналкил, алкокси, алкоксикарбонил, нитро, диалкилкарбамоил или циано, R3 представляет водород, галоген или алкокси, Неt1 является любой из групп формул [2]-[8], и Неt2 представляет пиридил, пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил (Неt2 может быть замещенным 1-3 одинаковыми или разными атомами галогена), или их соли. Примеры особенно предпочтительных среди представленных выше амидных производных включают следующие амидные производные (1)-(40) или их соли: (1) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (2) 3-йод-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (3) 3-хлор-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (4) 3-фтор-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (5) 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (6) 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (7) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(2-пиразинил)пиримидин-2-иламино]фенил}бензамид, (8) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид, (9) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид, (10) 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид, (11) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(1,2-дигидропиридазин-4-ил)пиримидин-2-иламино]-4-метилфенил}бензамид, (12) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридазинил)пиримидин-2-иламино]фенил}бензамид, (13) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (14) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиридин-2-иламино]фенил}бензамид, (15) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиридин-2-иламино]фенил}бензамид, (16) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[2-(3-пиридил)пиридин-6-иламино]фенил}бензамид, (17) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[3-(3-пиридил)пиридин-5-иламино]фенил}бензамид, (18) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[3-(3-пиридил)фениламино]фенил}бензамид, (19) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[2-(3-пиридил)пиразин-6-иламино]фенил}бензамид, (20) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[5-(3-пиридил)-1,2,4-триазин-3-иламино]фенил}бензамид, (21) 3-метил-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (22) 4-(4-метилпиперазин-1-илметил)-3-нитро-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (23) 3-метокси-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (24) 3,5-дибром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (25) 3,5-диметокси-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (26) 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (27) 3-бром-4-(4-этилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (28) 3-бром-4-[4-(н-пропил)пиперазин-1-илметил]-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (29) 3-бром-4-(N,N-диметиламинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (30) 3-бром-4-(N,N-диэтиламинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (31) 3-бром-4-(1-пирролидинилметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (32) 3-бром-4-(пиперидинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (33) 3-бром-4-(морфолинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (34) 3-бром-4-(цис-3,5-диметилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (35) 3-бром-4-(4-метилгексагидро-1Н-1,4-диазепин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (36) 3-бром-4-(1-пиперазинилметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (37) 4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (38) 4-(1-пиперазинилметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (39) 3-метоксикарбонил-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид и (40) 3-циано-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид. Соединение данного изобретения обладает подавляющей BCR-ABL тирозинкиназу активностью и пригодно в качестве терапевтического средства при таких заболеваниях, как хроническая миелогенная лейкемия, острая лимфобластная лейкемия и острая миелогенная лейкемия (см., например, непатентный документ 9). Соединение вышеприведенной формулы [1] в случае (В) описывалось в уровне техники (см., например, патентный документ 1 или 2), но специально не раскрыто в публикации. Соединение вышеприведенной формулы [1] в случае (А) также не описано. Данное изобретение далее будет подробно описано. Примеры «насыщенной циклической аминогруппы» включают 4-8-членную насыщенную циклическую группу, которая представляет насыщенную циклическую группу, имеющую, по меньшей мере, один атом азота в качестве атома, образующего цикл, а также может иметь от 1 до 3 одинаковых или разных членов, выбранных из группы, состоящей из атома азота, атома кислорода и атома серы. Когда атом, образующий кольцо циклического амина, является атомом азота или атомом серы, атом азота или атом серы может образовывать оксид. Примерами являются пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил и гексагидро-1Н-1,4-диазепинил. Эти заместители могут иметь присоединение по своему любому положению. Конкретно это означает, что «пирролидинил» включает все из 1-пирролидинила, 2-пирролидинила и 3-пирролидинила. «Алкил» включает линейные или разветвленные алкильные группы, имеющие от 1 до 10 атомов углерода, например метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторичный бутил, трет-бутил, н-пентил, изопентил, н-гексил, изогексил, н-гептил, изогептил, н-октил, н-нонил и н-децил. Линейные алкильные группы, имеющие от 1 до 3 атомов углерода, являются особенно предпочтительными. Алкильная часть «галогеналкила», алкоксикарбонила», «гидроксиалкила», «моноалкиламино», «диалкиламино», «моноалкилкарбамоила», «диалкилкарбамоила», «алкокси», алкоксиалкила» и «гидроксиалкила» включает вышеназванный алкил. «Галоген» включает, например, фтор, хлор, бром и йод. «Галогеналкил» включает моногалогеналкил, дигалогеналкил и тригалогеналкил, и галогеновая составляющая «галогеналкила» включает представленный выше галоген. «Галогеналкил» включает, например, трифторметил и 2,2,2-трифторэтил. «Ацил» включает ацильные группы, имеющие от 1 до 11 атомов углерода, например формил, ацетил, пропионил, бутирил, изобутирил, бензоил, 1-нафтоил и 2-нафтоил. «Пиридил» включает, например, 2-пиридил, 3-пиридил и 4-пиридил. «Пиримидинил» включает, например, 2-пиримидинил, 4-пиримидинил и 5-пиримидинил. «Пиразинил» включает, например, 2-пиразинил. «Пиридазинил» включает, например, 3-пиридазинил и 4-пиридазинил. «1,2-дигидропиридазинил» включает, например, 1,2-дигидропиридазин-3-ил и 1,2-дигидропиридазин-4-ил. Соединение данного изобретения может быть получено из известного соединения или промежуточного соединения, которое может быть с легкостью получено, например, следующим способом. При получении соединения согласно данному изобретению обычно, если исходное вещество содержит реакционноспособные заместители, которые не должны участвовать в данной реакции, его используют уже защищенным соответствующими защитными группами по существу известными методами, когда исходные вещества имеют заместители, которые не предназначены для реакции. После реакции защитные группы могут быть удалены известными методами. Процесс 1
где R1, R2, R3, Неt1 и Неt2 определены выше. Эта реакция является реакцией конденсации соединения [9] и соединения [10] и поэтому проводится по существу известными методами, используемыми для реакции конденсации. Соединение [1] может быть получено путем взаимодействия карбоновой кислоты в качестве соединения [10] или ее реакционноспособного производного с амином в качестве соединения [9]. Примеры реакционноспособного производного соединения [10] включают те соединения, которые обычно используют при реакции конденсации с образованием амида, например галогенангидрида кислоты (например, хлорангидрида кислоты, бромангидрида кислоты и т.д.), смешанного ангидрида кислоты, имидазолида и активного амида. При использовании карбоновой кислоты [10] используют конденсирующий агент (например, 1,1′-оксалилдиимидазол, 1-этил-3-(3-диметиламинопропил)карбодиимид, дициклогексилкарбодиимид, диэтилцианофосфонат, дифенилфосфорилазид, 2-хлор-1-метилпиридиния йодид и т.д.) и реакцию проводят при температуре от -20 до 100°С в присутствии или в отсутствие основания (например, органического основания, такого как триэтиламин, N,N-диизопропил-N-этиламин, N,N-диметиланилин, пиридин, 4-диметиламинопиридин, 1,8-диазабицикло[5.4.0]ундец-7-ен и т.д.). Растворитель конкретно не ограничивается, поскольку он не участвует в реакции, и его примеры включают простые эфиры, такие как тетрагидрофуран и диэтиловый эфир; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; нитрилы, такие как ацетонитрил и пропионитрил; углеводороды, такие как бензол и толуол; галогенированные углеводороды, такие как хлороформ и дихлорметан; и смеси этих растворителей. В этом случае могут быть также добавлены добавки (например, 1-гидроксибензотриазол, N-гидроксисукцинимид и т.д.). Время реакции меняется в зависимости от видов исходных веществ и конденсирующего агента и температуры реакции, но предпочтительно составляет от 30 минут до 24 часов. Количество соединения [10] и конденсирующего агента предпочтительно составляет от 1 до 3 моль на моль соединения [9]. При использовании галогенангидрида кислоты в качестве реакционноспособного производного соединения [10] реакцию проводят при температуре от -20 до 100°С, используя пиридиновый растворитель, такой как пиридин или 4-метилпиридин, или основание и растворитель, описанные выше. А также в качестве добавки может использоваться 4-диметиламинопиридин. Время реакции меняется в зависимости от вида галогенангидрида кислоты и температуры реакции, но предпочтительно составляет от 30 минут до 24 часов. Соединение [9] в качестве исходного вещества, где Неt1 является группой формулы [6], может быть получено способом, который описан в патентном документе 1. Соединение [9] в качестве исходного соединения, где Неt1 является группой формулы [4], [5] или [7], может быть получено следующим образом:
где Неt1 и Неt2, имеют значения, представленные выше, R4 и R5 представляют алкил или гидрокси, R6, R7 и R8 представляют алкил, и Х1 представляет галоген. Стадия 1 Эта реакция представляет собой реакцию перекрестного соединения с использованием соединения [11] и борорганического соединения [12] или оловоорганического соединения [13] и может быть проведена по существу известными методами. Например, эту реакцию проводят при 20-200°С в подходящем растворителе в присутствии палладиевого катализатора. В качестве палладиевого катализатора обычно используют тетракис(трифенилфосфин)палладий, дихлорбис(трифенилфосфин)палладий и дихлорбис(три-о-толилфосфин)палладий. Растворитель, используемый в реакции, конкретно не ограничивается, поскольку он не участвует в реакции, и примерами его являются простые эфиры, такие как тетрагидрофуран, 1,4-диоксан и 1,2-диметоксиэтан; спирты, такие как метанол и этанол; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; углеводороды, такие как бензол, толуол и ксилол; органические амины, такие как пиридин и триэтиламин; и смеси этих растворителей. При использовании соединения [12] существенным является добавление основания (например, гидроксида натрия, карбоната калия, трехзамещенного фосфата калия и т.д.). Время реакции меняется в зависимости от вида исходного вещества и температуры реакции, но предпочтительно составляет от 1 до 48 часов. Стадия 2 Эта реакция является реакцией восстановления ароматической нитрогруппы соединения [14] до аминогруппы и поэтому проводится по существу известными методами, используемыми при реакции восстановления. Данный метод включает, например, метод обработки цинком или оловом в кислых условиях. В соответствии с методом каталитического восстановления, например, гидрирование можно проводить, используя платину, никель Ренея, платину-на-угле (Pt-С), палладий-на-угле (Pd-С) или рутениевый комплекс в качестве катализатора. Кроме того, примерами являются метод с использованием сульфида, такого как дитионит натрия, и способ восстановления формиатом аммония или гидразином в присутствии металлического катализатора. Соединение [11] в качестве исходного соединения, где Неt1 является группой формулы [4], можно получить путем взаимодействия 2,4-дихлорпиридина (полученного, например, методом, описанным в непатентном документе 12) с 2-метил-5-нитроанилином, используя метод J. P. Wolfe et al. c использованием палладиевого катализатора (см. непатентные документы 10 и 11). Когда Неt1 является группой формулы [5], например, данное соединение может быть получено посредством взаимодействия 1-бром-3-иодбензола с 2-метил-5-нитроанилином. Когда Неt1 является группой формулы [7], например, данное соединение может быть получено посредством взаимодействия 2,6-дихлорпиразина с 2-метил-5-нитроанилином. Растворитель, используемый в реакции, специально не ограничивается, поскольку он не участвует в реакции, и примерами его являются простые эфиры, такие как тетрагидрофуран, 1,4-диоксан и 1,2-диметоксиэтан; углеводороды, такие как бензол, толуол и ксилол; и смеси из этих растворителей. Реакцию проводят при температуре от 70 до 100°С в присутствии основания. Примеры палладиевого катализатора включают трис(дибензилиденацетон)дипалладий (0), ацетат палладия(II) и три(о-толилфосфин)палладий(0). Количество палладия предпочтительно составляет от 0,5 до 4 мол.% от галогенированного арила. В качестве лиганда палладиевого катализатора, например, можно использовать 1,3-бис(дифенилфосфино)пропан, 1,1′-бис(дифенилфосфино)ферроцен и (±)-2,2′-бис(дифенилфосфино)-1,1′-бинафтил [(±)-БИНАФ; BINAP]. Примеры основания включают трет-бутоксид натрия, трет-бутоксид калия, карбонат цезия, карбонат калия и карбонат натрия. Время реакции меняется в зависимости от исходного вещества и температуры реакции, но предпочтительно составляет от 1 до 36 часов. Соединение [11], где Неt1 является группой [4], может быть также получено путем взаимодействия 2,4-дихлорпиридина с 2-метил-5-нитроанилином при 20-200°С в подходящем растворителе в присутствии или в отсутствие основания. Примеры основания включают пиридин, триэтиламин, N,N-диизопропил-N-этиламин, карбонат калия, гидрокарбонат натрия и гидроксид калия. Растворитель конкретно не ограничивается, поскольку он не участвует в реакции, и его примеры включают простые эфиры, такие как тетрагидрофуран, дибутиловый эфир и 1,4-диоксан; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; углеводороды, такие как бензол и толуол; спирты, такие как этиленгликоль и 2-метоксиэтанол; галогенированные углеводороды, такие как хлороформ и дихлорметан; диметилсульфоксид и смеси этих растворителей. Время реакции меняется в зависимости от исходного вещества и температуры реакции, но предпочтительно составляет от 1 до 24 часов. Соединение [14а] в качестве исходного соединения [соединение [14], где Неt1 является группой формулы [4]), может быть также получено следующим образом:
где R4, R5, R6, R7, R8, Неt2 и Х1 имеют значения, представленные выше, и Х2 представляет галоген. Стадия 1 Эта реакция является реакцией перекрестного соединения с использованием соединения [15] и борорганического соединения [12] или оловоорганического соединения [13] и может быть проведена таким же образом, который описан выше. Стадия 2 Соединение [17] получают галогенированием соединения [16]. Поэтому данную реакцию проводят по существу известными методами. Данную реакцию проводят с использованием оксихлорида фосфора, оксибромида фосфора, пентахлорида фосфора или пентабромида фосфора в растворителе или без него. Растворитель, используемый в реакции, специально не ограничивается, поскольку он не участвует в реакции, и примерами его являются простые эфиры, такие как тетрагидрофуран, дибутиловый эфир и 1,4-диоксан; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводороды, такие как хлороформ и дихлорметан; и смеси этих растворителей. Реакцию обычно проводят при температуре от комнатной до 130°С, и время реакции предпочтительно составляет от 20 минут до 24 часов. Стадия 3 Соединение [14а] может быть получено путем взаимодействия соединения [17] с соединением [18] с использованием вышеуказанного метода при использовании палладиевого катализатора (см., например, непатентные документы 10 и 11). Соединение [9а] [соединение [9], где Неt1 является группой формулы [4]), может быть получено путем взаимодействия соединения [17] с соединением [19] с использованием вышеприведенного метода при использовании палладиевого катализатора [см., например, непатентные документы 10 и 11) с получением соединения [20] и удаления защиты у соединения [20].
где Неt2 и Х2 имеют значения, представленные выше, и R9 представляет защитную группу. Стадия 1 Исходное соединение [19] может быть получено путем защиты 2,4-диаминотолуола с помощью подходящей защитной группы с использованием по существу известных методов. Примеры защитной группы включают ацильные производные, такие как бензоил, ацетил и формил; и производные уретанового типа, такие как бензилоксикарбонил, трет-бутоксикарбонил и 2,2,2-трихлорэтоксикарбонил. Соединение [20] можно получить путем взаимодействия соединения [17] с соединением [19], используя палладиевый катализатор. Стадия 2 При реакции удаления защиты у соединения [20] защитную группу ацильного типа удаляют путем гидролиза, используя кислоту или щелочь, или удаляют с помощью аммиачной воды или гидразина. Примеры кислоты, используемой при гидролизе, включают неорганические кислоты, такие как соляная кислота и серная кислота, и примеры основания включают неорганические основания, такие как гидроксид натрия и гидроксид калия. Примеры растворителя, используемого в реакции, включают спирты, такие как метанол и этанол; простые эфиры, такие как тетрагидрофуран и 1,4-диоксан; воду и смеси этих растворителей. Температура реакции находится в интервале от 0 до 100°С, и время реакции обычно составляет от нескольких минут до 24 часов. Когда защитная группа является производным уретанового типа, ее можно удалить гидрированием с использованием палладиевого катализатора или удалить обработкой соляной кислотой, трифторуксусной кислотой, триметилсилилйодидом или трифторидом бора, также в зависимости от вида защитной группы. Исходное соединение [9], где Неt1 является группой формулы [8], можно получить методом, описанным в сравнительном примере 18, представленным ниже. Соединение [10] в качестве исходного соединения может быть получено следующим образом:
где R1, R2 и R3 имеют значения, представленные выше, R10 представляет алкил, и Х3 представляет удаляемую группу, такую как Cl, Br, I, OTs или OMs. Стадия 1 Соединение [23] может быть получено конденсацией соединения [21] (которое можно получить, например, методом, описанным в непатентном документе 13) с помощью амина [22] (где удаляемая группа Х3 представляет такую удаляемую группу, как галоген, мезилат или тозилат). Эта реакция представляет собой реакцию нуклеофильного замещения алкилгалогенида и аминов и проводится по существу известными методами. Эту реакцию проводят в подходящем растворителе, используя избыток амина или в присутствии основания. Примеры предпочтительного основания включают пиридин, триэтиламин, N,N-диизопропил-N-этиламин, карбонат калия и гидрокарбонат натрия. Растворитель специально не ограничивается, поскольку он не участвует в реакции, и его примеры включают простые эфиры, такие как тетрагидрофуран и диэтиловый эфир; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; нитрилы, такие как ацетонитрил и пропионитрил; углеводороды, такие как бензол и толуол; спирты, такие как метанол и этанол; воду и смеси этих растворителей. Температура реакции обычно находится в интервале от 0°С до 100°С. Время реакции меняется в зависимости от исходного вещества и температуры реакции, но предпочтительно составляет от 30 минут до 24 часов. Стадия 2 Соединение [10] можно получить гидролизом соединения [23]. Реакцию обычно проводят в подходящем растворителе в присутствии кислоты или основания. Примеры кислоты, используемой при гидролизе, включают неорганические кислоты, такие как соляная кислота и серная кислота, и примеры основания включают неорганические основания, такие как гидроксид натрия и гидроксид калия. Примеры растворителя, используемого в реакции, включают спирты, такие как метанол и этанол; простые эфиры, такие как тетрагидрофуран и 1,4-диоксан; воду и смеси этих растворителей. Температура реакции находится обычно в интервале от 0 до 100°С, и время реакции составляет обычно от 30 минут до 24 часов. Процесс 2
где R1, R2, R3, Неt1 и Неt2 имеют определения, представленные выше, Х4 представляет Cl, Br, I или SR11, и R11 представляет алкил. Соединение [1] можно получить взаимодействием соединения [24] с соединением [25]. Реакцию проводят при температуре от 20 до 200°С в подходящем растворителе в присутствии или в отсутствие основания. Примеры основания включают пиридин, триэтиламин, N,N-диизопропил-N-этиламин, карбонат калия, гидрокарбонат натрия и гидроксид калия. Растворитель конкретно не ограничивается, поскольку он не участвует в реакции, и его примеры включают простые эфиры, такие как тетрагидрофуран, диэтиловый эфир и 1,4-диоксан; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; углеводороды, такие как бензол и толуол; спирты, такие как этиленгликоль и 2-метоксиэтанол; галогенированные углеводороды, такие как хлороформ и дихлорметан; диметилсульфоксид и смеси этих растворителей. Время реакции меняется в зависимости от исходного вещества и температуры реакции, но предпочтительно составляет от 1 до 24 часов. Соединение [24] в качестве исходного вещества может быть получено конденсацией 2,4-диаминотолуола с соединением [10] в соответствии с модифицированным процессом 1. Соединение [25] в качестве исходного вещества можно получить путем использования 2,6-дибромпиридина, когда Неt1 является группой формулы [2], 3,5-дибромпиридина, когда Неt1 является группой формулы [3], или 2,4-дихлорпиримидина, когда Неt1 является группой формулы [6] в соответствии с процессом 4, описанным ниже. Когда Неt1 является группой формулы [4], соединение [25] может быть также получено методом, описанным в приведенном выше процессе 1. Процесс 3
где R1, R2, R3 и Неt2 имеют значения, представленные выше. Соединение [1b] (соединение [1], где Неt1 является группой формулы [6]) может быть получено взаимодействием соединения [26] или его кислотно-аддитивной соли с соединением [27]. Данную реакцию проводят при температуре от 20 до 200°С в подходящем растворителе. Растворитель специально не ограничивается, поскольку он не участвует в реакции, и его примеры включают спирты, такие как метанол, этанол, 2-пропанол и 2-метоксиэтанол. Количество соединения [27] составляет от 1 до 2 моль и предпочтительно от 1 до 1,2 моль на моль соединения [26]. Время реакции меняется в зависимости от исходного вещества и температуры реакции, но предпочтительно составляет от 30 минут до 30 часов. При использовании кислотно-аддитивной соли соединения [26] реакция может быть проведена добавлением подходящего основания (например, карбоната калия, гидрокарбоната натрия, гидроксида натрия, гидроксида калия и т.д.). Соединение [26] в качестве исходного соединения может быть получено в виде свободной соли или кислотно-аддитивной соли путем взаимодействия соединения [24] с цианамидом по методу, описанному в документе (см., например, непатентный документ 14). Соединение [27] в качестве исходного вещества может быть получено, например, в соответствии с модифицированным методом, описанным в патентном документе 1. Процесс 4
где R1, R2, R3, R4, R5, R6, R7, R8, Неt1 и Неt2 имеют значения, представленные выше, Х5 представляет галоген. Эта реакция является реакцией перекрестного соединения с использованием соединения [28] и борорганического соединения [12] или оловоорганического соединения [13] и может быть проведена по существу известными методами. Например, эту реакцию проводят при температуре от 20 до 200°С в подходящем растворителе в присутствии палладиевого катализатора. В качестве палладиевого катализатора используют тетракис(трифенилфосфин)палладий, дихлорбис(трифенилфосфин)палладий и дихлорбис(три-о-толилфосфин)палладий. Растворитель, используемый в реакции, специально не ограничивают, поскольку он не участвует в реакции, и его примеры включают простые эфиры, такие как тетрагидрофуран, 1,4-диоксан и 1,2-диметоксиэтан; спирты, такие как метанол и этанол; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид; углеводороды, такие как бензол, толуол и ксилол; органические амины, такие как пиридин и триэтиламин; и смеси этих растворителей. При использовании соединения [12] существенно добавление основания (например, гидроксида натрия, карбоната калия, трехзамещенного фосфата калия и т.д.). Время реакции меняется в зависимости от исходного вещества и температуры реакции, но предпочтительно составляет от 1 до 48 часов. Соединение [28] в качестве исходного соединения может быть получено путем взаимодействия соединения [24] с 4-гидрокси-2-(метилтио)пиридином, когда Неt1 является группой формулы [4], или взаимодействием соединения [24] с 4-гидрокси-2-(метилтио)пиримидином и обработки продукта реакции оксихлоридом фосфора (см., например, непатентный документ 15), когда Неt1 является группой формулы [6], или взаимодействием по методу, описанному в документе (см., например, непатентный документ 16) с использованием соединения [24] и 2,4-дихлорпиримидина, когда Неt1 является группой формулы [6]. Соединение данного изобретения можно использовать в качестве лекарственного средства в виде свободного основания, однако его можно использовать также в виде фармацевтически приемлемой соли, полученной по существу известными методами. Эти соли включают соли минеральных кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота и фосфорная кислота, и соли органических кислот, таких как уксусная кислота, лимонная кислота, виннокаменная кислота, малеиновая кислота, янтарная кислота, фумаровая кислота, п-толуолсульфоновая кислота, бензолсульфоновая кислота и метансульфоновая кислота. Гидрохлорид амидного производного по данному изобретению, например, может быть получен растворением амидного производного в спиртовом растворе, этилацетатном растворе или эфирном растворе хлористого водорода. Как показано в примерах испытания, описанных далее, соединение данного изобретения обладает высокой ингибирующей активностью в отношении BCR-ABL тирозинкиназы по сравнению с пиримидиновым производным, описанным в патентном документе 1. Поэтому лекарственное средство данного изобретения пригодно в качестве профилактического или терапевтического средства для лечения заболеваний, связанных с активностью BCR-ABL тирозинкиназы, например хронической миелогенной лейкемии, острой лимфобластной лейкемии и острой миелогенной лейкемии. Когда соединение данного изобретения применяют в качестве лекарственного средства, его можно применять для лечения млекопитающих, включая людей, или само по себе, или в виде фармацевтической композиции, содержащей данное соединение и в фармацевтически приемлемом нетоксичном и инертном носителе в соотношении, например, от 0,1 до 99,5% или предпочтительно от 0,5 до 90%. Используют одно или более из вспомогательных средств для изготовления препаратов, таких как наполнители или твердый, полутвердый и жидкий разбавитель. Желательно применять фармацевтическую композицию в стандартной дозированной форме. Фармацевтическую композицию данного изобретения можно вводить внутривенно, перорально, непосредственно в целевую ткань, местно (например, трансдермально) или ректально. Само собой разумеется, что применима дозированная форма, пригодная для любого из способов введения, описанных выше. Желательно вводить препарат перорально. Желательно устанавливать дозировку соединения как ингибитора BCR-ABL тирозинкиназы или терапевтического средства для лечения хронической миелогенной лейкемии с учетом состояния пациента, возраста, веса тела и особенности и тяжести заболевания и других факторов, таких как путь введения; но обычно для взрослых пациентов дозой соединения данного изобретения является количество в интервале от 0,1 до 1000 мг/индивидууму в сутки и предпочтительно от 1 до 500 мг/индивидууму в сутки. В некоторых случаях достаточно количество ниже этого интервала, и, наоборот, в других случаях необходимо большее количество. Его можно вводить путем разделения общей дозы на два или три введения в день. НАИЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ Данное изобретение далее будет описано более подробно с помощью сравнительных примеров, примеров, примеров испытания и примеров изготовления лекарственных форм соединения данного изобретения, которыми, однако, данное изобретение не ограничивается. Сравнительный пример 1 3-Бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Стадия 1 Этил 3-бром-4-метилбензоат 10,00 г 3-Бром-4-метилбензойной кислоты суспендировали в 100 мл этанола и добавляли 2,7 мл концентрированной серной кислоты и затем смесь нагревали с обратным холодильником в течение 22 часов. После того как растворитель отгоняли при пониженном давлении, остаток смешивали с ледяной водой, нейтрализовали насыщенным водным раствором гидрокарбоната натрия (рН 8) и затем экстрагировали этилацетатом. Экстракт промывали водой и затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении с получением 10,99 г целевого соединения в виде коричневого масляного продукта. 1H-ЯМР (CDCl3) Стадия 2 Этил 3-бром-4-(бромометил)бензоат Это соединение получали модифицированным методом, описанным в документе (J. Med. Chem., 2000, 43(8), 1508-1518). 10,0 г Этил 3-бром-4-метилбензоата, полученного на стадии 1, растворяли в 125 мл четыреххлористого углерода и после добавления 6,83 г N-бромсукцинимида и 80 г бензоилпероксида раствор нагревали с обратным холодильником при облучении светом лампы накаливания (1500 Вт) в течение 8 часов. После удаления нерастворенных веществ фильтрованием фильтрат разбавляли 500 мл дихлорметана. Раствор промывали по очереди водой и насыщенным водным раствором гидрокарбоната натрия, а затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении с получением 13,02 г неочищенного продукта в виде коричневого масляного продукта. 1H-ЯМР (CDCl3) Стадия 3 Этил 3-бром-4-(4-метилпиперазин-1-илметил)бензоат 11,40 г Этил 3-бром-4-(бромметил)бензоата, полученного на стадии 2, растворяли в 114 мл безводного тетрагидрофурана и после добавления 5,3 г карбоната калия по каплям в течение 10 минут добавляли раствор 2,86 г N-метилпиперазина в 10 мл тетрагидрофурана при перемешивании в атмосфере аргона при комнатной температуре. После перемешивания при комнатной температуре в течение 4 часов нерастворенные вещества удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 7,53 г целевого соединения в виде коричневого масляного продукта. 1H-ЯМР (CDCl3) Стадия 4 3-Бром-4-(4-метилпиперазин-1-илметил)бензойной кислоты дигидрохлорид 2,00 г Этил 3-бром-4-(4-метилпиперазин-1-илметил)бензоата, полученного на стадии 3, растворяли в 40 мл метанола и после добавления 8,8 мл 1 N водного раствора гидроксида натрия смесь нагревали с обратным холодильником в течение 1 часа. После того как растворитель отгоняли, остаток растворяли в 40 мл воды. Раствор промывали 40 мл эфира, и водный слой подкисляли (рН 2) 1 N соляной кислотой при охлаждении льдом. После того как воду отгоняли, операцию добавления 50 мл толуола к остатку с последующим азеотропным удалением воды повторяли три раза с получением 2,56 г неочищенного продукта в виде бесцветных кристаллов. 1H-ЯМР (D2O) Стадия 5 3-Бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид 1,50 г дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензойной кислоты, полученного на стадии 4, суспендировали в 6,3 мл тионилхлорида с последующим перемешиванием при нагревании в течение 24 часов. Реакционный раствор охлаждали на воздухе и выпавшие в осадок кристаллы собирали фильтрованием и затем промывали диэтиловым эфиром с получением 1,34 г неочищенного продукта в виде бесцветного кристаллического вещества. Температура плавления: 229-231°С (с разложением) 1H-ЯМР (D2O) Сравнительный пример 2 3-Йод-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, как и в сравнительном примере 1, за исключением того, что 3-йод-4-метилбензойную кислоту использовали вместо 3-бром-4-метилбензойной кислоты на стадии 1, получали бледно-желтое кристаллическое вещество. Температура плавления: 218-220°С (с разложением) 1H-ЯМР (D2O) Сравнительный пример 3 3-Хлор-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, как и в сравнительном примере 1, за исключением того, что 3-хлор-4-метилбензойную кислоту использовали вместо 3-бром-4-метилбензойной кислоты на стадии 1, получали бесцветные кристаллы. Температура плавления: 245-247°С (с разложением) 1H-ЯМР (D2O) Сравнительный пример 4 3-Фтор-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, как и в сравнительном примере 1, за исключением того, что 3-фтор-4-метилбензойную кислоту использовали вместо 3-бром-4-метилбензойной кислоты на стадии 1, получали бесцветное кристаллическое вещество. Температура плавления: 242-244°С (с разложением) 1H-ЯМР (D2O) Сравнительный пример 5 4-(4-Метилпиперазин-1-илметил)-3-трифторметилбензоилхлорида дигидрохлорид Таким же образом, как и в сравнительном примере 1, за исключением того, что 4-метил-3-трифторбензойную кислоту использовали вместо 3-бром-4-метилбензойной кислоты на стадии 1, получали бледно-коричневое кристаллическое вещество. Температура плавления: 214-216°С (с разложением) 1H-ЯМР (D2O) Сравнительный пример 6 4-Метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]анилин Стадия 1 3-(Диметиламино)-1-(5-пиримидинил)-2-пропен-1-он Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). 6,01 г диметилацеталя N,N-диметилформамида добавляли к 1,54 г 5-ацетилпиримидина (Khim. Geterotsikl. Soedim., 1981, (7), 958-962) и смесь нагревали с обратным холодильником в течение 15 часов. После того как реакционный раствор охлаждали на воздухе, добавляли небольшое количество диизопропилового эфира и выпавшие в осадок кристаллы собирали фильтрованием с получением 1,52 г целевого соединения в виде красновато-коричневого кристаллического вещества. Температура плавления: 133-135°С 1H-ЯМР (CDCl3) Стадия 2 1-(2-Метил-5-нитрофенил)гуанидин К 135 г нитрата 1-(2-метил-5-нитрофенил)гуанидина (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) непосредственно добавляли 21 г гидроксида натрия в 1,0 л холодного водного раствора с последующим перемешиванием при комнатной температуре в течение 10 минут. Кристаллическое вещество отфильтровывали, достаточно промывали водой и затем сушили сжатым воздухом при 60°С с получением 102 г целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 135-142°С 1H-ЯМР (ДМСО-d6) Стадия 3 1-Метил-4-нитро-2-[4-(5-пиримидинил)пиримидин-2-иламино]бензол К 1,51 г 3-(диметиламино)-1-(5-пиримидинил)-2-пропен-1-она, полученного на стадии 1, добавляли 1,66 г 1-(2-метил-5-нитрофенил)гуанидина, полученного на стадии 2, с последующим перемешиванием при 120°С в течение 2 часов. К отвердевшему реакционному раствору добавляли 2-пропанол и кристаллическое вещество собирали фильтрованием, а затем промывали по очереди 2-пропанолом и диэтиловым эфиром с получением 1,95 г целевого соединения в виде бледно-коричневого кристаллического вещества. Температура плавления: 200-203°С 1H-ЯМР (ДМСО-d6) Стадия 4 4-Метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]анилин Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). 1,95 г 1-метил-4-нитро-2-[4-(5-пиримидинил)пиримидин-2-иламино]бензола, полученного на стадии 3, суспендировали в 300 мл метанола и после добавления 0,50 г 10% палладия-на-угле смесь гидрировали при 30°С при 4 атм в течение 18 часов. Катализатор удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 0,60 г целевого соединения в виде желтого аморфного вещества. 1H-ЯМР (CDCl3) Сравнительный пример 7 4-Метил-3-[4-(2-пиразинил)пиримидин-2-иламино]анилин Стадия 1 3-(Диметиламино)-1-(2-пиразинил)-2-пропен-1-он Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). К 5,00 г 2-ацетилпиразина добавляли 5,37 г диметилацеталя N,N-диметилформамида и смесь нагревали с обратным холодильником в течение 19 часов. Реакционный раствор охлаждали на воздухе и выпавшее в осадок кристаллическое вещество растворяли в этилацетате и затем концентрировали при пониженном давлении. После добавления небольшого количества диэтилового эфира выпавшие в осадок кристаллы собирали фильтрованием и затем промывали по очереди диэтиловым эфиром и диизопропиловым эфиром с получением 5,20 г целевого соединения в виде коричневого кристаллического вещества. 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[4-(2-пиразинил)пиримидин-2-иламино]бензол Это соединение получали модифицированными методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). 2,00 г 3-(Диметиламино)-1-(2-пиразинил)-2-пропен-1-она, полученного на стадии 1, и 2,90 г нитрата 1-(2-метил-5-нитрофенил)гуанидина (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) суспендировали в 23 мл 2-пропанола и после добавления 0,50 г гидроксида натрия смесь нагревали с обратным холодильником в течение 20 часов. После охлаждения реакционного раствора на воздухе выпавшие в осадок кристаллы собирали фильтрованием с получением 3,25 г неочищенного кристаллического вещества. Неочищенное кристаллическое вещество растворяли в хлороформе-метаноле (2:1) и нерастворившиеся вещества удаляли фильтрованием, а затем фильтрат концентрировали при пониженном давлении с получением 1,93 г целевого соединения в виде охристого кристаллического вещества. Температура плавления: 207-210°С 1H-ЯМР (ДМСО-d6) Стадия 3 4-Метил-3-[4-(2-пиразинил)пиримидин-2-иламино]анилин Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). 1,00 г 1-метил-4-нитро-2-[4-(2-пиразинил)пиримидин-2-иламино]бензола, полученного на стадии 2, суспендировали в 50 мл метанола и после добавления 100 мг 10% палладия-на-угле смесь гидрировали при комнатной температуре при 3 атм в течение 14 часов и затем гидрировали при 3,4 атм в течение 4 часов. Катализатор удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 0,49 г целевого соединения в виде желтого аморфного вещества. 1H-ЯМР (CDCl3) Сравнительный пример 8 3-[4-(6-Хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилин Стадия 1 5-Ацетил-2-хлорпиридин 1,84 г Размолотого хлорида магния суспендировали в 20 мл толуола и по очереди добавляли 9,4 мл триэтиламина и 4,46 г диэтилмалоната. После перемешивания при комнатной температуре в течение 1,5 часов каплями добавляли суспензию 4,84 г 6-хлорникотиноилхлорида в 10 мл толуола в течение 20 минут с последующим перемешиванием при комнатной температуре в течение 2 часов. После нейтрализации 60 мл 1 N соляной кислоты водный слой отделяли. Водный слой дополнительно экстрагировали диэтиловым эфиром и органические слои объединяли, а затем растворитель отгоняли при пониженном давлении. К полученному неочищенному кристаллическому веществу добавляли диметилсульфоксид-воду (25 мл-1 мл) с последующим перемешиванием с нагреванием до 150-160°С в течение 2 часов. Реакционный раствор охлаждали на воздухе и добавляли воду, а затем выпавшие в осадок кристаллы собирали фильтрованием. Выпавшее в осадок кристаллическое вещество растворяли в этилацетате, и раствор промывали по очереди водой и насыщенным водным раствором гидрокарбоната натрия и затем сушили над безводным сульфатом магния. Полученное неочищенное кристаллическое вещество промывали диизопропиловым эфиром и затем собирали фильтрованием с получением 2,74 г целевого соединения в виде полупрозрачного кристаллического вещества. Температура плавления: 101-102°С 1H-ЯМР (CDCl3) Стадия 2 1-(6-Хлорпиридин-3-ил)-3-(диметиламино)-2-пропен-1-он Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). К 2,68 г 5-ацетил-2-хлорпиридина, полученного на стадии 1, добавляли 2,26 г диметилацеталя N,N-диметилформамида и смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения на воздухе реакционный раствор непосредственно очищали колоночной хроматографией на силикагеле. Полученное неочищенное кристаллическое вещество промывали диэтиловым эфиром и затем собирали фильтрованием с получением 1,87 г целевого соединения в виде желтого кристаллического вещества. Температура плавления: 122-123°С 1H-ЯМР (CDCl3) Стадия 3 2-[4-(6-Хлорпиридин-3-ил)пиримидин-2-иламино]-1-метил-4-нитробензол К 1,83 г 1-(6-хлорпиридин-3-ил)-3-(диметиламино)-2-пропен-1-она, полученного на стадии 2, и 1,69 г 1-(2-метил-5-нитрофенил)гуанидина, полученного на стадии 2 сравнительного примера 6, добавляли 18 мл 2-пропанола и смесь нагревали с обратным холодильником в течение 16 часов. После охлаждения реакционного раствора на воздухе выпавшее в осадок кристаллическое вещество собирали фильтрованием и промывали диэтиловым эфиром. Полученное неочищенное кристаллическое вещество очищали колоночной хроматографией на силикагеле с получением 0,91 г целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 210-212°С 1H-ЯМР (ДМСО-d6) Стадия 4 3-[4-(6-Хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилин К 842 мг 2-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-1-метил-4-нитробензола, полученного на стадии 3, добавляли 6 мл концентрированной соляной кислоты и добавляли раствор 2,78 г дигидрата хлорида олова (II) в 4 мл концентрированной соляной кислоты при перемешивании с нагреванием до 55°С. Смесь постепенно нагревали до 100°С и дополнительно перемешивали с нагреванием до 100°С в течение 15 минут. Реакционный раствор охлаждали на воздухе и добавляли воду, а затем подщелачивали 10% водным раствором гидроксида натрия. После добавления хлороформа и перемешивания в течение некоторого времени нерастворимые вещества удаляли фильтрованием и водный слой отделяли. Водный слой дополнительно экстрагировали хлороформом и органические слои объединяли и после осушивания над безводным сульфатом магния растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением неочищенного продукта. Неочищенный продукт кристаллизовали добавлением диэтилового эфира, и кристаллическое вещество собирали фильтрованием с получением 680 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 117-118°С 1H-ЯМР (CDCl3) Сравнительный пример 9 3-[4-(5-Бромпиридин-3-ил)пиримидин-2-иламино]-4-метиланилин Стадия 1 5-Бромникотиноилхлорид К 5,00 г 5-бромникотиновой кислоты добавляли 74 мл тионилхлорида и смесь нагревали с обратным холодильником в течение 6 часов. После того как растворитель отгоняли при пониженном давлении, кристаллическое вещество промывали диизопропиловым эфиром и собирали фильтрованием с получением 4,09 г целевого соединения в виде бесцветного кристаллического вещества. Температура плавления: 72-74°С 1H-ЯМР (CDCl3) Стадия 2 3-Ацетил-5-бромпиридин 1,24 г размолотого хлорида магния суспендировали в 13 мл толуола и по очереди добавляли 6,2 мл триэтиламина и 2,93 г диэтилмалоната. После перемешивания при комнатной температуре в течение 1,5 часов каплями добавляли суспензию 4,08 г 5-бромникотиноилхлорида, полученного на стадии 1, в 10 мл толуола в течение 15 минут с последующим перемешиванием при комнатной температуре в течение 2 часов. После нейтрализации 40 мл 1N соляной кислоты водный слой отделяли. Водный слой экстрагировали диэтиловым эфиром и органические слои объединяли, а затем растворитель отгоняли при пониженном давлении. К полученному масляному продукту добавляли диметилсульфоксид-воду (17 мл-0,7 мл) с последующим перемешиванием при нагревании до 150-160°С в течение 2 часов. Реакционный раствор охлаждали на воздухе и добавляли воду, а затем выпавшее в осадок кристаллическое вещество собирали фильтрованием. Выпавшее в осадок кристаллическое вещество растворяли в этилацетате, промывали по очереди водой и насыщенным водным раствором гидрокарбоната натрия, а затем сушили над безводным сульфатом магния. Добавляли 0,60 г активированного угля (Kyoryoku Shirasagi MOIWY433) и после стояния в течение 10 минут активированный уголь удаляли фильтрованием, фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 0,89 г целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 87-89,5°С 1H-ЯМР (CDCl3) Стадия 3 1-(5-Бромпиридин-3-ил)-3-(диметиламино)-2-пропен-1-он Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). К 859 мг 3-ацетил-5-бромпиридина, полученного на стадии 2, добавляли 563 мг диметилацеталя N,N-диметилформамида и смесь нагревали с обратным холодильником в течение одного часа. После охлаждения на воздухе реакционный раствор непосредственно очищали колоночной хроматографией на силикагеле. Полученное кристаллическое неочищенное вещество промывали диэтиловым эфиром и затем собирали фильтрованием с получением 860 мг целевого соединения в виде желтого кристаллического вещества. Температура плавления: 131-131,5°С 1H-ЯМР (CDCl3) Стадия 4 2-[4-(5-Бромпиридин-3-ил)пиримидин-2-иламино]-1-метил-4-нитробензол К 833 мг 1-(5-Бромпиридин-3-ил)-3-(диметиламино)-2-пропен-1-она, полученного на стадии 3, и 634 мг 1-(2-метил-5-нитрофенил)гуанидина, полученного на стадии 2 сравнительного примера 6, добавляли 7 мл 2-пропанола и смесь нагревали с обратным холодильником в течение 17 часов. После того как реакционный раствор высыхал на воздухе, выпавшее в осадок кристаллическое вещество собирали фильтрованием и промывали диэтиловым эфиром с получением 823 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 206-208°С 1H-ЯМР (ДМСО-d6) Стадия 5 3-[4-(5-Бромпиридин-3-ил)пиримидин-2-иламино]-4-метиланилин К 807 мг 2-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-1-метил-4-нитробензола, полученного на стадии 4, добавляли 5 мл концентрированной соляной кислоты и добавляли раствор 2,36 г дигидрата хлорида олова (II) в 3,5 мл концентрированной соляной кислоты при перемешивании с нагреванием до 55°С. Смесь постепенно нагревали до 100°С и дополнительно перемешивали с нагреванием до 100°С в течение 15 минут. Реакционный раствор охлаждали на воздухе и добавляли воду, а затем подщелачивали 10% водным раствором гидроксида натрия. После добавления хлороформа и перемешивания в течение некоторого времени нерастворимые вещества удаляли фильтрованием и водный слой отделяли. Водный слой дополнительно экстрагировали хлороформом и органические слои объединяли и после высушивания над безводным сульфатом магния растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением неочищенного продукта. Неочищенный продукт кристаллизовали добавлением диэтилового эфира-этилацетата и кристаллическое вещество собирали фильтрованием с получением 528 мг целевого соединения в виде желтого кристаллического вещества. Температура плавления: 129,5-130°С 1H-ЯМР (CDCl3) Сравнительный пример 10 3-[4-(1,2-Дигидропиридазин-4-ил)пиримидин-2-иламино]-4-метиланилин Стадия 1 4-Ацетилпиридазин К 3,55 г калиевой соли моноэтилового эфира малоновой кислоты и 2,21 г хлорида магния добавляли 12 мл N,N-диметилформамида и смесь перемешивали при нагревании до 60°С в течение 4 часов (реакционный раствор 1). Отдельно готовили реакционный раствор (реакционный раствор 2) перемешиванием 2,07 г 4-пиридазинкарбоновой кислоты (J. Heterocyclic Chem., 1990, 27, 579-582) и 2,95 г 1,1′-карбонилбис-1Н-имидазола в 12 мл N,N-диметилформамида при комнатной температуре в течение 4 часов и реакционный раствор добавляли к реакционному раствору 1 с последующим перемешиванием при комнатной температуре в течение 26 часов. К реакционному раствору добавляли диэтиловый эфир и смесь нейтрализовали 50 мл 1N соляной кислоты. Водный слой отделяли и водный слой дополнительно экстрагировали четыре раза диэтиловым эфиром. Органические слои объединяли и после высушивания над безводным сульфатом магния растворитель отгоняли при пониженном давлении. К полученному масляному продукту добавляли диметилсульфоксид-воду (5 мл-0,4 мл) с последующим перемешиванием с нагреванием до 150-160°С в течение 2 часов. Растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле. Полученное неочищенное кристаллическое вещество промывали диизопропиловым эфиром и затем собирали фильтрованием с получением 429 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 66,5-67,5°С 1H-ЯМР (CDCl3) Стадия 2 3-(Диметиламино)-1-(4-пиридазинил)-2-пропен-1-он Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). К 410 мг 4-ацетилпиридазина, полученного на стадии 1, добавляли 440 мг диметилацеталя N,N-диметилформамида и смесь нагревали с обратным холодильником в течение одного часа. После охлаждения на воздухе реакционный раствор непосредственно очищали колоночной хроматографией на силикагеле. Полученное кристаллическое неочищенное вещество промывали диэтиловым эфиром и затем собирали фильтрованием с получением 341 мг целевого соединения в виде оранжевого кристаллического вещества. Температура плавления: 136-138°С 1H-ЯМР (CDCl3) Стадия 3 1-Метил-4-нитро-2-[4-(4-пиридазинил)пиримидин-2-иламино]бензол К 327 мг 3-(диметиламино)-1-(4-пиридазинил)-2-пропен-1-она, полученного на стадии 2, и 359 мг 1-(2-метил-5-нитрофенил)гуанидина, полученного на стадии 2 сравнительного примера 6, добавляли 4 мл 2-пропанола и смесь нагревали с обратным холодильником в течение 22 часов. Реакционный раствор охлаждали на воздухе и выпавшее в осадок кристаллическое вещество собирали фильтрованием, а затем по очереди промывали 2-пропанолом и диэтиловым эфиром с получением 437 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 243-245°С 1H-ЯМР (ДМСО-d6) Стадия 4 3-[4-(1,2-Дигидропиридазин-4-ил)пиримидин-2-иламино]-4-метиланилин К 413 мг 1-метил-4-нитро-2-[4-(4-пиридазинил)пиримидин-2-иламино]бензола, полученного на стадии 3, добавляли 3 мл концентрированной соляной кислоты и раствор 1,51 г дигидрата хлорида олова (II) в 2 мл концентрированной соляной кислоты при перемешивании с нагреванием до 55°С. Смесь постепенно нагревали до 100°С и дополнительно перемешивали при нагревании до 100°С в течение 25 минут. Реакционный раствор охлаждали на воздухе и после добавления воды раствор подщелачивали 10% водным раствором гидроксида натрия. После добавления хлороформа и перемешивания в течение некоторого времени нерастворимые вещества удаляли фильтрованием и водный слой отделяли. Водный слой дополнительно экстрагировали хлороформом и органические слои объединяли и после высушивания над безводным сульфатом магния растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 38 г целевого соединения в виде бледно-желтого аморфного вещества. 1H-ЯМР (CDCl3) Сравнительный пример 11 4-Метил-3-[4-(3-пиридазинил)пиримидин-2-иламино]анилин Стадия 1 3-(Диметиламино)-1-(3-пиридазинил)-2-пропен-1-он Это соединение получали модифицированным методом, описанным в документе (Japanese Unexamined Patent Publication (Kokai) No. 6-87834). К 762 мг 3-ацетилпиридазина (Arzneim.-Forsch./Drug Res., 1989, 39(2), 1196-1201) добавляли 818 мг диметилацеталя N,N-диметилформамида и смесь нагревали с обратным холодильником в течение 1,5 часов. После охлаждения на воздухе реакционный раствор непосредственно очищали колоночной хроматографией на силикагеле. Полученное неочищенное кристаллическое вещество промывали диизопропиловым эфиром и затем собирали фильтрованием с получением 945 мг целевого соединения в виде желтовато-коричневого кристаллического вещества. Температура плавления: 102-105°С 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[4-(3-пиридазинил)пиримидин-2-иламино]бензол 800 мг 3-(Диметиламино)-1-(3-пиридазинил)-2-пропен-1-она, полученного на стадии 1, и 876 мг 1-(2-метил-5-нитрофенил)гуанидина, полученного на стадии 2 сравнительного примера 6, перемешивали при нагревании до 120°С в течение 3 часов. Отвердевший реакционный раствор кристаллизовали добавлением 2-пропанола и затем промывали по очереди 2-пропанолом и диэтиловым эфиром с получением 1,21 г целевого соединения в виде темно-коричневого кристаллического вещества. Температура плавления: 275-277°С 1H-ЯМР (CF3COOD) Стадия 3 4-Метил-3-[4-(3-пиридазинил)пиримидин-2-иламино]анилин 754 мг 1-метил-4-нитро-2-[4-(3-пиридазинил)пиримидин-2-иламино]бензола, полученного на стадии 2, суспендировали в 40 мл метанола и добавляли 4,21 г дитионата натрия и 3,05 г гидрокарбоната натрия и затем смесь нагревали с обратным холодильником в течение 5 часов. После того как реакционный раствор охлаждался на воздухе, нерастворимые вещества удаляли фильтрованием и растворитель отгоняли при пониженном давлении. К остатку добавляли воду и хлороформ с отделением водного слоя, а затем водный слой экстрагировали три раза хлороформом. Органические слои объединяли, промывали по очереди водой, насыщенным раствором соли и затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 247 мг целевого соединения в виде желтого масляного продукта. 1H-ЯМР (CDCl3) Сравнительный пример 12 4-Метил-3-[4-(3-пиридил)пиридин-2-иламино]анилин Стадия 1 2-[(4-Хлор)пиридин-2-иламино]-1-метил-4-нитробензол Это соединение получали модифицированным методом, описанным в документе (J. Org. Chem., 1996, 61, 7240-7241). К 2,00 г 2,4-дихлорпиридина (Recl. Trav. Chim. Pays-Bas., 1950, 69, 673-699), 2,26 г 2-метил-5-нитроанилина, 121 мг ацетата палладия (II), 336 мг (±)-2,2′-бис(дифенилфосфино)-1,1′-бинафтила [(±)-BINAP] и 6,16 г карбоната цезия добавляли 120 мл толуола и затем смесь перемешивали при нагревании до 70°С в течение 23 часов в атмосфере аргона. После того как нерастворимые вещества удаляли фильтрованием, растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 2,11 г неочищенного продукта. Неочищенный продукт промывали диэтиловым эфиром с получением 1,22 г целевого соединения в виде желтого кристаллического вещества. Температура плавления: 130-133°С. 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[4-(3-пиридил)пиридин-2-иламино]бензол К 20 мл дезаэрированных тетрагидрофурана-воды (1:1) добавляли по очереди 264 мг 2-[(4-хлор)пиридин-2-иламино]-1-метил-4-нитробензола, полученного на стадии 1, 162 мг диэтил(3-пиридил)борана, 470 мг карбоната калия и 173 мг тетракис(трифенилфосфин)палладия (0) и смесь перемешивали при нагревании до 80°С в течение 44 часов в атмосфере аргона. Реакционный раствор разбавляли этилацетатом, чтобы отделить водный слой, а затем водный слой дополнительно экстрагировали три раза этилацетатом. Органические слои объединяли, промывали по очереди водой и насыщенным раствором соли и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 247 мг неочищенного продукта. Неочищенный продукт кристаллизовали добавлением хлороформа-метанола и затем собирали фильтрованием с получением 143 мг целевого соединения в виде оранжевого кристаллического вещества. Температура плавления: 170-173°С 1H-ЯМР (CDCl3) Стадия 3 4-Метил-3-[4-(3-пиридил)пиридин-2-иламино]анилин К 126 мг 1-метил-4-нитро-2-[4-(3-пиридил)пиридин-2-иламино]бензола, полученного на стадии 2, добавляли 1 мл концентрированной соляной кислоты и добавляли раствор 465 мг дигидрата хлорида олова (II) в 1 мл концентрированной соляной кислоты с перемешиванием при нагревании до 60°С. Смесь постепенно нагревали до 100°С и дополнительно перемешивали при нагревании до 100°С в течение 40 минут. После того как реакционный раствор охлаждался на воздухе, добавляли воду и раствор подщелачивали 10% водным раствором гидроксида натрия. Раствор экстрагировали три раза этилацетатом и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Полученное неочищенное кристаллическое вещество промывали небольшим количеством хлороформа и затем собирали фильтрованием с получением 93 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 183-186°С 1H-ЯМР (CDCl3) Сравнительный пример 13 4-Метил-3-[4-(5-пиримидинил)пиридин-2-иламино]анилин Стадия 1 1-Метил-4-нитро-2-[4-(5-пиримидинил)пиридин-2-иламино]бензол Таким же образом, что и в сравнительном примере 12 (стадия 2), за исключением того, что дигидрокси(5-пиримидинил)боран использовали вместо диэтил(3-пиридил)борана, получали целевое соединение. Неочищенное кристаллическое вещество, полученное путем очистки колоночной хроматографией на силикагеле, промывали диэтиловым эфиром. Желтое кристаллическое вещество Температура плавления: 230-232°С 1H-ЯМР (ДМСО-d6) Стадия 2 4-Метил-3-[4-(5-пиримидинил)пиридин-2-иламино]анилин 163 мг 1-Метил-4-нитро-2-[4-(5-пиримидинил)пиридин-2-иламино]бензол, полученный на стадии 1, растворяли в 32 мл тетрагидрофуране-метаноле (1:1) и добавляли 98 мг 10% палладия-на-угле. Кроме того, добавляли 284 мг формиата аммония и смесь нагревали с обратным холодильником на бане с температурой 90°С в течение 40 минут. Катализатор удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. К остатку добавляли воду и этилацетат с отделением водного слоя. Водный слой дополнительно экстрагировали этилацетатом. Органические слои объединяли и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 149 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 179-180°С 1H-ЯМР (ДМСО-d6) Сравнительный пример 14 4-Метил-3-[2-(3-пиридил)пиридин-6-иламино]анилин Стадия 1 2-Бром-6-(3-пиридил)пиридин Это соединение получали модифицированным методом, описанным в документе (Chem. Pharm. Bull., 1985, 33(11), 4755-4763). К 40 мл тетрагидрофурана по очереди добавляли 1,76 г диэтил(3-пиридил)борана, 5,92 г 2,6-дибромпиридина, 1,99 г тетра-н-бутиламмония бромида, 692 мг тетракис(трифенилфосфин)палладия (0) и 1,87 г измельченного гидроксида калия и смесь нагревали с обратным холодильником в течение трех часов в атмосфере аргона. После охлаждения на воздухе реакционный раствор разбавляли этилацетатом, и нерастворимые вещества удаляли фильтрованием. Растворитель из фильтрата отгоняли при пониженном давлении, к остатку добавляли этилацетат и насыщенный раствор соли с отделением водного слоя. Органический слой сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 1,26 г целевого соединения в виде бледно-желтого кристаллического вещества. 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[2-(3-пиридил)пиридин-6-иламино]бензол Это соединение получали модифицированным методом, описанным в документе (J. Org. Chem., 2000, 65, 1144-1157). К 940 мг 2-бром-6-(3-пиридил)пиридина, полученного на стадии 1, 730 мг 2-метил-5-нитроанилина, 37 мг трис(дибензилиденацетон)дипалладия (0), 75 мг (±)-2,2′-бис(дифенилфосфино)-1,1′-бинафтила [(±)-BINAP] и 1,82 мг карбоната цезия добавляли 12 мл толуола и смесь перемешивали при нагревании до 110°С в течение 24 часов в атмосфере аргона. После охлаждения на воздухе реакционный раствор разбавляли этилацетатом и нерастворимые вещества удаляли фильтрованием. Растворитель из фильтрата отгоняли при пониженном давлении и остаток кристаллизовали добавлением диэтилового эфира. Полученное кристаллическое вещество собирали фильтрованием и затем промывали этилацетатом-диэтиловым эфиром с получением 646 мг целевого соединения в виде желтого кристаллического вещества. Температура плавления: 148-150°С 1H-ЯМР (CDCl3) Стадия 3 4-Метил-3-[2-(3-пиридил)пиридин-6-иламино]анилин 500 мг 1-Метил-4-нитро-2-[2-(3-пиридил)пиридин-6-иламино]бензола, полученного на стадии 2, растворяли в 10 мл этанола и добавляли 1,05 г цинка (порошок), 430 мг хлорида аммония и 0,46 мл уксусной кислоты и затем смесь перемешивали при нагревании до 80°С в течение 30 минут. Катализатор удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. К остатку добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия с отделением водного слоя. Водный слой дополнительно три раза экстрагировали этилацетатом. Органические слои объединяли и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении с получением 114 мг целевого соединения в виде бледно-желтого аморфного вещества. 1H-ЯМР (CDCl3) Сравнительный пример 15 4-Метил-3-[3-(3-пиридил)пиридин-5-иламино]анилин Стадия 1 3-Бром-5-(3-пиридил)пиридин Таким же образом, что и в сравнительном примере 14 (стадия 1), за исключением того, что 3,5-дибромпиридин использовали вместо 2,6-дибромпиридина, получали целевое соединение. Бесцветное кристаллическое вещество 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[3-(3-пиридил)пиридин-5-иламино]бензол Это соединение получали модифицированным методом, описанным в документе (J. Org. Chem., 1996, 61, 7240-7241). К 25 мг (±)-2,2′-бис(дифенилфосфино)-1,1′-бинафтила [(±)-BINAP] добавляли 2 мл толуола и (±)-BINAP растворяли перемешиванием при нагревании до 80°С в атмосфере аргона. Раствор охлаждали до комнатной температуры и добавляли 6 мг ацетата палладия (II) и, перемешивая в течение одной минуты, добавляли 620 мг 3-бром-5-(3-пиридил)пиридин, полученный на стадии 1, 482 мг 2-метил-5-нитроанилина, 1,20 г карбоната цезия и 2 мл (всего 4 мл) толуола с последующим перемешиванием при 80°С в течение 18 часов и дополнительным нагреванием до 100°С в течение 24 часов в атмосфере аргона. После охлаждения на воздухе реакционный раствор разбавляли этилацетатом и нерастворимые вещества удаляли фильтрованием. Растворитель из фильтрата отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с получением 108 мг целевого соединения в виде желтого кристаллического вещества. Температура плавления: 195-198°С 1H-ЯМР (CDCl3) Стадия 3 4-Метил-3-[3-(3-пиридил)пиридин-5-иламино]анилин Таким же образом, что и в сравнительном примере 12 (стадия 3), за исключением того, что 1-метил-4-нитро-2-[3-(3-пиридил)пиридин-5-иламино]бензол, полученный на стадии 2, использовали вместо 1-метил-4-нитро-2-[4-(3-пиридил)пиридин-2-иламино]бензола, получали целевое соединение. Остаток, полученный концентрированием при пониженном давлении, дополнительно не очищали. Бледно-коричневый маслянистый продукт 1H-ЯМР (CDCl3) Сравнительный пример 16 4-Метил-3-[3-(3-пиридил)фениламино)анилин Стадия 1 2-(3-Бромфениламино)-1-метил-4-нитробензол Это соединение получали модифицированным методом, описанным в документе (J. Org. Chem., 2000, 65, 1144-1157). К 1,00 г 1-бром-3-йодбензола, 591 мг 2-метил-5-нитроанилина, 32 мг трис-(дибензилиденацетон)дипалладия (0), 66 мг (±)-2,2′-бис(дифенилфосфино)-1,1′-бинафтила [(±)-BINAP] и 1,61 г карбоната цезия добавляли 14 мл толуола и смесь перемешивали при нагревании до 100°С в течение 36 часов в атмосфере аргона. После охлаждения на воздухе нерастворимые вещества удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 256 мг целевого соединения в виде оранжевого кристаллического вещества. Температура плавления: 114-116°С 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[3-(3-пиридил)фениламино]бензол Таким же образом, что и в сравнительном примере 12 (стадия 2), за исключением того, что 2-(3-бромфениламино)-1-метил-4-нитробензол, полученный на стадии 1, использовали вместо 2-[(4-хлор)пиридин-2-иламино]-1-метил-4-нитробензола, получали целевое соединение. Полученный продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Желтое кристаллическое вещество. Температура плавления: 162-165°С 1H-ЯМР (ДМСО-d6) Стадия 3 4-Метил-3-[3-(3-пиридил)фениламино]анилин Таким же образом, что и в сравнительном примере 12 (стадия 3), за исключением того, что 1-метил-4-нитро-2-[3-(3-пиридил)фениламино]бензол, полученный на стадии 2, использовали вместо 1-метил-4-нитро-2-[4-(3-пиридил)пиридин-2-иламино]-бензола, получали целевое соединение. Остаток, полученный концентрированием при пониженном давлении, дополнительно не очищали. Бледно-желтый маслянистый продукт 1H-ЯМР (CDCl3) Сравнительный пример 17 4-Метил-3-[2-(3-пиридил)пиразин-6-иламино)анилин Стадия 1 2-[(2-Хлор)пиразин-6-иламино)-1-метил-4-нитробензол Таким же образом, что и в сравнительном примере 12 (стадия 1), за исключением того, что 2,6-дихлорпиразин использовали вместо 2,4-дихлорпиридина, получали целевое соединение. Желтое кристаллическое вещество 1H-ЯМР (CDCl3) Стадия 2 1-Метил-4-нитро-2-[2-(3-пиридил)пиразин-6-иламино]бензол К 64 мл дезаэрированных тетрагидрофурана-воды (1:1) добавляли по очереди 790 мг 2-[(2-хлор)пиразин-6-иламино]-1-метил-4-нитробензола, полученного на стадии 1, 406 мг дигидрокси(3-пиридил)борана, 1,41 г карбоната калия и 520 мг тетракис(трифенилфосфин)палладия (0) и смесь нагревали с обратным холодильником на бане с температурой 100°С в течение 3 часов в атмосфере аргона. Добавляли 32 ил тетрагидрофурана-воды (1:1) и смесь дополниетльно нагревали с обратным холодильником в течение 3 часов и затем давали постоять при комнатной температуре в течение ночи. Выпавшие в осадок нерастворимые вещества собирали фильтрованием, экстрагировали и промывали метанолом, а затем растворитель из фильтрата отгоняли при пониженном давлении. К остатку добавляли диэтиловый эфир и после перемешивания кристаллическое вещество собирали фильтрованием и затем промывали метанолом с получением 270 мг целевого соединения в виде аморфного вещества. 1H-ЯМР (ДМСО-d6) Стадия 3 4-Метил-3-[2-(3-пиридил)пиразин-6-иламино]анилин 107 мг 1-метил-4-нитро-2-[2-(3-пиридин)пиразин-6-иламино]бензол, полученный на стадии 2, растворяли в 10 мл метанола (часть не растворялась, а суспендировалась) и добавляли 16 мг 10% палладия-на-угле. Кроме того, добавляли 221 мг формиата аммония с последующим перемешиванием с нагреванием на водяной бане с температурой 50°С в течение 15 часов. Катализатор удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. К остатку добавляли воду и этилацетат с отделением водного слоя. Водный слой дополнительно эктрагировали этилацетатом. Органические слои объединяли и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении с получением 95 мг целевого соединения. 1H-ЯМР (CDCl3) Сравнительный пример 18 4-Метил-3-[5-(3-пиридил)-1,2,4-триазин-3-иламино]анилин Стадия 1 3-Метилтио-5-(3-пиридил)-1,2,4-триазин Сначала получали (3-пиридил)глиоксаля гидробромид модифицированным методом, описанным в документе (Heterocycles, 1990, 31(12), 2163-2172). 5,00 г гидробромида 3-(бромацетил)пиридина (J. Heterocyclic Chem., 1969, 6(6), 891-900) суспендировали в 30 мл метанола и добавляли 3,40 г N-оксида пиридина при перемешивании с охлаждением льдом с последующим перемешиванием при комнатной температуре в течение 26 часов после удаления ледяной бани. Это соединение использовали для последущей реакции без выделения. Затем получали 3-метилтио-5-(3-пиридил)-1,2,4-триазин модифицированным методом, описанным в документе (J. Med. Chem., 1979, 22(6), 671-677). К вышеуказанному раствору добавляли 4,18 г гидрохлорида S-метилтиосемикарбазида (Heterocycles, 1979, 12(6), 745-749) и 1,51 г гидрокарбоната натрия при перемешивании с охлаждением льдом и добавляли 6 мл воды и после медленного возвращения температуры к комнатной температуре смесь перемешивали при комнатной температуре в течение 57 часов. Реакционный раствор подщелачивали добавлением холодного водного насыщенного раствора гидрокарбоната натрия, дважды экстрагировали этилацетатом и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 1,37 г неочищенного продукта. Неочищенный продукт промывали теплым диизопропиловым эфиром с получением 1,13 г целевого соединения в виде бледного желтовато-зеленого кристаллического вещества. Температура плавления: 98-102°С 1H-ЯМР (CDCl3) Стадия 2 3-Метилсульфинил-5-(3-пиридил)-1,2,4-триазин 3,00 г 3-Метилтио-5-(3-пиридил)-1,2,4-триазина, полученного на стадии 1, растворяли в 50 мл дихлорметана и добавляли 5,76 г 70% м-хлорбензойной кислоты при перемешивании с охлаждением льдом. После перемешивания при охлаждении льдом в течение 20 минут температура возвращалась к комнатной температуре и добавляли 1,5 г сульфата магния и 10 г NH-силикагеля (Chromatorex NH-DM1020, производимый Fuji Silysia Chemical Co., Ltd.). После перемешивания при комнатной температуре в течение 10 минут нерастворимые вещества удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 1,65 г неочищенного продукта. Неочищенный продукт промывали 2-пропанолом-диэтиловым эфиром с получением 1,07 целевого соединения в виде бледно-коричневого кристаллического вещества. Температура плавления: 150-152°С 1Н-ЯМР (CDCl3) Стадия 3 N-(5-Амино-2-метилфенил)ацетамид 3,00 г N-(2-метил-5-нитрофенил)ацетамида (Can. J. Chem., 1984, 62, 1292-1296) суспендировали в 100 мл этанола и добавляли 600 мг 10% палладия-на-угле и затем смесь гидрировали при комнатной температуре при давлении 4 атм в течение 3 часов. Катализатор удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении с получением 2,50 г неочищенного продукта. Неочищенный продукт промывали теплым раствором диизопропилового эфира с получением 2,37 г целевого соединения в виде бледно-зеленого кристаллического вещества. Температура плавления: 136-139°С 1Н-ЯМР (D2О) Стадия 4 4-Метил-3-[5-(3-пиридил)-1,2,4-триазин-3-иламино]анилин 671 мг N-(5-Амино-2-метилфенил)ацетамида, полученного на стадии 3, растворяли в 40 мл тетрагидрофурана и добавляли 180 мг 60% гидрида натрия при перемешивании с охлаждением льдом. После перемешивания при охлаждении льдом в течение 5 минут температуру возвращали к комнатной температуре с последующим перемешиванием в течение 30 минут и добавляли 900 мг 3-метилсульфинил-5-(3-пиридил)-1,2,4-триазина, полученного на стадии 2. После перемешивания при комнатной температуре в течение 3,5 часов реакционной раствор смешивали с ледяной водой, экстрагировали дважды дихлорметаном и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 733 мг 4-метил-3-{N-ацетил-N-[5-(3-пиридил)-1,2,4-триазин-3-ил]амино}анилина в качестве промежуточного соединения. Указанное выше соединение растворяли в 10 мл метанола и добавляли 2,0 мл 1N водного раствора гидроксида натрия с последующим перемешиванием при комнатной температуре в течение 45 минут. Реакционный раствор смешивали с водой, дважды экстрагировали этилацетатом и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 478 мг целевого соединения в виде желтовато-коричневого аморфного вещества. 1H-ЯМР (CDCl3) Сравнительный пример 19 3-Метил-4-(4-метилпиперазин-1-илметил)бензоилхлорида гидрохлорид Стадия 1 1-(4-Метокси-2-метилбензоил)-4-метилпиперазин К 3,32 г 4-метокси-2-метилбензойной кислоты, 5,75 г гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида и 4,05 г 1-гидроксибензотриазола добавляли 10 мл N,N-диметилформамида. При перемешивании при комнатной температуре по очереди каплями добавляли раствор 2,00 г N-метилпиперазина в 10 мл N,N-диметилформамида и раствор 1,52 г триэтиламина в 10 мл N,N-диметилформамида с последующим перемешиванием при комнатной температуре в течение 15 часов. После того как растворитель отгоняли при пониженном давлении к остатку добавляли насыщенный водный раствор гидрокарбоната натрия и раствор дважды экстрагировали этилацетатом. После высушивания над безводным сульфатом магния растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 4,25 г целевого соединения в виде бледно-желтого маслянистого продукта. 1Н-ЯМР (CDCl3) Стадия 2 1-(4-Гидрокси-2-метилбензоил)-4-метилпиперазин 4,89 г 1-(4-Метокси-2-метилбензоил)-4-метилпиперазин, полученный на стадии 1, растворяли в 150 мл дихлорметана и каплями добавляли раствор 9,87 г трибромида бора в 100 мл дихлорметана при охлаждении льдом. После перемешивания при охлаждении льдом в течение одного часа температуру возвращали к комнатной температуре и смесь дополнительно перемешивали в течение 15 часов. Реакционный раствор охлаждали льдом и подщелачивали добавлением 50 мл воды и 150 мл насыщенного водного раствора гидрокарбоната натрия и затем нерастворимые вещества удаляли фильтрованием. Фильтрат экстрагировали хлороформом и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 2,20 г целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 167-168°С 1H-ЯМР (CDCl3) Стадия 3 3-Метил-4-(4-метилпиперазин-1-илметил)фенол 1,96 г 1-(4-Гидрокси-2-метилбензоил)-4-метилпиперазина, полученного на стадии 2, растворяли в 35 мл тетрагидрофурана и добавляли 0,317 г литийалюмогидрида несколькими порциями при перемешивании с охлаждением льдом. После перемешивания при комнатной температуре в течение 4 часов смесь снова охлаждали льдом и и добавляли 0,317 г литийалюмогидрида несколькими порциями при перемешивании с охлаждением льдом с последующим перемешиванием при комнатной температуре в течение 15 часов. Реакционный раствор охлаждали льдом и добавляли тетрагидрофуран и после разложения литияалюмогидрида нерастворимые вещества удаляли фильтрованием. Растворитель из фильтрата отгоняли, и остаток кристаллизовали добавлением ацетона с получением 1,10 г целевого соединения в виде бесцветного кристаллического вещества. Температура плавления: 174-176°С 1Н-ЯМР (CDCl3) Стадия 4 3-Метил-4-(4-метилпиперазин-1-илметил)фенилтрифторметансульфонат 660 мг 3-Метил-4-(4-метилпиперазин-1-илметил)фенола, полученного на стадии 3, растворяли в 6,6 мл пиридина и добавляли 1,86 г безводной трифторметансульфоновой кислоты при перемешивании с охлаждением льдом с последующим перемешиванием при комнатной температуре в течение 12 часов. Реакционный раствор смешивали с ледяной водой, экстрагировали три раза этилацетатом и затем промывали водой. После высушивания над безводным сульфатом магния растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 535 мг целевого соединения в виде бледно-желтого маслянистого продукта. 1H-ЯМР (CDCl3) Стадия 5 Метил 3-метил-4-(4-метилпиперазин-1-илметил)бензоат 705 мг 3-Метил-4-(4-метилпиперазин-1-илметил)фенил трифторметансульфоната, полученного на стадии 4, растворяли в смеси растворителей из 8,40 мл диметилсульфоксида, 4,96 мл метанола, 2,68 мл 1,2-дихлорэтана и 0,76 мл триэтиламина, а затем добавляли 62,8 мг 1,3-бис(дифенилфосфино)пропана и 34,2 мг ацетата палладия (II). При перемешивании при комнатной температуре реакционный раствор барботировали газообразным моноксидом углерода в течение 5 минут и раствор дополнительно нагревали с обратным холодильником в течение одного часа при барботировании газообразным моноксидом углерода. После охлаждения на воздухе к реакционному раствору добавляли воду и этилацетат и нерастворимые вещества удаляли фильтрованием, а затем фильтрат экстрагировали этилацетатом. Экстракт промывали водой и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 392 мг целевого соединения в виде бледно-желтого маслянистого продукта. 1H-ЯМР (CDCl3) Стадия 6 3-Метил-4-(4-метилпиперазин-1-илметил)бензойной кислоты дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадия 4), за исключением того, что метил 3-метил-4-(4-метилпиперазин-1-илметил)бензоат, полученный на стадии 5, использовали вместо этил 3-бром-4-(4-метилпиперазин-1-илметил)бензоата, получали целевое соединение. Бесцветное кристаллическое вещество Стадия 7 3-Метил-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадия 5), за исключением того, что 3-метил-4-(4-метилпиперазин-1-илметил)бензойной кислоты дигидрохлорид, полученный на стадии 6, использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензойной кислоты, получали целевое соединение. Бесцветное кристаллическое вещество Сравнительный пример 20 4-(4-Метилпиперазин-1-илметил)-3-нитробензоилхлорида дигидрохлорид Стадия 1 Этил 4-(бромметил)-3-нитробензоат Таким же образом, что и в сравнительном примере 1 (стадия 1), за исключением того, что 4-(бромметил)-3-нитробензойную кислоту использовали вместо 3-бром-4-метилбензойной кислоты, получали целевое соединение. Желтый маслянистый продукт 1Н-ЯМР (CDCl3) Стадия 2 Этил 4-(4-метилпиперазин-1-илметил)-3-нитробензоат Таким же образом, что и в сравнительном примере 1 (стадия 3), за исключением того, что этил 4-(бромметил)-3-нитробензоат, полученный на стадии 1, использовали вместо 3-бром-4-(бромметил)бензоата, получали целевое соединение. Желтое кристаллическое вещество Температура плавления: 92-94°С 1Н-ЯМР (CDCl3) Стадия 3 4-(4-Метилпиперазин-1-илметил)-3-нитробензойной кислоты дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадия 4), за исключением того, что этил 4-(4-метилпиперазин-1-илметил)-3-нитробензоат, полученный на стадии 2, использовали вместо этил 3-бром-4-(4-метилпиперазин-1-илметил)бензоата, получали целевое соединение. Бледно-коричневое кристаллическое вещество Температура плавления: 180°С (с разложением) 1Н-ЯМР (D2О) Стадия 4 4-(4-Метилпиперазин-1-илметил)-3-нитробензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадия 5), за исключением того, что 4-(4-метилпиперазин-1-илметил)-3-нитробензойной кислоты дигидрохлорид, полученный на стадии 3, использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензойной кислоты, получали целевое соединение. Бледно-коричневое кристаллическое вещество Температура плавления: 190°С (с разложением) 1Н-ЯМР (D2О) Сравнительный пример 21 3-Метокси-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадии 2-5), за исключением того, что метил 3-метокси-4-метилбензоат использовали вместо этил 3-бром-4-метилбензоата на стадии 2, получали целевое соединение. Бесцветное кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 22 3,5-дибром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадии 2-5), за исключением того, что метил 3,5-дибром-4-метилбензоат использовали вместо этил 3-бром-4-метилбензоата на стадии 2, получали целевое соединение. Бледно-оранжевое кристаллическое вещество 1H-ЯМР (D2O) Сравнительный пример 23 3,5-Диметокси-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что 3,5-диметокси-4-метилбензойную кислоту использовали вместо 3-бром-4-метилбензойной кислоты на стадии 1, получали целевое соединение. Бледно-желтое кристаллическое вещество 1H-ЯМР (D2O) Сравнительный пример 24 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Стадия 1 Этил 3-(N,N-диметилкарбамоил)-4-метилбензоат Это соединение получали модифицированным методом, описанным в документе (Org. Lett., 2002, 4, 2849-2851). 1,00 г этил 3-йод-4-метилбензоата (промежуточное соединение из сравнительного примера 2) растворяли в 30 мл N,N-диметилформамида и добавляли 23 мг трис(дибензилиденацетон)дипалладия (0). При перемешивании при комнатной температуре добавляли 643 мкл оксихлорида фосфора с последующим перемешиванием с нагреванием до 120°С в течение 12 часов в атмосфере аргона. Реакционный раствор смешивали с насыщенным водным раствором гидрокарбоната натрия, дважды экстрагировали этилацетатом, промывали насыщенным раствором соли и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 338 мг целевого соединения в виде коричневого маслянистого продукта. 1Н-ЯМР (CDCl3) Стадия 2 Этил 4-(бромметил)-3-(N,N-диметилкарбамоил)бензоат Таким же образом, что и в сравнительном примере 1 (стадия 2), за исключением того, что этил 3-(N,N-диметилкарбамоил)-4-метилбензоат, полученный на стадии 1, использовали вместо этил 3-бром-4-метилбензоата, получали целевое соединение. Желтый маслянистый продукт 1H-ЯМР (CDCl3) Стадия 3 Этил 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензоат Таким же образом, что и в сравнительном примере 1 (стадия 3), за исключением того, что этил 4-(бромметил)-3-(N,N-диметилкарбамоил)бензоат, полученный на стадии 2, использовали вместо этил 3-бром-4-бромметилбензоата, получали целевое соединение. Коричневый маслянистый продукт 1H-ЯМР (CDCl3) Стадия 4 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензойной кислоты дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадия 4), за исключением того, что этил 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензоат, полученный на стадии 3, использовали вместо этил 3-бром-4-(4-метилпиперазин-1-илметил)бензоата получали целевое соединение. Бледно-желтое аморфное вещество 1Н-ЯМР (D2О) Стадия 5 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадия 5), за исключением того, что 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензойной кислоты дигидрохлорид, полученный на стадии 4, использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)бензойной кислоты дигидрохлорида, получали целевое соединение. Бледно-оранжевое кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 25 3-Бром-4-(4-этилпиперазин-1-илметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что N-этилпиперазин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бледно-коричневое кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 26 3-Бром-4-[4-(н-пропил)пиперазин-1-илметил]бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что N-(н-пропил)пиперазин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бесцветное кристаллическое вещество 1Н-ЯМР(D2О) Сравнительный пример 27 3-Бром-4-(N,N-диметиламинометил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что диметиламин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бесцветное кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 28 3-Бром-4-(N,N-диэтиламинометил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что диэтиламин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бесцветное кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 29 3-Бром-4-(1-пирролидинилметил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что пирролидин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бледно-коричневое кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 30 3-Бром-4-(пиперидинометил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что пиперидин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бесцветное кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 31 3-Бром-4-(морфолинометил)бензоилхлорида дигидрохлорид Таким же образом, что и в сравнительном примере 1, за исключением того, что морфолин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Розовое кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 32 3-Бром-4-(цис-3,5-диметилпиперазин-1-илметил)бензойной кислоты дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадии 1-4), за исключением того, что цис-2,6-диметилпиперазин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Бесцветное кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 33 3-Бром-4-(4-метилгексагидро-1Н-1,4-диазепин-1-илметил)бензойной кислоты дигидрохлорид Таким же образом, что и в сравнительном примере 1 (стадии 1-4), за исключением того, что 4-метилгексагидро-1Н-1,4-диазепин использовали вместо N-метилпиперазина на стадии 3, получали целевое соединение. Желтое кристаллическое вещество 1Н-ЯМР (D2О) Сравнительный пример 34 3-Бром-4-(4-(трет-бутоксикарбонил)пиперазин-1-илметил)бензойная кислота Стадия 1 Этил 3-бром-4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]бензоат 1,00 г Этил 3-бром-4-(бромметил)бензоата (промежуточное соединение из сравнительного примера 1) растворяли в 10 мл безводного тетрагидрофурана, и после добавления 473 мг карбоната калия каплями добавляли 467 мг N-(трет-бутоксикарбонил)пиперазина при перемешивании при комнатной температуре в атмосфере аргона. После перемешивания при комнатной температуре в течение 20 часов нерастворимые вещества удаляли фильтрованием и растворитель из фильтрата отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 918 мг целевого соединения в виде бледно-желтого маслянистого продукта. 1Н-ЯМР (CDCl3) Стадия 2 3-Бром-4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]бензойная кислота 898 мг Этил 3-бром-4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]бензоата, полученного на стадии 1, растворяли в 5 мл метанола и добавляли 3,2 мл 1N водного раствора гидроксида натрия. После перемешивания при комнатной температуре в течение 3 часов смесь нейтрализовали медленным добавлением 3,2 мл 1N соляной кислоты. Выпавшее в осадок кристаллическое вещество собирали фильтрованием, промывали водой и затем сушили при пониженном давлении с получением 796 мг целевого соединения в виде бесцветного кристаллического вещества. Температура плавления: 204-205°С (с разложением) 1Н-ЯМР (ДМСО-d6) Сравнительный пример 35 4-[4-(трет-Бутоксикарбонил)пиперазин-1-илметил]-3-трифторметилбензойная кислота Таким же образом, что и в сравнительном примере 34, за исключением того, что 4-(бромметил)-3-трифторметилбензоат (промежуточное соединение из сравнительного примера 5) использовали вместо этил 3-бром-4-(бромметил)бензоата на стадии 1, получали целевое соединение. Бесцветное кристаллическое вещество Температура плавления: 126-134°С 1Н-ЯМР (CDCl3) Пример 1 3-Бром-4-(4-метилпиперазин-1-илметил)-N-(4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил)бензамид 0,74 г 4-Метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) растворяли в 27 мл безводного пиридина и добавляли 920 мг 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида (сравнительный пример 1) с последующим перемешиванием при комнатной температуре в течение 14 часов. Реакционный раствор смешивали с ледяной водой и насыщенным водным раствором гидрокарбоната натрия, а затем экстрагировали этилацетатом. Экстракт промывали водой и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 1,48 г неочищенного продукта. Неочищенный продукт кристаллизовали добавлением хлороформа-диэтилового эфира (1:1) и кристаллическое вещество собирали фильтрованием с получением 1,05 г целевого соединения в виде бесцветного кристаллического вещества. Температура плавления: 202-203°С (с разложением) Элементный анализ (для С29Н30BrN7О·0,9Н2О) Вычислено(%): С, 59,17; Н, 5,44; N, 16,65 Найдено (%): С, 59,16; Н, 5,21; N, 16,64 Пример 2 3-Йод-4-(4-метилпиперазин-1-илметил)-N-(4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил)бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что 3-йод-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид (сравнительный пример 2) использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида и что реакцию проводили при комнатной температуре в течение 24 часов и получающееся кристаллическое вещество перекристаллизовывали из метанола. Бесцветное кристаллическое вещество Температура плавления: 199-200°С (с разложением) Элементный анализ (для С29Н30IN7О) Вычислено(%): С, 56,23; Н, 4,88; N, 15,83 Найдено (%): С, 56,13; Н, 4,94; N, 15,80 Пример 3 3-Хлор-4-(4-метилпиперазин-1-илметил)-N-(4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил)бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что 3-хлор-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид (сравнительный пример 3) использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида и что реакцию проводили при комнатной температуре в течение 24 часов. Бесцветное кристаллическое вещество Температура плавления: 193-194°С (с разложением) Элементный анализ (для С29Н30ClN7О·0,6Н2О) Вычислено(%): С, 64,64; Н, 5,84; N, 18,20 Найдено (%): С, 64,62; Н, 5,60; N, 18,23 Пример 4 3-Фтор-4-(4-метилпиперазин-1-илметил)-N-(4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил)бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что 3-фтор-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорид (сравнительный пример 4) использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида и что реакцию проводили при комнатной температуре в течение 22 часов, и неочищенный кристаллический продукт, полученный очисткой с помощью колоночной хроматографии на силикагеле, промывали хлороформом-диэтиловым эфиром (1:1). Бесцветное кристаллическое вещество Температура плавления: 197-199°С (с разложением) Элементный анализ (для С29Н30FN7О·0,3Н2О) Вычислено(%): С, 67,37; Н, 5,97; N, 18,96 Найдено (%): С, 67,36; Н, 5,96; N, 18,93 Пример 5 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-(4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил)бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что 4-(4-метилпиперазин-1-илметил)-3-трифторметилбензоилхлорида дигидрохлорид (сравнительный пример 5) использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида, и что реакцию проводили при комнатной температуре в течение 22 часов, и что получающееся кристаллическое вещество промывали диэтиловым эфиром. Бесцветное кристаллическое вещество Температура плавления: 182-183°С (с разложением) Элементный анализ (для С30Н30F3N7О·0,3Н2О) Вычислено(%): С, 63,55; Н, 5,44; N, 17,29 Найдено (%): С, 63,43; Н, 5,37; N, 17,29 Пример 6 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-(4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил)бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что 4-(4-метилпиперазин-1-илметил)-3-трифторметилбензоилхлорида дигидрохлорид (сравнительный пример 5), использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида и 4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]анилин (сравнительный пример 6) использовали вместо 4-метил-3-[4-(3-пиридинил)пиримидин-2-иламино]анилина и что реакцию проводили при комнатной температуре в течение 20 часов, и неочищенное кристаллическое вещество, полученное очисткой колоночной хроматографией на силикагеле, промывали диэтиловым эфиром. Бледно-желтое кристаллическое вещество Температура плавления: 231-233°С (с разложением) Элементный анализ (для С29Н29F3N8О·0,2Н2О) Вычислено(%): С, 61,52; Н, 5,23; N, 19,79 Найдено (%): С, 61,37; Н, 5,24; N, 19,81 Пример 7 3-Бром-4-(4-метилпиперазин-1-илметил)-N-(4-метил-3-[4-(2-пиразинил)пиримидин-2-иламино]фенил)бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что 4-метил-3-[4-(2-пиразинил)пиримидин-2-иламино]анилин (сравнительный пример 7) использовали вместо 4-метил-3-[4-(3-пиридинил)пиримидин-2-иламино]анилина и что реакцию проводили при комнатной температуре в течение 18 часов. Бледно-желтое кристаллическое вещество Температура плавления: 213-214°С (с разложением) Элементный анализ (для С28Н29BrN8О) Вычислено(%): С, 58,64; Н, 5,10; N, 19,54 Найдено (%): С, 58,41; Н, 5,11; N, 19,24 Пример 8 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид 629 мг 3-[4-(6-Хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина (сравнительный пример 8) суспендировали в 7 мл ацетонитрила и затем по очереди добавляли 24 мг 4-диметиламинопиридина и 1,15 мл N,N-диизопропил-N-этиламина. При охлаждении льдом добавляли пятью порциями 979 мг 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида дигидрохлорида (сравнительный пример 1) с последующим перемешиванием при комнатной температуре в течение одного часа после удаления ледяной бани. Реакционный раствор смешивали с водой, экстрагировали хлороформом и затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле. Полученное неочищенное кристаллическое вещество по очереди промывали этилацетатом и диэтиловым эфиром и кристаллическое вещество собирали фильтрованием с получением 939 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 219-222°С (с разложением) Элементный анализ (для С29Н29BrClN7О) Вычислено(%): С, 57,39; Н, 4,82; N, 16,15 Найдено (%): С, 57,07; Н, 4,75; N, 16,09 Пример 9 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метиланилин (сравнительный пример 9) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина, и неочищенное кристаллическое вещество, полученное очисткой колоночной хроматографией на силикагеле, перекристаллизовывали из этилацетата-диэтилового эфира. Бледно-желтое кристаллическое вещество Температура плавления: 194-195°С (с разложением) Элементный анализ (для С29Н29Br2N7О·0,3Н2О) Вычислено(%): С, 53,03; Н, 4,54; N, 14,93 Найдено (%): С, 53,07; Н, 4,53; N, 14,70 Пример 10 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метиланилин (сравнительный пример 9) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина, и дигидрохлорид 4-(4-метилпиперазин-1-илметил)-3-трифторметилбензоилхлорида (сравнительный пример 5) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением диизопропилового эфира-этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 171-173°С (с разложением) Элементный анализ (для С30Н29BrF3N7О·0,7Н2О) Вычислено(%): С, 55,17; Н, 4,69; N, 15,01 Найдено (%): С, 55,16; Н, 4,57; N, 14,94 Пример 11 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(1,2-дигидропиридазин-4-ил)пиримидин-2-иламино]-4-метилфенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 3-[4-(1,2-дигидропиридазин-4-ил)пиримидин-2-иламино]-4-метиланилин (сравнительный пример 10) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что экстрагирование проводили этилацетатом, и остаток, полученный очисткой с помощью колоночной хроматографии на силикагеле, промывали диизопропиловым эфиром. Бледно-желтое аморфное вещество Элементный анализ (для С28Н31BrN8О·0,8{(СН3)2СН}2О) Вычислено(%): С, 59,94; Н, 6,47; N, 17,05 Найдено (%): С, 59,51; Н, 6,30; N, 16,80 Пример 12 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридазинил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридазинил)пиримидин-2-иламино]анилин (сравнительный пример 11) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что неочищенное кристаллическое вещество, полученное путем очистки колоночной хроматографией на силикагеле, промывали этилацетатом-хлороформом. Бледно-желтое кристаллическое вещество Температура плавления: 185-187°С (с разложением) Элементный анализ (для С28Н29BrN8О·0,1Н2О) Вычислено(%): С, 58,28; Н, 5,13; N, 19,42 Найдено (%): С, 58,24; Н, 5,00; N, 19,48 Пример 13 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид 150 мг 4-Метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]анилин (сравнительный пример 6) растворяли в 4 мл N,N-диметилформамида, а затем по очереди добавляли 255 мг дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензойной кислоты (сравнительный пример 1) и 109 мг триэтиламина. При перемешивании суспензии при комнатной температуре по очереди добавляли 106 мг диэтилцианофосфоната и 55 мг триэтиламина с последующим перемешиванием при комнатной температуре в течение 3 часов. После того как растворитель отгоняли при пониженном давлении, остаток смешивали с водой и насыщенным водным раствором гидрокарбоната натрия и затем экстрагировали хлороформом. Экстракт промывали водой и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 240 мг неочищенного продукта. Неочищенный продукт растворяли в хлороформе-метаноле и добавляли 2-пропанол с последующим концентрированием при пониженном давлении. Выпавшее в осадок кристаллическое вещество собирали фильтрованием и промывали по очереди 2-пропанолом и диэтиловым эфиром с получением 147 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 238-240°С (с разложением) Элементный анализ (для С28Н29BrN8О·0,1Н2О) Вычислено(%): С, 58,46; Н, 5,12; N, 19,48 Найдено (%): С, 58,21; Н, 5,02; N, 19,30 Пример 14 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиридин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиридин-2-иламино]анилин (сравнительный пример 12) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что неочищенное кристаллическое вещество, полученное путем очистки колоночной хроматографией на силикагеле, промывали этилацетатом-хлороформом-метанолом. Бледно-желтое кристаллическое вещество Температура плавления: 244-245°С (с разложением) Элементный анализ (для С30Н31BrN6О·0,6Н2О) Вычислено(%): С, 61,88; Н, 5,57; N, 14,43 Найдено (%): С, 61,71; Н, 5,49; N, 14,13 Пример 15 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиридин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиримидинил)пиридин-2-иламино]анилин (сравнительный пример 13) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что неочищенное кристаллическое вещество, полученное путем очистки колоночной хроматографией на силикагеле, промывали этилацетатом-диэтиловым эфиром. Бледно-желтое кристаллическое вещество Температура плавления: 244-246°С Элементный анализ (для С29Н30BrN7О·0,2Н2О·0,2СН3СООС2Н2) Вычислено(%): С, 60,28; Н, 5,43; N, 16,51 Найдено (%): С, 60,12; Н, 5,40; N, 16,28 Пример 16 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[2-(3-пиридил)пиридин-6-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[2-(3-пиридил)пиридин-6-иламино]анилин (сравнительный пример 14) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что неочищенное кристаллическое вещество, очищенное колоночной хроматографией на силикагеле, дополнительно не промывали. Бледно-желтое аморфное вещество Элементный анализ (для С30Н31BrN6О·1,5Н2О) Вычислено(%): С, 60,20; Н, 5,73; N, 14,04 Найдено (%): С, 60,39; Н, 5,55; N, 13,00 FAB-МС(Поз.) m/z 571, (Нег.) m/z 569 Пример 17 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[3-(3-пиридил)пиридин-5-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[3-(3-пиридил)пиридин-5-иламино]анилин (сравнительный пример 15) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что экстрагирование проводили этилацетатом и маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата-диэтилового эфира. Бледно-желтое кристаллическое вещество Температура плавления: 139-141°С Элементный анализ (для С30Н31BrN6О·1,2Н2О) Вычислено(%): С, 60,75; Н, 5,68; N, 14,17 Найдено (%): С, 60,96; Н, 5,62; N, 13,98 Пример 18 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[3-(3-пиридил)фениламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[3-(3-пиридил)пиридин-5-иламино]анилин (сравнительный пример 16) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что экстрагирование проводили этилацетатом, и маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата-гексана. Бледно-коричневое кристаллическое вещество Температура плавления: 174-175°С Элементный анализ (для С31Н32BrN5О) Вычислено(%): С, 65,26; Н, 5,65; N, 12,28 Найдено (%): С, 65,12; Н, 5,73; N, 12,19 Пример 19 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[2-(3-пиридил)пиразин-6-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[2-(3-пиридил)пиразин-6-иламино]анилин (сравнительный пример 17) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что экстрагирование проводили этилацетатом, и аморфное вещество, очищенное колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Желтое кристаллическое вещество Температура плавления: 192-193°С Элементный анализ (для С29Н30BrN7О·0,25Н2О) Вычислено(%): С, 60,37; Н, 5,33; N, 16,99 Найдено (%): С, 60,58; Н, 5,35; N, 16,76 Пример 20 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[5-(3-пиридил)-1,2,4-триазин-3-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[5-(3-пиридил)-1,2,4-триазин-3-иламино]анилин (сравнительный пример 18) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и что аморфное вещество, очищенное колоночной хроматографией на силикагеле, кристаллизовали добавлением 2-пропанола. Желтое кристаллическое вещество Температура плавления: 219-221°С Элементный анализ (для С28Н29BrN8О·1,2Н2О·0,1СН3СН(ОН)СН3) Вычислено(%): С, 56,55; Н, 5,40; N, 18,64 Найдено (%): С, 56,58; Н, 5,00; N, 18,27 Пример 21 3-Метил-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-метил-4-(4-метилпиперазин-1-илметил)бензоилхлорида (сравнительный пример 19) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 192-193°С Элементный анализ (для С30Н33N7О) Вычислено(%): С, 70,98; Н, 6,55; N, 19,31 Найдено (%): С, 70,79; Н, 6,67; N, 19,39 Пример 22 4-(4-Метилпиперазин-1-илметил)-3-нитро-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 4-(4-метилпиперазин-1-илметил)-3-нитробензоилхлорида (сравнительный пример 20) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что экстрагирование проводили этилацетатом, и маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата-диэтилового эфира. Бледно-желтое кристаллическое вещество Температура плавления: 184-186°С Элементный анализ (для С29Н30N8О3·0,7Н2О) Вычислено(%): С, 63,19; Н, 5,74; N, 20,33 Найдено (%): С, 63,38; Н, 5,57; N, 20,00 Пример 23 3-Метокси-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-метокси-4-(4-метилпиперазин-1-илметил)бензоилхлорида сравнительный пример 21) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата и затем промывали диэтиловым эфиром. Бледно-желтое кристаллическое вещество Температура плавления: 171-172°С (с разложением) Элементный анализ (для С30Н33N7О2·0,6Н2О) Вычислено(%): С, 67,42; Н, 6,45; N, 18,35 Найдено (%): С, 67,23; Н, 6,36; N, 18,19 Пример 24 3,5-Дибром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3,5-дибром-4-(4-метилпиперазин-1-илметил)бензоилхлорида (сравнительный пример 22) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата и затем промывали диэтиловым эфиром. Бледно-желтое кристаллическое вещество Температура плавления: 227-229°С Элементный анализ (для С29Н29Br2N7О·0,1Н2О·0,35СН3СО2С2Н5) Вычислено(%): С, 53,38; Н, 4,72; N, 14,33 Найдено (%): С, 53,02; Н, 4,74; N, 14,09 Пример 25 3,5-Диметокси-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3,5-диметокси-4-(4-метилпиперазин-1-илметил)-3-бензоилхлорида (сравнительный пример 23) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что масляный продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Желтое кристаллическое вещество Температура плавления: 201-214°С (с разложением) Элементный анализ (для С31Н35N7О3·0,5Н2О) Вычислено(%): С, 66,17; Н, 6,45; N, 17,43 Найдено (%): С, 65,91; Н, 6,42; N, 17,42 Пример 26 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 1, за исключением того, что дигидрохлорид 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)бензоилхлорида (сравнительный пример 24) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что масляный продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Оранжевое кристаллическое вещество Температура плавления: 210-214°С (с разложением) Элементный анализ (для С32Н36N8О2·0,6Н2О) Вычислено(%): С, 66,79; Н, 6,52; N, 19,47 Найдено (%): С, 66,41; Н, 6,17; N, 19,36 Пример 27 3-Бром-4-(4-этилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-(4-этилпиперазин-1-илметил)бензоилхлорида (сравнительный пример 25) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 202-203°С Элементный анализ (для С30Н32BrN7О·0,25Н2О) Вычислено(%): С, 60,97; Н, 5,54; N, 16,59 Найдено (%): С, 60,96; Н, 5,54; N, 16,32 Пример 28 3-Бром-4-[4-(н-пропил)пиперазин-1-илметил]-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-[4-(н-пропил)пиперазин-1-илметил]бензоилхлорида (сравнительный пример 26) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 204-205°С Элементный анализ (для С31Н34BrN7О·0,4Н2О) Вычислено(%): С, 61,26; Н, 5,77; N, 16,13 Найдено (%): С, 61,48; Н, 5,66; N, 15,79 Пример 29 3-Бром-4-(N,N-диметиламинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-(N,N-диметиламинометил)бензоилхлорида (сравнительный пример 27) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бесцветное кристаллическое вещество Температура плавления: 154-155°С Элементный анализ (для С26Н25BrN6О) Вычислено(%): С, 60,35; Н, 4,87; N, 16,24 Найдено (%): С, 60,20; Н, 4,97; N, 16,13 Пример 30 3-Бром-4-(N,N-диэтиламинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-(N,N-диэтиламинометил)бензоилхлорида (сравнительный пример 28) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 172-173°С Элементный анализ (для С28Н29BrN6О) Вычислено(%): С, 61,65; Н, 5,36; N, 15,41 Найдено (%): С, 61,35; Н, 5,36; N, 15,35 Пример 31 3-Бром-4-(1-пирролидинилметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-(1-пирролидинилметил)бензоилхлорида (сравнительный пример 29) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 195-196°С Элементный анализ (для С28Н27BrN6О) Вычислено(%): С, 61,88; Н, 5,01; N, 15,46 Найдено (%): С, 61,68; Н, 5,12; N, 15,11 Пример 32 3-Бром-4-(пиперидинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-(пиперидинометил)бензоилхлорида (сравнительный пример 30) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 158-159°С Элементный анализ (для С29Н29BrN6О) Вычислено(%): С, 62,48; Н, 5,24; N, 15,07 Найдено (%): С, 62,23; Н, 5,25; N, 14,83 Пример 33 3-Бром-4-(морфолинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 8, за исключением того, что 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилин (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) использовали вместо 3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метиланилина и дигидрохлорид 3-бром-4-(морфолинометил)бензоилхлорида (сравнительный пример 31) использовали вместо дигидрохлорида 3-бром-4-(4-метилпиперазин-1-илметил)бензоилхлорида и что маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этилацетата. Бледно-желтое кристаллическое вещество Температура плавления: 179-180°С Элементный анализ (для С28Н29BrN6О2) Вычислено(%): С, 60,11; Н, 4,86; N, 15,02 Найдено (%): С, 59,94; Н, 4,93; N, 14,96 Пример 34 3-Бром-4-(цис-3,5-диметилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали модифицированным методом, описанным в документе (Synthesis, 1982, 288-291). К 356 мг 4-метил-3-[4-(3-пиридил)пиридин-2-иламино]анилина (Japanese Unexamined Patent Publication (Kokai) No. 6-87834) и 770 мг дигидрохлорида 3-бром-4-(цис-3,5-диметилпиперазин-1-илметил)бензойной кислоты (сравнительный пример 32) по очереди добавляли 7 мл дихлорметана и 715 мкл триэтиламина. При перемешивании при комнатной температуре добавляли 446 мг фенил N-фенилфосфорамидохлоридата (Synthesis, 1982, 288-291) с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор смешивали с водой и затем дважды экстрагировали хлороформом. Экстракт промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали по очереди колоночной хроматографией на силикагеле и колоночной хроматографией на NH-силикагеле и полученный маслянистый продукт кристаллизовали из этилацетата. Кристаллическое вещество промывали диэтиловым эфиром с получением 259 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 204-205°С Элементный анализ (для С30Н32BrN7О) Вычислено(%): С, 61,43; Н, 5,50; N, 16,72 Найдено (%): С, 61,19; Н, 5,48; N, 16,49 Пример 35 3-Бром-4-(4-метилгексагидро-1Н-1,4-диазепин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в примере 34, за исключением того, что дигидрохлорид 3-бром-4-(4-метилгексагидро-1Н-1,4-диазепин-1-илметил)бензойной кислоты (сравнительный пример 33) использовали вместо дигидрохлорида 3-бром-4-(цис-3,5-диметилпиперазин-1-илметил)бензойной кислоты. Бледно-желтое кристаллическое вещество Температура плавления: 156-157°С Элементный анализ (для С30Н32BrN7О) Вычислено(%): С, 61,43; Н, 5,50; N, 16,72 Найдено (%): С, 61,13; Н, 5,43; N, 16,39 Пример 36 3-Бром-4-(1-пиперазинилметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид Сначала 3-бром-4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]-N-(4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил)бензамид получали по модификации метода, описанного в документе (Synthesis, 1982, 288-291). Таким же образом, что и в сравнительном примере 34, за исключением того, что 4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]анилин (сравнительный пример 6) использовали вместо 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилина и 3-бром-4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]бензойную кислоту (сравнительный пример 34) использовали вместо дигидрохлорида 3-бром-4-(цис-3,5-диметилпиперазин-1-илметил)бензойной кислоты и что экстрагирование проводили этилацетатом и экстракт очищали только колоночной хроматографией на силикагеле, а затем использовали для последующей реакции без дополнительной очистки. Затем получали 3-бром-4-(1-пиперазинилметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид. К 187 мг неочищенного продукта реакции, представленного выше, добавляли 1,5 мл трифторуксусной кислоты с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор подщелачивали добавлением 10% водного раствора гидроксида натрия и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH-силикагеле и кристаллизовали добавлением этилацетата. Кристаллическое вещество промывали теплым этилацетатом с получением 49 мг целевого соединения в виде бледно-желтого кристаллического вещества. Бледно-желтое кристаллическое вещество Температура плавления: 225-228°С (с разложением) Элементный анализ (для С27Н27BrN8О·0,3Н2О) Вычислено(%): С, 57,41; Н, 4,92; N, 19,84 Найдено (%): С, 57,53; Н, 5,11; N, 18,92 FAB-МС(Поз.) m/z 559 Пример 37 4-[4-(Трет-бутоксикарбонил)пиперазин-1-илметил]-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид Это соединение получали таким же образом, что и в сравнительном примере 34, за исключением того, что 4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]анилин (сравнительный пример 6) использовали вместо 4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]анилина и 4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]-3-трифторметилбензойную кислоту (сравнительный пример 35) использовали вместо дигидрохлорида 3-бром-4-(цис-3,5-диметилпиперазин-1-илметил)бензойной кислоты и что экстрагирование проводили этилацетатом, и маслянистый продукт, очищенный колоночной хроматографией на силикагеле, кристаллизовали добавлением этанолом, а затем промывали по очереди этанолом и диэтиловым эфиром. Бледно-желтое кристаллическое вещество Температура плавления: 188-191°С 1Н-ЯМР (CDCl3) Пример 38 4-(1-Пиперазинилметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид К 1,00 г 4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида (пример 37) добавляли 8 мл трифторуксусной кислоты с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор подщелачивали добавлением 10% водного раствора гидроксида натрия и добавляли дихлорметан. Выпавшее в осадок кристаллическое вещество собирали фильтрованием с получением 530 мг неочищенного кристаллического вещества. Фильтрат экстрагировали дважды дихлорметаном, сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении с получением 370 мг неочищенного кристаллического вещества. 900 мг обоих неочищенных кристаллических веществ объединяли, очищали колоночной хроматографией на NH-силикагеле, а затем кристаллизовали добавлением изопропанола. Кристаллическое вещество промывали по очереди этилацетатом и диэтиловым эфиром с получением 258 мг целевого соединения в виде бледно-желтого кристаллического вещества. Бледно-желтое кристаллическое вещество Температура плавления: 208-211°С Элементный анализ (для С28Н27F3N8О) Вычислено(%): С, 61,31; Н, 4,96; N, 20,43 Найдено (%): С, 61,03; Н, 5,01; N, 20,33 Пример 39 3-Метоксикарбонил-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид 100 мг 3-Йод-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамида (пример 2), 1 мг дихлорбис(трифенилфосфин)палладия (II) и 20 мг гидрокарбоната натрия суспендировали в 5 мл безводного метанола и суспензию нагревали с обратным холодильником на бане с температурой 80°С в течение 2 часов при барботаже реакционного раствора газообразным моноксидом углерода. После охлаждения на воздухе к реакционному раствору добавляли воду и этилацетат и нерастворимые вещества удаляли фильтрованием, а затем фильтрат экстрагировали этилацетатом. Экстракт промывали водой и сушили над безводным сульфатом магния, а затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле и кристаллизовали добавлением этилацетата-диэтилового эфира с получением 64 мг целевого соединения в виде бесцветного кристаллического вещества. Температура плавления: 159-161°С (с разложением) Элементный анализ (для С31Н33N7О3·0,2Н2О) Вычислено(%): С, 67,06; Н, 6,06; N, 17,66 Найдено (%): С, 66,77; Н, 6,03; N, 17,68 Пример 40 3-Циано-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид 200 мг 3-Йод-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамида (пример 2), 35 мг тетракис(трифенилфосфин)палладия (0) и 45 мг 60% цианида цинка суспендировали в 2 мл безводного N,N-диметилформамида с последующим перемешиванием при нагревании до 80°С в течение 24 часов. После сушки на воздухе реакционный раствор смешивали с насыщенным водным раствором гидрокарбоната натрия и дважды экстрагировали хлороформом. Экстракт сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле и кристаллизовали добавлением этанола с получением 34 мг целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 191-193°С (с разложением) Элементный анализ (для С30Н30N8О·0,5Н2О) Вычислено(%): С, 68,29; Н, 5,92; N, 21,24 Найдено (%): С, 68,05; Н, 5,99; N, 21,12 Пример 41 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида гидрохлорид 5,00 г 3-Бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида (пример 13) суспендировали в 250 мл 50% водного раствора этанола и добавляли 9,24 мл 1N соляной кислоты с последующим перемешиванием с нагреванием на горячей бане при 80°С с получением гомогенного раствора. Реакционный раствор фильтровали при нагревании и растворитель отгоняли из фильтрата при пониженном давлении. Остаток отгоняли при нагревании в 30 мл этанола и затем оставляли стоять при комнатной температуре в течение одного дня. Выпавшее в осадок кристаллическое вещество собирали фильтрованием и промывали этанолом с получением 5,13 г целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 184-186°С (с разложением) Элементный анализ (для С28Н29BrN8О·1,0НСl·2,0Н2О) Вычислено(%): С, 52,06; Н, 5,31; N, 17,35 Найдено (%): С, 51,72; Н, 5,17; N, 17,21 Пример 42 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида гидрохлорид Это соединение получали таким же образом, что и в примере 41, за исключением того, что 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-(4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил)бензамид (пример 6) использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида. Бледно-желтое кристаллическое вещество Температура плавления: 244-246°С (с разложением) Элементный анализ (для С29Н29F3N8О·1,0НСl·0,8Н2О) Вычислено(%): С, 56,78; Н, 5,19; N, 18,27 Найдено (%): С, 56,80; Н, 4,96; N, 18,49 Пример 43 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамида гидрохлорид Это соединение получали таким же образом, что и в примере 41, за исключением того, что 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил)бензамид (пример 10) использовали вместо 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида. Бледно-желтое кристаллическое вещество Температура плавления: 184-187°С Элементный анализ (для С30Н29BrF3N7О·1,0НСl·1,0Н2О) Вычислено(%): С, 51,85; Н, 4,64; N, 14,11 Найдено (%): С, 51,78; Н, 4,74; N, 13,92 Пример 44 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамида метансульфонат 7,00 г 4-(4-Метилпиперазин-1-илметил)-3-трифторметил-N-(4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил)бензамида (пример 6) суспендировали в 70 мл метанола и каплями добавляли раствор 1,20 г метансульфоновой кислоты в 3 мл метанола с последующим перемешиванием при нагревании на масляной бане при 50°С в течение 10 минут. Реакционный раствор смешивали с 700 мг активированного угля (Kyoryoku Shirasagi MOIWY433) и затем нагревали с обратным холодильником в течение 30 минут. Реакционный раствор фильтровали при нагревании и растворитель из фильтрата отгоняли при пониженном давлении. Остаток растворяли при нагревании в 10 мл метанола и затем оставляли стоять при комнатной температуре в течение 10 минут. В результате весь реакционный раствор отвердевал. Данный раствор кристаллизовали добавлением изопропанола с получением 7,20 г целевого соединения в виде бледно-желтого кристаллического вещества. Температура плавления: 171-173°С Элементный анализ (для С29Н29F3N8О·1,0СН3SO3Н·1,0Н2О) Вычислено(%): С, 53,25; Н, 5,21; N, 16,56 Найдено (%): С, 53,04; Н, 5,39; N, 16,74 Пример испытания 1 Ингибирующее рост клеток действие Клетки К562 и клетки U937 (куплены из American Type Culture Collection) культивировали в среде RPMI-1640 (производимой Sigma), содержащей 10% (об./об.) телячьей эмбриональной сыворотки (FCS; ТЭС) (производимой Sigma) (RPMI-1640/FCS). Клетки К562 и клетки U937 засевали при плотности 5000 клеток/100 мкл/ячейку, соответственно. Планшету инкубировали в термостате с повышенным содержанием СО2 в течение ночи. Испытуемое лекарственное вещество готовили с помощью диметилсульфоксида (ДМСО) (производимого Nacalai Tesque) с концентрацией в 1000 раз выше, чем концентрация для испытания (0, от 0,00001 до 1 мкМ). Полученный раствор разбавляли в 500 раз в среде RPMI-1640/FCS и затем добавляли 100 мкл разбавителя в ячейку. Планшету инкубировали в термостате с повышенным содержанием СО2. Через 72 часа в каждую ячейку добавляли 20 мкл жидкости Cell counting Kit-8 (5 ммоль/л WST-8, 0,2 ммоль/л 1-метокси-PMS, 150 ммоль/л NaCl) (производимой Dojindo). После реакции с развитием цвета в термостате с повышенным содержанием СО2 в течение 3 часов определяли поглощение формазана, производимого путем восстановления WST-8 при 450 нм, используя многоуровневый счетчик ARVOsx (производимый Wallac). Значение IC50 (ПК50) (в мкМ) рассчитывали по следующей формуле: Степень ингибирования роста клеток = 100 – (поглощение ячейками с лекарственным средством/поглощение ячейками с 0,1% ДМСО)·100 После логарифмически-логистического преобразования, которое давало 50%-ную степень ингибирования роста клеток, определяемую как IC50, высчитывали по методу наименьших квадратов. Результаты представлены в таблице 1. В качестве контрольного лекарственного средства использовали 4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид (см. патентный документ 1).
Как видно из результатов, представленных в таблице 1, соединения данного изобретения обладают превосходным ингибирующим действием в отношении BCR-ABL тирозинкиназы. Клетки К562, использованные в примере испытания 1, были положительными в отношении BCR-ABL клеток, которые были собраны из плеврального выпота пациента с хронической миелогенной лейкемией на поздней стадии, который подвергся острой трансформации. Клетки U937 были злокачественными отрицательными в отношении BCR-ABL клетками, которые были взяты от пациента с гистиоцитной лимфомой. Как видно по отношению ингибирования клеточного роста (клетки U937/клетки К562), соединения данного изобретения являются лекарственными средствами, обладающими более высокой безопасностью, чем контрольное лекарственное вещество. Кроме того, соединения данного изобретения обладают ингибирующим рост клеток действием в несколько сотен раз более сильным по сравнению с контрольным лекарственным средством, и поэтому можно ожидать, что они проявят соответствующее ингибирующее рост клеток действие не только на ранее известные мутантные киназы, но также на мутантные киназы, которые будут обнаружены в будущем. Соответственно соединения данного изобретения очень полезны в качестве терапевтического средства для лечения таких заболеваний, как хроническая миелогенная лейкемия, острая лимфобластная лейкемия и острая миелогенная лейкемия. Пример испытания 2 Ингибирующее действие на самофосфорилирование у мутантной (Е255К) BCR-ABL Клетки 293Т (приобретенные из АТСС) культивировали в модифицированной Дульбекко (Dulbecco) среде Игла (производимой Sigma), содержащей 10% ТЭС (DMEM/FCS). Клетки засевали в количестве 5 мл покрытые поли-L-лизином 6 см чашки, так чтобы получилось 1,2·106 клеток/ячейку. Чашку инкубировали в термостате с повышенным уровнем СО2 в течение ночи. Используя липофектаминовый реагент (производимый Invitrogen), 2 мкг Е255К вектор экспрессии мутантного bcr-abl гена трансфицировали в клетки. Через 16 часов после трансфицирования в каждую ячейку добавляли 5 мкл приготовленного в ДМСО (производимом Nacalai tesque) испытуемого лекарственного средства, таким образом получали концентрацию, в 1000 раз более высокую. Чашку инкубировали в термостате с повышенным уровнем СО2 в течение 2 часов. После обработки трипсином клетки собирали в 15 мл центрифужную пробирку. Пробирку центрифугировали при 1000 об/мин при комнатной температуре в течение одной минут. После удаления среды добавляли 50 мкл раствора для лизиса клеток. Клетки подвергали цитолизу с помощью миксера. Пробирку оставляли стоять при 4°С в течение 15 минут и переносили в 1,5 мл пробирку. Пробирку центрифугировали при 12000 об/мин при 4°С в течение 15 минут. Лизат клеток собирали в другую 1,5 мл пробирку. Концентрацию белка определяли по методу ВСА. Образцы лизата клеток, содержащие 5 мкл белка, анализировали с помощью электрофореза в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE). После элетрофореза белок переносили на найлоновый фильтр (Hybond-P) влажным методом при 4°С в течение ночи. Найлоновый фильтр подвергали воздействию 10 мл ФБФР(PBS)/0,1% твин-20, содержащих 0,2 мкг/мл антител против фосфорилирования тирозинкиназы (РY99) (производимый Toyobo) при комнатной температуре в течение одного часа. Найлоновый фильтр промывали три раза ФБФР, а затем подвергали воздействию 10 мл ФБФР/0,1% твин-20, содержащих 0,4 мкг/мл конъюгированных с АП против мышиного IgG (производимого Cell Signaling) при комнатной температуре в течение одного часа. После того как найлоновый фильтр отмывали ФБФР четыре раза, определяли самофосфорилирование р210 BCR-ABL с помощью дающего окрашивание реагента щелочной фосфатазы. Результаты представлены следующим образом: +++: почти полное ингибирование фосфорилирования; ++: около половины ингибирования; +: слабое ингибирование; и -: нет ингибирования. В качестве контрольного лекарственного средства использовали 4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид (см. патентный документ 1).
Как видно из результатов, представленных в таблице 2, соединения данного изобретения обладают эффектом ингибирования самофосфорилирования в отношении Е255К мутантной BCR-ABL тирозинкиназы. Таким образом, можно ингибировать рост клеток, вызванный мутантной киназой. В частности, контрольное лекарственное средство не оказывает ингибирующего действия, и поэтому очевидно, что это действие специфично для соединений данного изобретения. Кроме того, соединения данного изобретения обладают также сильным ингибирующим самофосфорилирование действием в отношении Е255К мутантной BCR-ABL тирозинкиназы, на которую невозможно оказать ингибирующее самофосфорилирование действие некоторыми контрольными лекарственными средствами, и поэтому можно ожидать, что они проявят адекватное ингибирующее самофосфорилирование действие в отношении мутантных киназ, которые будут обнаружены в будущем. Соответственно соединения данного изобретения очень полезны в качестве терапевтического средства для лечения таких заболеваний, как хроническая миелогенная лейкемия, острая лимфобластная лейкемия, острая миелогенная лейкемия.
Смешанный порошок данного состава прессуют обычным способом и формуют с получением пероральных таблеток.
Смешанный порошок данного состава прессуют обычным способом и формуют с получением пероральных таблеток. ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ Как описано выше, так как соединение данного изобретения является соединением, обладающим высокой ингибирующей BCR-ABL тирозинкиназу активностью, фармацевтическая композиция, содержащая соединение данного изобретения в качестве активного ингредиента, пригодна как ингибитор BCR-ABL тирозинкиназы. Терапевтическое средство для лечения хронической миелогенной лейкемии, терапевтическое средство для лечения острой миелогенной лейкемии и терапевтическое средство для лечения острой лимфобластной лейкемии у млекопитающих, включая людей.
Формула изобретения
1. Амидное производное, представленное следующей формулой [1] в любом из следующих случаев (А) и (В) или его соль
(A) R1 представляет 5-7 членную насыщенную циклическую группу, имеющую 1-2 атома азота в качестве атома, образующего цикл (насыщенная циклическая аминогруппа может быть замещена 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из C1-10алкила, С1-10алкоксикарбонила), моноC1-10алкиламино или диС1-10алкиламино; R2 представляет C1-10алкил, галоген, галогенС1-10алкил, C1-10алкокси, C1-10алкоксикарбонил, нитро, моноС1-10алкилкарбамоил, диС1-10алкилкарбамоил или циано; R3 представляет водород, галоген или С1-10алкокси, Het1 представляет любую из групп следующих формул [2]-[8]
Het2 представляет пиридил, пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил [Het2 может быть замещенным 1-3 одинаковыми или разными заместителями, выбранными из галогена), кроме соединения, в котором R1 является (i) пирролидинилом, пиперидинилом, пиперазинилом или морфолинилом, каждый из которых может быть замещен 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из алкила, алкоксикарбонила, галогена, галогеналкила, гидроксиалкила, амино, моноалкиламино, диалкиламино, карбамоила, моноалкилкарбамоила и диалкилкарбамоила, (ii) моноалкиламино или (iii) диалкиламино, Het1 является группой формулы [6], а Het2 является пиразинилом или пиридилом, который может быть замещенным алкилом; (В) R1 представляет 4-метилпиперазин-1-ил, 1-пирролидинил, пиперидино, 4-этилпиперазин-1-ил, 4-н-пропилпиперазин-1-ил, цис-3,5-диметилпиперазин-1-ил, морфолино, диметиламино или диэтиламино; R2 представляет метил, галоген, трифторметил, метокси, метоксикарбонил, нитро, диметилкарбамоил или циано; R3 представляет водород, бром или метокси; Het1 представляет группу формулы [6]; Het2 представляет 3-пиридил. 2. Амидное производное по п.1 или его соль, в котором R1 представляет 5-7 членную насыщенную циклическую группу, имеющую 1-2 атома азота в качестве атома, образующего цикл (насыщенная циклическая аминогруппа может быть замещена 1-3 одинаковыми или разными заместителями, выбранными из группы, состоящей из С1-10алкила, С1-10алкокосикарбонила), моноС1-10алкиламино или диС1-10алкиламино, R2 представляет C1-10алкил, галоген, галоген C1-10алкил, C1-10алкокси, C1-10алкоксикарбонил, нитро, диС1-10алкилкарбамоил или циано, R3 представляет водород, галоген или С1-10алкокси, Het1 является любой из групп формул [2]-[8], и Het2 представляет пиридил, пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил (Het2 может быть замещен 1-3 одинаковыми или разными атомами галогена). 3. Амидное производное по п.1, которое является соединением, выбранным из группы, состоящей из следующих ниже соединений (1)-(40), или его соль: (1) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (2) 3-йод-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (3) 3-хлор-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (4) 3-фтор-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (5) 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (6) 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил) пиримидин-2-иламино]фенил}бензамид, (7) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(2-пиразинил)пиримидин-2-иламино]фенил}бензамид, (8) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(6-хлорпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил} бензамид, (9) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил} бензамид, (10) 4-(4-метилпиперазин-1-илметил)-3-трифторметил-N-{3-[4-(5-бромпиридин-3-ил)пиримидин-2-иламино]-4-метилфенил}бензамид, (11) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{3-[4-(1,2-дигидропиридазин-4-ил)пиримидин-2-иламино]-4-метилфенил}бензамид, (12) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридазинил)пиримидин-2-иламино]фенил}бензамид, (13) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (14) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиридин-2-иламино]фенил}бензамид, (15) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(5-пиримидинил)пиридин-2-иламино]фенил}бензамид, (16) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[2-(3-пиридил)пиридин-6-иламино]фенил}бензамид, (17) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[3-(3-пиридил)пиридин-5-иламино]фенил}бензамид, (18) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[3-(3-пиридил)фениламино]фенил}бензамид, (19) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[2-(3-пиридил)пиразин-6-иламино]фенил}бензамид, (20) 3-бром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[5-(3-пиридил)-1,2,4-триазин-3-иламино]фенил}бензамид, (21) 3-метил-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (22) 4-(4-метилпиперазин-1-илметил)-3-нитро-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (23) 3-метокси-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (24) 3,5-дибром-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (25) 3,5-диметокси-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (26) 3-(N,N-диметилкарбамоил)-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (27) 3-бром-4-(4-этилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (28) 3-бром-4-[4-(н-пропил)пиперазин-1-илметил]-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (29) 3-бром-4-(N,N-диметиламинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (30) 3-бром-4-(N,N-диэтиламинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (31) 3-бром-4-(1-пирролидинилметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (32) 3-бром-4-(пиперидинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (33) 3-бром-4-(морфолинометил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (34) 3-бром-4-(цис-3,5-диметилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (35) 3-бром-4-(4-метилгексагидро-1Н-1,4-диазепин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид, (36) 3-бром-4-(1-пиперазинилметил)-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (37) 4-[4-(трет-бутоксикарбонил)пиперазин-1-илметил]-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (38) 4-(1-пиперазинилметил)-3-трифторметил-N-{4-метил-3-[4-(5-пиримидинил)пиримидин-2-иламино]фенил}бензамид, (39) 3-метоксикарбонил-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид и (40) 3-циано-4-(4-метилпиперазин-1-илметил)-N-{4-метил-3-[4-(3-пиридил)пиримидин-2-иламино]фенил}бензамид. 4. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении BCR-ABL тирозинкиназы, содержащая в качестве активного ингредиента амидное производное по любому одному из пп.1-3 или его соль и фармацевтически приемлемый нетоксичный и инертный носитель. 5. Ингибитор BCR-ABL тирозинкиназы, содержащий амидное производное по любому одному из пп.1-3 или его соль в качестве активного ингредиента и, необязательно, фармацевтически приемлемый нетоксичный и инертный носитель. 6. Терапевтическое средство для лечения хронической миелогенной лейкемии, содержащее амидное производное по любому одному из пп.1-3 или его соль в качестве активного ингредиента и, необязательно, фармацевтически приемлемый нетоксичный и инертный носитель. 7. Терапевтическое средство для лечения острой лимфобластной лейкемии, содержащее амидное производное по любому одному из пп.1-3 или его соль в качестве активного ингредиента и, необязательно, фармацевтически приемлемый нетоксичный и инертный носитель. 8. Терапевтическое средство для лечения острой миелогенной лейкемии, содержащее амидное производное по любому одному из пп.1-3 или его соль в качестве активного ингредиента и необязательно, фармацевтически приемлемый нетоксичный и инертный носитель. Приоритет по признакам: Признаки, касающиеся R1, R2, R3 и Het1 раскрыты в приоритетных заявках №№2002-189269 от 28.06.2002, 2002-305146 от 18.10.2002 и 2002-377937 от 26.12.2002, кроме следующих: для случая А где признак «R2 представляет нитрогруппу» раскрыт только в заявках №№2002-305146 от 18.10.2002 и 2002-377937 от 26.12.2002; и далее для случая А R1 представляет насыщенную циклическую аминогруппу, замещенную C1-10алкоксикарбонилом; R3 представляет галоген или С1-10алкоксигруппу, Het1 представляет формулы 2-5, 7 и 8; для случая В R1 представляет 1-пирролидинил, пиперидино, 4-этилпиперазин-1-ил, 4-н-пропилпиперазин-1-ил, цис-3,5-диметилпиперазин-1-ил, морфолино, диметиламино или диэтиламино; R2 представляет метил, метокси, метоксикарбонил, нитро, диметилкарбамоил или циано; R3 представляет водород, бром или метокси; дата приоритета соответствует дате подачи заявки в Российское патентное ведомство. Признак Het2 (для случая А), представляющий пиридил, пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил, значения «3-пиридил и 5-пиримидинил, пиразинил, пиридазинил или 1,2-дигидропиридазинил и Het2 может быть замещенным 1-3 одинаковыми или разными заместителями, выбранными из галогена» раскрыт во всех приоритетных заявках. Признак «4-пиримидинил, который может быть замещенным 1-3 одинаковыми или разными заместителями, выбранными из галогена» раскрыт в заявках №№2002-305146 от 18.10.2002 и 2002-377937 от 26.12.2002.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
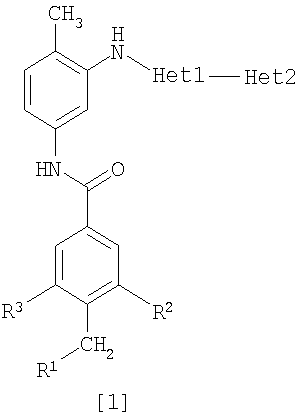

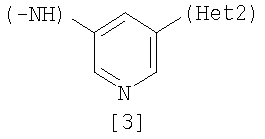
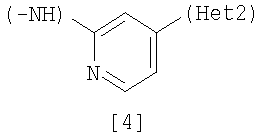
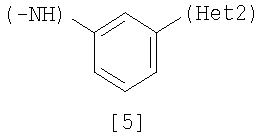
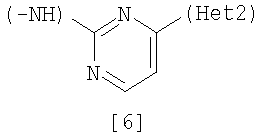
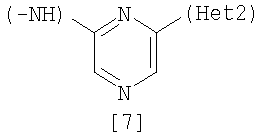
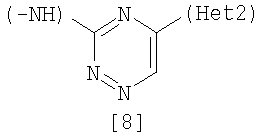
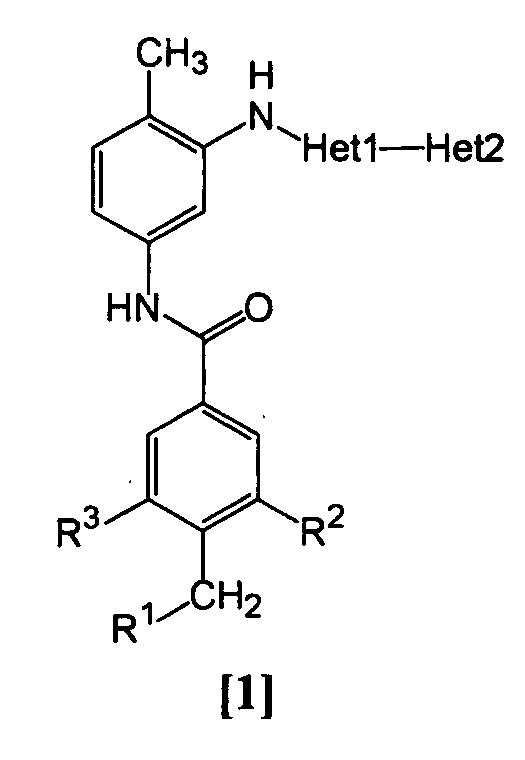
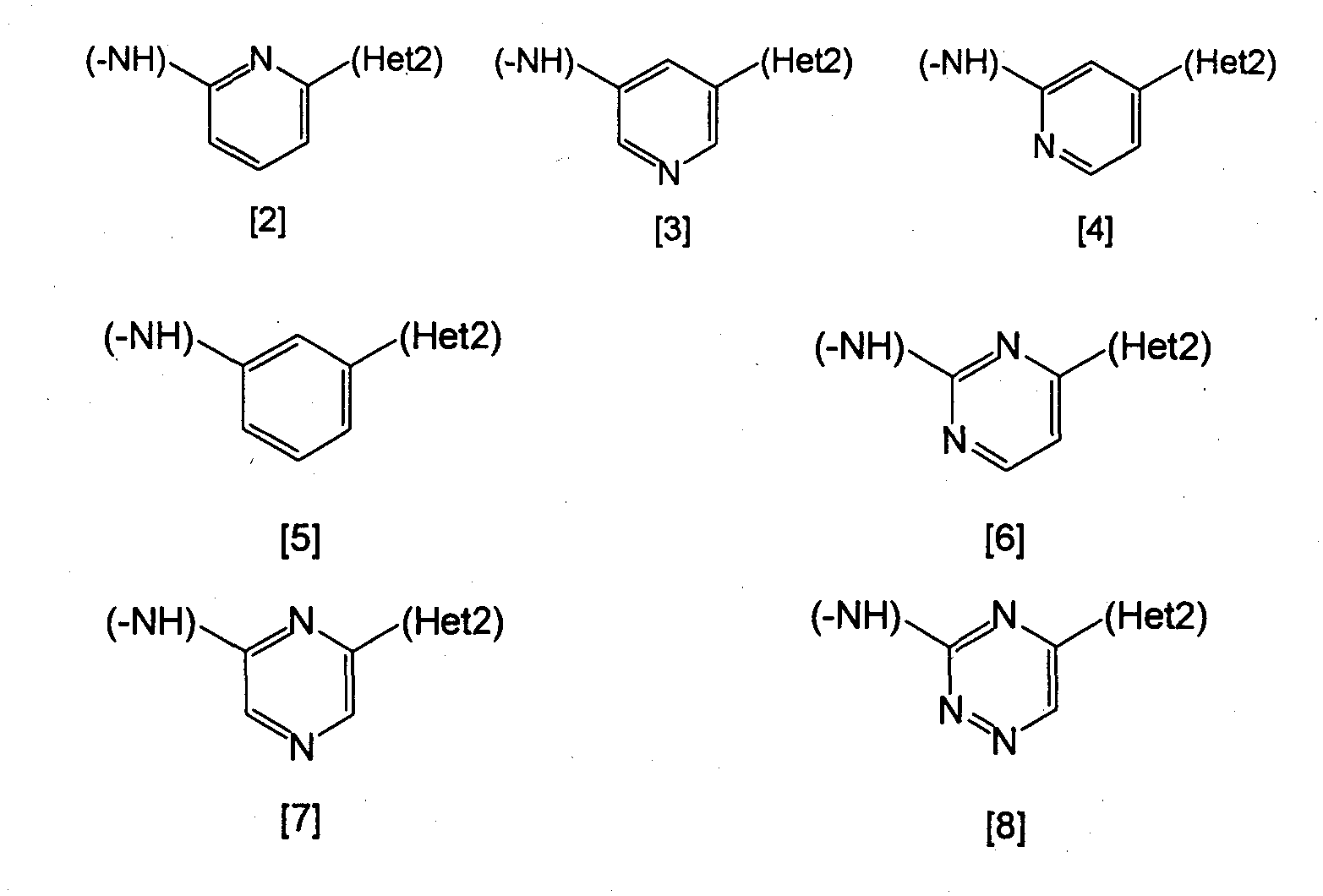
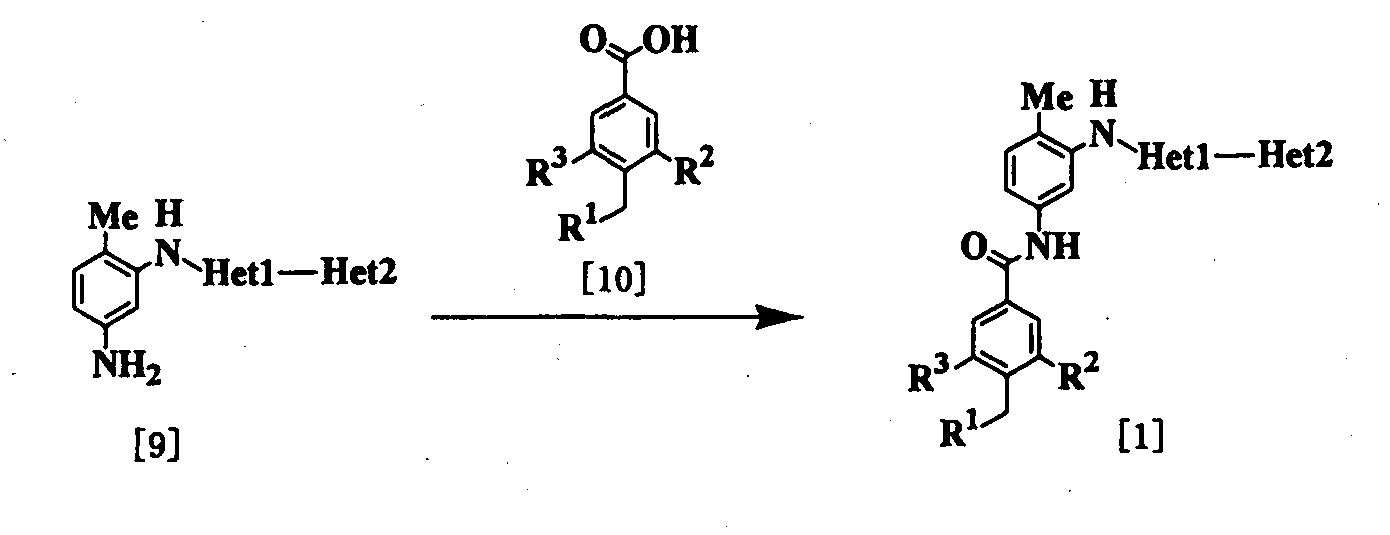
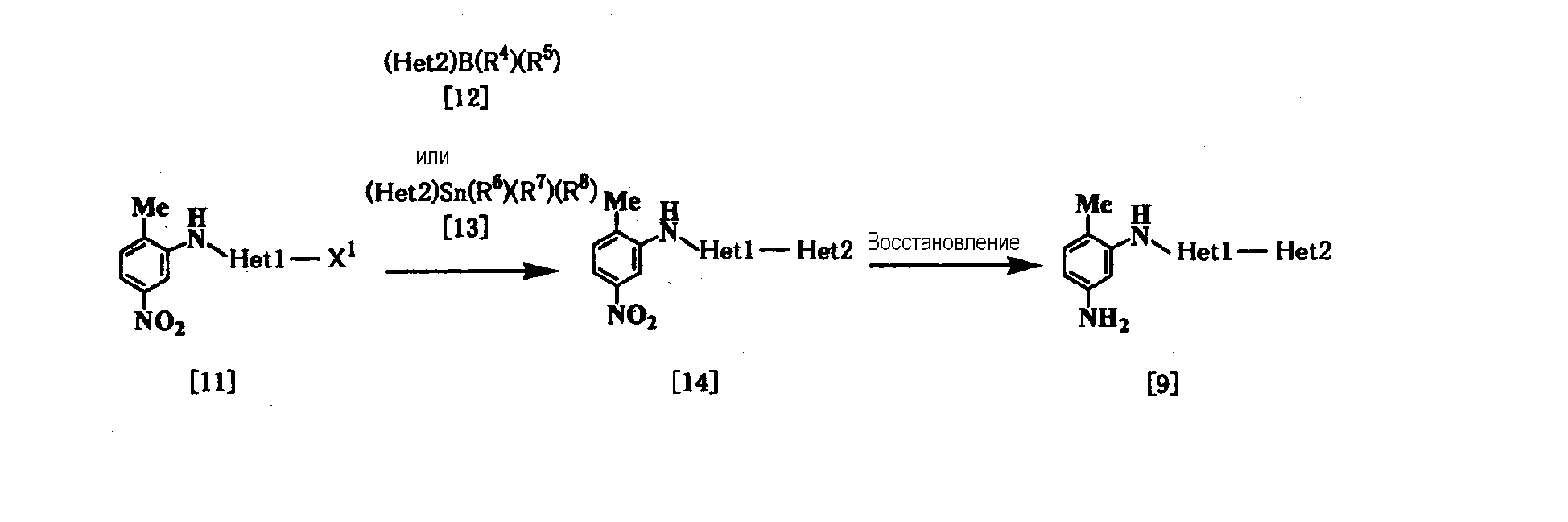
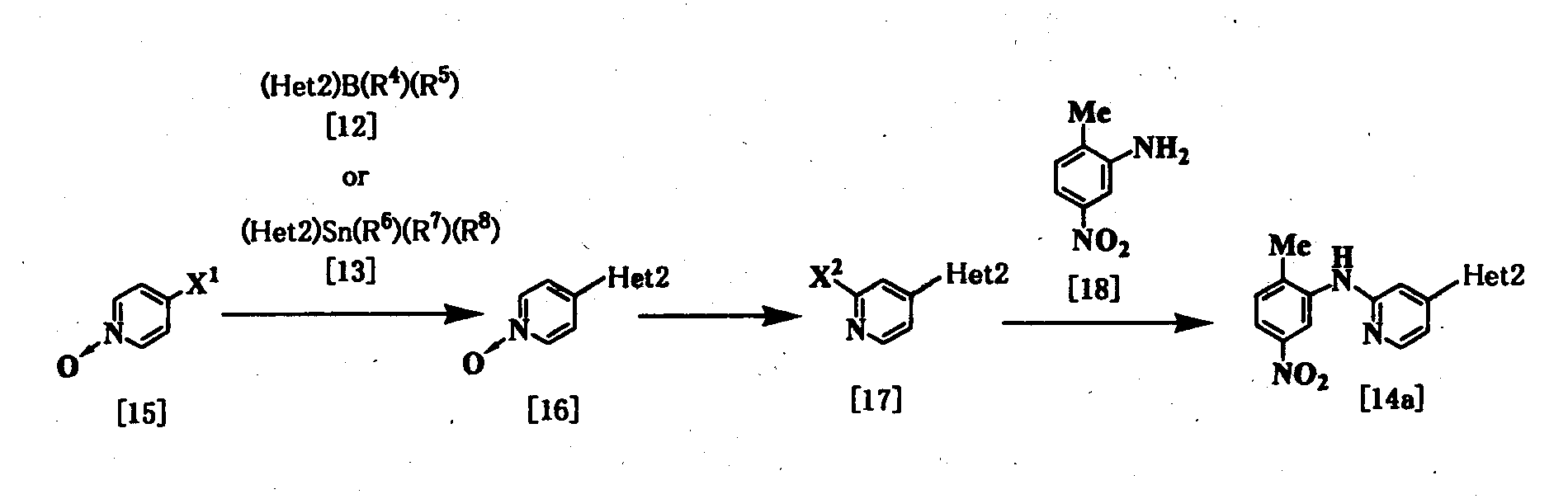
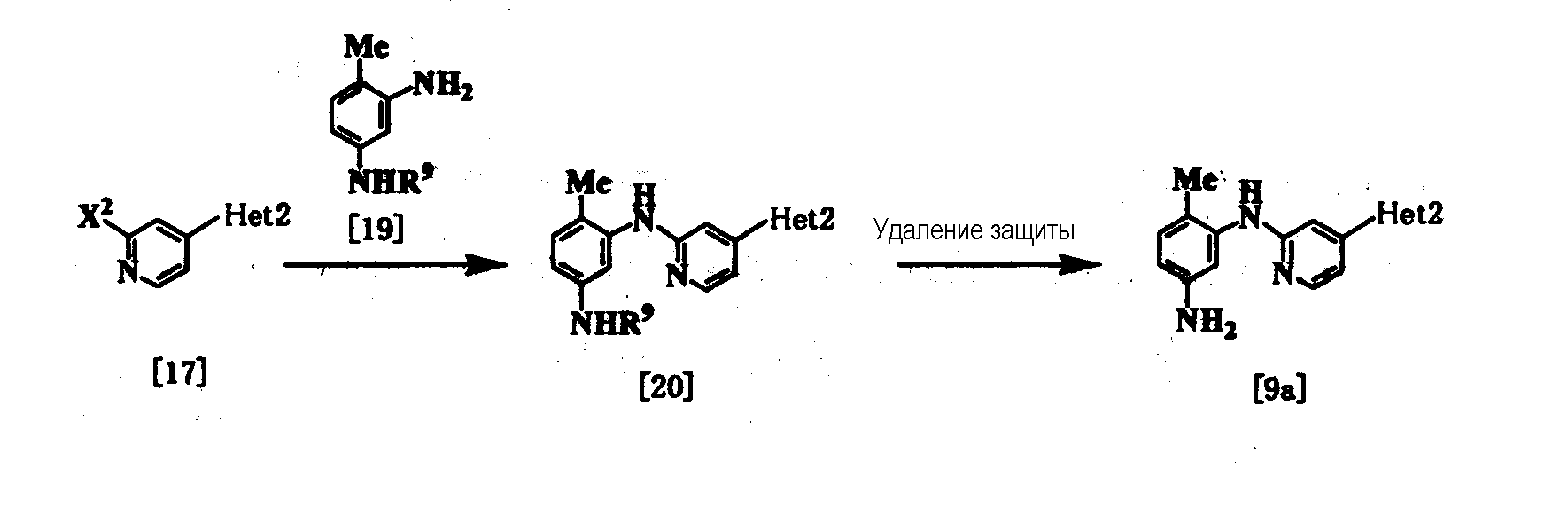
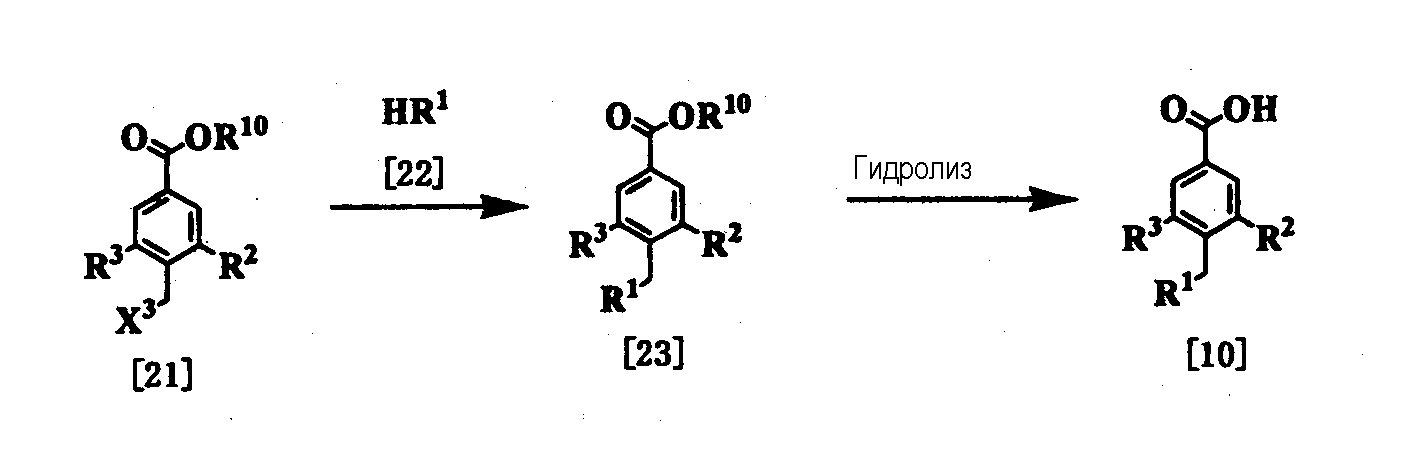
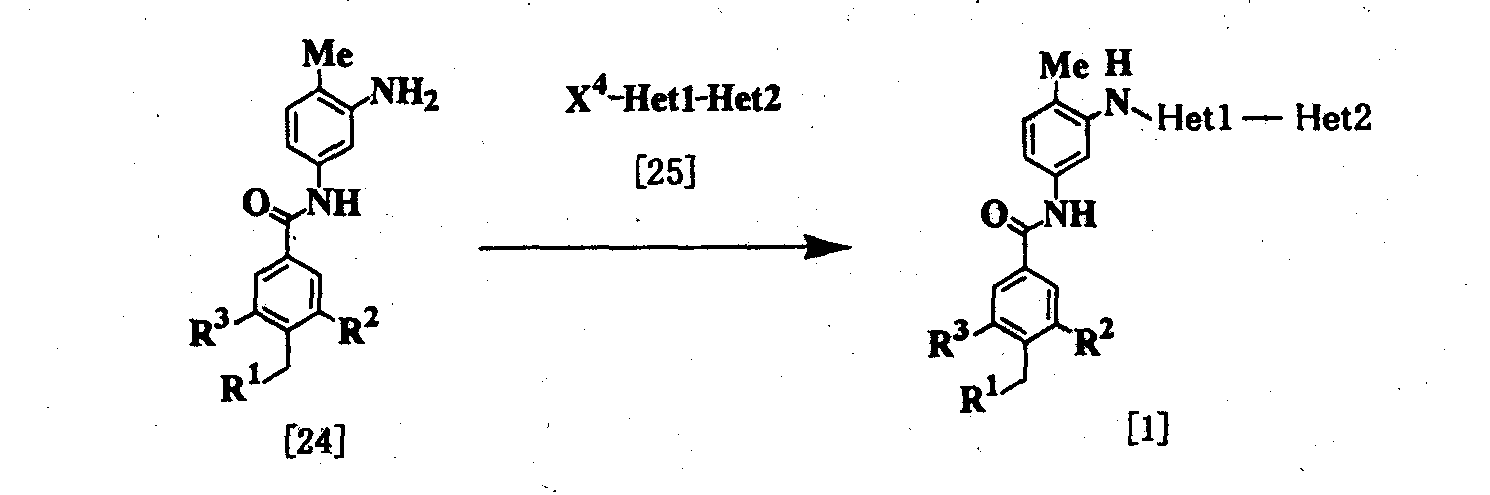
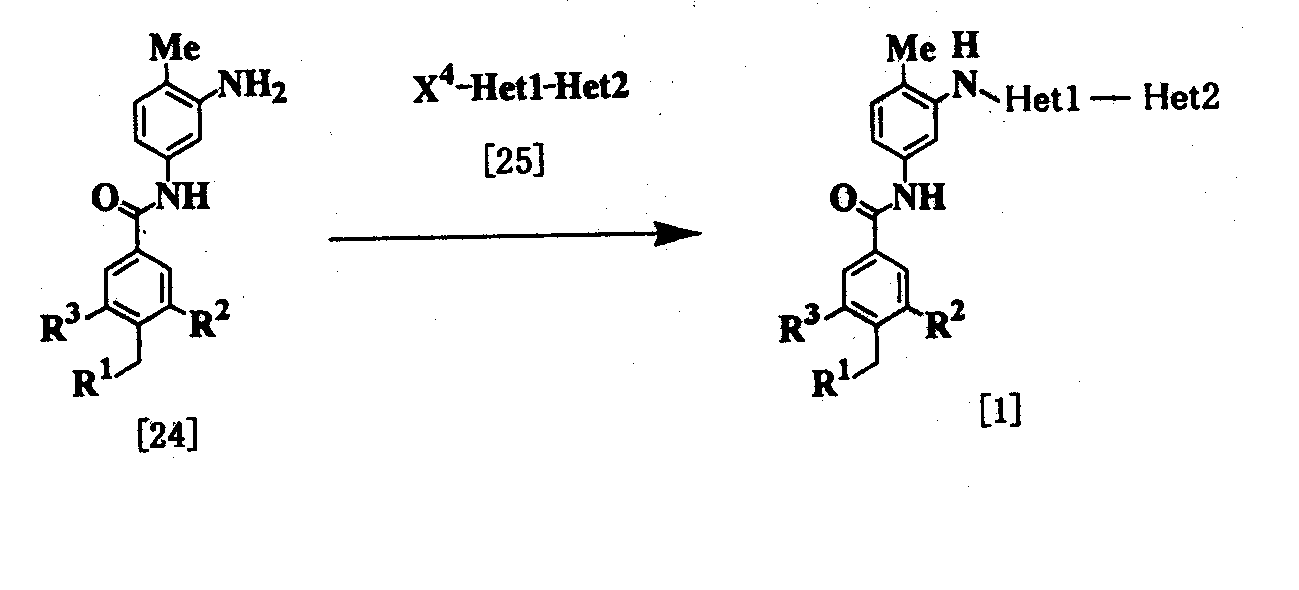
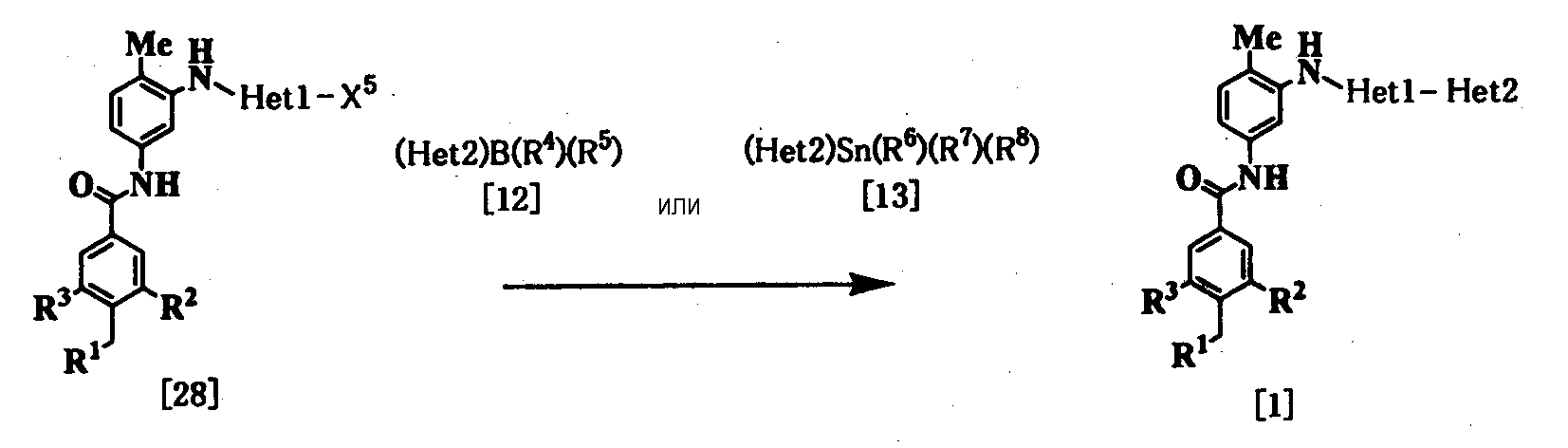
 : 1,39 (3H, т), 2,45 (3H, с), 4,37 (2H, кв.), 7,29 (1H, дд), 7,87 (1H, дд), 8,20 (1H, д).
: 1,39 (3H, т), 2,45 (3H, с), 4,37 (2H, кв.), 7,29 (1H, дд), 7,87 (1H, дд), 8,20 (1H, д). : 3,17 (3H, с), 7,58 (1H, дд), 8,67 (1H, дд), 8,97 (1H, дд), 9,47 (1H, д), 9,85 (1H, с).
: 3,17 (3H, с), 7,58 (1H, дд), 8,67 (1H, дд), 8,97 (1H, дд), 9,47 (1H, д), 9,85 (1H, с).