Патент на изобретение №2159245
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ
(57) Реферат: Описываются новые производные аза-антрациклинона общей формулы 1, где X1 и X2 означают C = O; X3 выбран из C = O, CHOH или C = N(R9), где R9 представляет собой гидрокси; R1, R2 и R3 означают водород; R4 означает водород или C1-6 алкоксил; R5 и R8 означают гидроксил; R6 означает водород или RB – CH2-, где RB означает фенил, незамещенный или замещенный C1-6 алкоксилом, пиридинил или группу формулы RC – CH = CH-, где RC означает водород; R7 означает метил, или их фармацевтически пригодные соли. Соединения применимы для лечения амилоидоза. Описываются содержащие их фармацевтические композиции. 2 с. и 3 з.п. ф-лы, 3 табл. 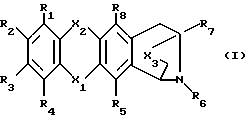 
Настоящее изобретение относится к новым производным аза-антрациклинона, их применению для лечения амилоидоза, способам их получения и содержащим их фармацевтическим композициям. Взаимосвязь между амилоидозом, клеточной смертью и утратой тканевых функций, по-видимому, имеет место в случае различных типов нарушений, включая нейродегенеративные. В этой связи предотвращение образования амилоида и/или индукция его дегенерации может иметь важное терапевтическое значение для всех патологических нарушений, связанных с амилоидозом, включая AL амилоидоз и нейродегенеративные заболевания типа болезни Альцгеймера. Настоящее изобретение предлагает новые аза-антрациклиноны и их применение для лечения амилоидоза. Новые соединения характеризуются присутствием гетероциклического кольца с внутренним мостиком, присоединенного к антрахиноновой системе. Новый класс молекул назван антразалиноном и родительское соединение, обозначенное как антразалон, может рассматриваться как относящееся к 8-аза-антрациклинонам 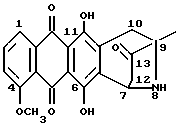 Антразалон Более конкретно настоящее изобретение предлагает производное антразалинона согласно формуле 1 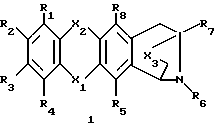 где X1 и X2 выбраны независимо из: C=O, C=NH и CH2; X3 выбран из CH2, C=O, CHOH  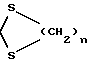 где n = 2 или 3, и C=N(R9), где R9 представляет собой гидрокси- или аминоарильную группу; R1, R2, R3 и R4 выбраны независимо из: водорода, гидроксила, C1-16 алкила, C1-16 алкоксила, C3-8 циклоалкоксила, галогена, аминогруппы, которая может оставаться незамещенной или быть моно- или дизамещенной ацильной, трифторацильной, аралкильной или арильной группой и OSO2(R10), где R10 представляет собой алкил или арил; R5 и R8 выбраны независимо из: водорода, гидроксила, C1-16 алкоксила, галогена, аминогруппа, которая может оставаться незамещенной или быть моно- или дизамещенной ацильной, трифторацильной, аракильной или арильной группой и OSO2(R10), где R10 имеет значения, указанные выше; R6 выбран из: водорода, RB-CH2-, где RB представляет собой арильную или гетероциклильную группу, или группу с формулой RC-CH=CH-, где RC представляет собой водород или C1-5 алкил; C1-16 алкила, C2-8 алкенила, C3-8 циклоалкила, ацила формулы -C(R11)=O, где R11 выбран из: водорода, C1-16 алкила, C3-8 циклоалкила, гидроксиалкила, гетероциклила, арила аралоксиалкила ацилоксиалкила и остатка природной аминокислоты, например, глицина, цистеина, фенилаланина или лейцина, или синтетической аминокислоты, или остатка ди- или трипептида, например, Gly-Gly, Gly-Phe, Gly-Leu или Gly-Phe-Leu, Gly-Leu-Phe; и R7 выбран из водорода, метила, CH2OH, CH2O-R12, где R12 представляет собой группу тетрагидропиранила (ТГП) или сахарид формулы 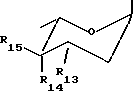 в которой R13 представляет собой амино- или аминоацильную группу, R14 и R15 оба являются водородами, или один из R14 или R15 представляет собой водород, а другой из R14 или R15 представляет гидрокси- или алкокси-, или галоген, или группу OSO2(R10) как указывалось выше, CH2-O-Ph-(амино), где аминогруппа может быть незамещенной или моно- или дизамещенной алкильной, ацильной, трифторацильной, аралкильной или арильной группой; и CH2-амино, где аминогруппа является моно- или дизамещенной алкильной, ацильной, трифторацильной, аралкильной или арильной группой, или аминогруппа входит в состав гетероцикла, например, пиперидинового, пирролидинового или морфолинового кольца, необязательно замещенного C1-16 алкилом или C1-16 алкилокси-, или арилоксигруппой, или его фармацевтически пригодной солью. Предпочтительными соединениями формулы 1 являются те, в которых: X1 и X2 выбраны независимо из: C=O и C=NH; X3 выбран из CH2, C=O, CHOH и C=N(R9), где R9 представляет собой гидрокси- или аминоарильную группу; R1, R2, R3 и R4 выбраны независимо из: водорода, гидроксила, C1-4 алкоксила, C3-8 циклоалкоксила, O-мезила (O-SO2CH3), аминогруппы и аминобензила; R5 и R6 выбраны независимо из: водорода, гидроксила, C1-4 алкоксила, галогена, аминогруппы, аминобензила и аминотрифторацетила; R6 выбран из: водорода, RB-CH2-, где RB представляет собой группы, указанные выше, C1-10 алкила, C2-6 алкенила, ацила формулы – C(R12)=O, где R12 выбран из: C1-10 алкила, гидроксиалкила, гетероциклила, арила аралоксиалкила, ацилоксиалкила и остатка природной аминокислоты, например, глицина, цистеина, фенилаланина, лейцина, или синтетической аминокислоты, или остатка ди- или трипептида, например, Gly-Gly, Gly-Phe, Gly-Leu, Gly-Phe-Leu, Gly-Leu-Phe; и R7 выбран из водорода, метила, CH2OH, CH2O-R12, где R12 представляет собой группу тетрагидропиранила (ТГП) или сахарид формулы 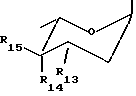 в которой R13 представляет собой амино- или аминотрифторацетильную или аминоацетильную группы, R15 является водородом, и R14 представляет собой гидрокси или иод, или O-мезил, CH2-O-Ph-NH-COR, где R представляет собой алкил, аралкил или арил, CH2-амино, где аминогруппа входит в состав гетероцикла, например, пиперидинового, пирролидинового, морфолинового или дигидропиридинового кольца, возможно замещенного C1-10 алкилом или C1-5 алкилокси- или арилоксигруппой. Более предпочтительными соединениями формулы 1 являются те, в которых: X1 и X2 выбраны независимо из: C=O и C=NH; X3 выбран из CH2, C=O и CHOH, R1, R2, R3 и R4 выбраны независимо из водорода, гидроксила, метила, метоксигруппы, O-мезилата, аминогруппы, аминобензила, фтора и хлора; R5 и R8 выбраны независимо из: водорода, гидроксила, метоксигруппы, этоксигруппы, аминогруппы и аминотрифторацетила; R6 выбран из: водорода, бензила, аллила, 3,4-диметоксибензила, пиридинметила, (N-метил-дигидропиридин)-метила, никотила, глицила и изолейцила; и R7 выбран из водорода, метила, CH2OH, CH2O-R12, где R12 представляет собой группу тетрагидропиранила (ТГП) или сахарид формулы 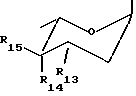 в которой R13 представляет собой амино- или аминотрифторацетильную, или аминоацетильную группы, R15 является водородом, и R14 представляет собой иод и CH2-амино, где аминогруппа входит в состав морфолинового кольца. Еще более предпочтительными соединениями формулы 1 являются те, в которых: X1 и X2 оба являются C=O; X3 является C=O; R1, R2, R3 каждый представляют собой водород, а R4 является водородом, гидрокси- или метоксигруппой; R5 и R8 выбраны независимо из водорода, гидроксила, метоксигруппы и аминогруппы; R6 выбран из водорода, пиридинметила, (N-метил-дигидропиридин)-метила, никотила, глицила и изолейцила; и R7 является метилом. “Алкильная” группа представляет собой C1-C16 алкильную группу. C1-C16 алкильная группа включает алкильные группы как линейной, так и разветвленной цепи. Предпочтительно, чтобы C1-16 алкильная группа представляла собой C1-C12 алкильную группу, такую как гексил, изогексил, гептил, октил, нонил, децил, ундецил и додецил группу или их разветвленные изомеры. Предпочтительно, чтобы C1-C12 алкильная группа представляла собой C1-C6 алкильную группу или C1-C5 алкильную группу, такую как метил, этил, пропил, изопропил, бутил, т-бутил, изобутил или пентил группу или их разветвленные изомеры. Алкильные группы, описанные выше, могут иметь заместители в одном или двух положениях, например, галогенный заместитель, такой как фтор, хлор, бром или иод, CF3, заместители в виде алкокси-, арил-, алкил-арил-, галоарил-, циклоалкил- или алкилциклоалкил-групп. Применяемый здесь термин “алкенил” включает радикалы как прямой, так и разветвленной цепи из 8 углеродных атомов, например, аллил, бутенил, гексенил, октенил. Применяемый здесь термин “циклоалкил” обозначает циклоалкильную группу, имеющую от 3 до 8 углеродных атомов, предпочтительно от 3 до 5 углеродных атомов. Примеры включают циклопропил, циклопентил, циклопентилметил, циклогептил, циклооктил. Гетероциклы являются от 3 до 6-членными, например, 3, 4, 5 или 6-членными насыщенными или ненасыщенными гетероциклами, содержащими по крайней мере один гетероатом, выбранный из O, S и N, которые могут быть присоединены ко второй от 5- до 6-членной, насыщенной или ненасыщенной гетероциклильной группе или к указанной циклоалкильной группе, или к арильной группе, как описывается ниже в данной работе. Примерами гетероциклильных групп являются пирролил, имидазолил, триазолил, тетразолил, оксазолил, тиазолил, тиадиазолил, тиенил, пиридинил, дигидропиридинил, пиперидинил, пиперазинил, пиразинил, пиримидинил, пиранил, пиридазинил, фуранил, пиразолил, изотиазолил, изоазолил, морфолинил, тиопиранил, бензотиенил, бензотиазолил или бензоксазолил. Такие группы могут замещаться гидрокси-, первичными, вторичными амино или четвертичными аминогруппами, радикалами, присоединенными к вторичным или четвертичным аминогруппам, например, C1-C12 прямой или разветвленной алкильными группами, фенил-, бензил-, алкокси-, фенокси- или бензилоксигруппами или атомами галогенов. Применяемый здесь термин “арил” включает как моноциклические, так и бициклические ароматические группы, содержащие от 6 до 10 углеродных атомов в циклической структуре, такие как фенил, нафтил, замещенный фенил или замещенный нафтил, в которых заместителем как фенила, так и нафтила может быть, например, C1-6алкил, галоген или C1-6алкоксигруппа. Применяемый здесь термин “галоген” обозначает фтор, хлор, бром или иод. Применяемый здесь термин “аралкил” относится к алкильным группам, имеющим, как обсуждалось ранее, арильный заместитель, как указывалось выше, например, бензил, 3,4-диметоксибензил, фенетил, дифенилметил и трифенилметил. Применяемый здесь термин “ароил” относится к группе с формулой – COAr, где Ar обозначает “арильную” группу, как указывалось ранее. Применяемый здесь термин “алкокси” или “арилокси” включает любую из описанных выше алкильных или аралкильных групп с присоединенным атомом кислорода. Применяемый здесь термин “алкоксиалкил” обозначает любой алкил из описанных выше, связанный с любой из указанных выше алкоксигрупп, например, с этоксипропильной группой. Применяемый здесь термин “арилоксиалкил” обозначает любой алкил из описанных выше, связанный с арилом, как обсуждалось выше, через атом кислорода, например, феноксиэтил. Применяемый здесь термин “аралоксиалкил” обозначает аралкил, обсуждаемый выше, присоединенный к алкилу, как описывалось выше, через атом кислорода, например, бензилоксиэтил. Применяемый здесь термин “ацилоксиалкил” обозначает C1-10 ацильную группу, присоединенную к алкильной группе, как указывалось выше, через атом кислорода, например, ацетоксиметил. Применяемый здесь термин “гидроксиалкил” обозначает алкильную группу, как обсуждалось выше, присоединенную к гидроксильной группе, например, гидроксиэтил. Ацильная группа обычно представляет собой C1-C10 ацильную группу, например, C1-C6 ацильную группу, такую как метаноил, этаноил, н-пропаноил, и-пропаноил, н-бутаноил, t-бутаноил, вторбутаноил, пентаноил или гексаноил группы. Данное изобретение включает также все возможные изомеры соединений формулы 1 и их меси, например, диастереоизомерные смеси и рацемические смеси. Таким образом, стереоцентры в 7-ом и 9-ом положении могут быть в R или S конфигурации (или в обеих конфигурациях, то есть присутствовать как смесь стереоизомеров). Обычно гликозидная связь сахаридов может быть в  или или  конфигурации (или в обоих, т.е. представлять смесь стереоизомеров). В настоящем изобретении предлагаются также соли тех соединений формулы 1, которые имеют группы, пригодные для образования солей, особенно соли соединений, имеющих карбоксильную группу или основную группу (то есть аминогруппу).
Настоящее изобретение включает соли производных антразалинона формулы 1. Соли обычно представляют собой физиологически допустимые или фармацевтически приемлемые соли, например, соли щелочных металлов и соли щелочноземельных металлов (то есть соли натрия, калия, лития, кальция и магния), соли аммония и соли пригодных органических аминов или аминокислот (то есть соли аргинина, прокаина), и аддитивные соли, образующиеся из приемлемых органических или неорганических кислот, например, соляной кислоты, серной кислоты, карбоновой кислоты, и органических сульфокислот (то есть уксусной, трифторуксусной, р-толуосульфокислоты).
Соединения формулы 1, в которых R6 представлен RB-CH2 группой, могут быть получены: (a) взаимодействием соединения формулы 2 конфигурации (или в обоих, т.е. представлять смесь стереоизомеров). В настоящем изобретении предлагаются также соли тех соединений формулы 1, которые имеют группы, пригодные для образования солей, особенно соли соединений, имеющих карбоксильную группу или основную группу (то есть аминогруппу).
Настоящее изобретение включает соли производных антразалинона формулы 1. Соли обычно представляют собой физиологически допустимые или фармацевтически приемлемые соли, например, соли щелочных металлов и соли щелочноземельных металлов (то есть соли натрия, калия, лития, кальция и магния), соли аммония и соли пригодных органических аминов или аминокислот (то есть соли аргинина, прокаина), и аддитивные соли, образующиеся из приемлемых органических или неорганических кислот, например, соляной кислоты, серной кислоты, карбоновой кислоты, и органических сульфокислот (то есть уксусной, трифторуксусной, р-толуосульфокислоты).
Соединения формулы 1, в которых R6 представлен RB-CH2 группой, могут быть получены: (a) взаимодействием соединения формулы 2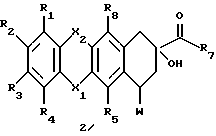 где X1, X2 и R1-R7, как описано выше, и W представляет собой удаляемую группу, с амином формулы H2N-CH2-RB, где RB, как указано выше, давая соединение формулы 1, где R6 представляет собой RB-CH2-; (b) по желанию, превращением полученного соединения формулы 1 в другое соединение формулы 1; и/или (c) по желанию, путем превращения соединения формулы 1 в его фармацевтически приемлемую соль. Пригодные группы W включают O-сахарид, такой как производные O-дауносаминила, O-ацил, такой как, например, O-трифторацетил или O-(п-нитробензоил) или O-этоксикарбонил, O-ацеталь, такой как O-ТГП. Предпочтительные амины формулы NH2-CH2-RB включают алкилариламины, например, бензиламин, 3,4-диметоксибензиламин или пиридиниметиламин. Соединение формулы 2 обычно реагирует с амином формулы NH2-CH2-RB, как указывалось выше. Амин обычно присутствует в 1-10-кратном избытке. Реакция может проходить в совместимом с ней органическом растворителе, таком как метиленхлорид или пиридин. Может присутствовать такое органическое основание, как пиридин. Реакция может продолжаться в течение от 6 до 48 часов, обычно при температуре от -10oC до комнатной (то есть около 20oC). Предпочтительно применять 4-кратный избыток амина формулы NH2-CH2-RB. Растворителем обычно является пиридин. Предпочтительными условиями реакции являются комнатная температура и длительность в течение от 12 до 24 часов. Необходимо подчеркнуть, что эта реакция является новой в области химии антрациклинов и антрациклинонов. Производные антрациклинона, в которых R6 представляет собой водород, могут быть получены, например, путем деблокирования соответствующего NCH2-R’B производного, где R’B представляет собой 3,4-диметоксифенильную или винильную группы. Деблокирование обычно достигается путем окисления, например, обработкой 5,6-дициано-1,4-бензохиноном (ДБХ). Реакция может протекать в присутствии подходящего для этой цели растворителя. Предпочтительно применение эквивалентного количества ДБХ. Предпочтительно, чтобы растворитель представлял собой смесь метиленхлорида и воды (обычно в объемном отношении 20:1). Реакция обычно протекает при комнатной температуре в течение от 1 до 6 часов. Производные антразалинона формулы 1 могут быть дополнительно видоизменены в различные S-N-замещенные производные с помощью стандартных химических процедур. Например, 8-N-алкил, -алкенил, -циклоалкил антразалиноны формулы 1 предпочтительно получают путем взаимодействия соединения формулы 1, в котором R6 представляет собой водород, с группой формулы R6-X, где R6 представляет собой C1-C16 алкил, C2-C8 алкенил или C3-C8 циклоалкил, а X является удаляемой группой, такой как галоген, O-SO2CH3, O-SO2CF3 или O-SO2-C6H4CH3. Предпочтительно, чтобы X был галогеном. Более предпочтительно, чтобы X был иодом. Может присутствовать подходящий растворитель. Предпочтительно применение 2-20-кратного избытка R6-X. Предпочтительно, чтобы реакция протекала в органическом растворителе, таком как метиленхлорид или диметилформамид. Реакция обычно проходит при температуре от 40 до 80oC в течение от 4 до 24 часов. N-ацил-антразалиноны формулы 1 получают предпочтительно путем взаимодействия соединения формулы 1, в котором R6 является водородом, с ацильным производным формулы R11-CO-Hal или (R11-CO)2O, где R11 представляет собой указанные выше радикалы, и Hal является галогеном, предпочтительно хлором. Предпочтительно применять 2-20-кратный избыток ацильного производного. Обычно присутствует растворитель, например, органический растворитель, такой как метиленхлорид или диметилформамид. Предпочтительно, чтобы реакция протекала при температуре от -10 до 40oC в течение от 1 до 24 часов. В дополнительном примере N-ацил-антразалиноны формулы 1 могут быть получены путем взаимодействия антразалинона формулы 1, в котором R6 является водородом, с производным кислоты формулы R11-COOH в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид или 2-этокси-1-этоксикарбонил-1,2-дигидрохинон (ЭЭДХ) в безводном органическом растворителе. Предпочтителен 1-4-кратный избыток кислоты. Предпочтительно, чтобы безводным органическим растворителем был диметилформамид. Обычно используется эквивалентное количество ЭЭДХ. Реакция обычно протекает при комнатной температуре в течение 15 часов. Соединения формулы 1, в которых R6 представляет собой остаток аминокислоты или ди-, трипептида, могут быть получены аналогичным образом в соответствии с условиями конденсации, известными в химии пептидов. C-13-карбонильная группа может быть также восстановлена до C-13-дигидро или видоизменена до гидразона и затем восстановлена с получением C-13-дезоксопроизводных путем процедур, уже известных из химии антрациклинов. Например, для того чтобы получить C-13-дигидропроизводные формулы 1, антразалинон формулы 1 (X3=CO) вступает во взаимодействие с восстанавливающим агентом в органическом растворителе при температуре от -10oC до комнатной в течение от 5 до 30 минут. Предпочтительные условия охватывают растворение агликонов формулы 1, как указывалось выше, в безводном метиленхлориде и обработку его 5-10-кратным избытком боргидрида тетраметиламмония при комнатной температуре в течение 5 минут. Соединения формулы 2 доступны из природных источников или могут быть получены путем известных синтетических методов исходя из известных антрациклинов и антрациклинонов. Например, 7-O-сахарид, в котором сахаром является дауносаминил, может быть получен как производное такого натурального источника, как даунорубицин, или может быть получен путем его же синтетической модификации. Другие агликоны, видоизмененные в позиции C-7, могут быть получены путем хорошо известных процедур. Например, 7-O-ТГП производные формулы 2 (соединения, в которых W является ТГП) легко получить путем взаимодействия агликона формулы 3 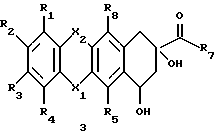 с дигидропираном в органическом растворителе и в присутствии кислотного катализатора при комнатной температуре в течение от 1 до 4 часов. Предпочтительно, чтобы соединение формулы 3 растворялось в метиленхлориде и реагировало с 4-мя эквивалентами дигидропирана в присутствии каталитического количества п-толуолсульфокислоты при комнатной температуре в течение 2 часов, 7-O-ТГП производное выделяется путем промывания реакционной смеси водным раствором гидрокарбоната натрия и водой и последующим удалением растворителя в условиях пониженного давления. 7-O-ацил производные формулы 2 могут быть получены путем взаимодействия соединения формулы 3 с подходящей карбоновой кислотой, ангидридом кислоты или ацилхлоридом в органическом растворителе и в присутствии основания при температуре от -10oC до комнатной в течение от 1 до 6 часов. Например, 7-O-ацетил производное формулы 2 (W=O-COCH3) может быть получено путем взаимодействия соединения формулы 3 с уксусным ангидридом в органическом растворителе, таком как метиленхлорид, и в присутствии органического основания, такого как пиридин. Соединение выделяется путем осаждения сырого материала в неполярном растворителе, таком как гексан. Некоторые исходные материалы для получения соединений формулы 1 известны, другие могут быть получены аналогичным образом, исходя из известных антрациклинов или антрациклинонов путем известных процедур. Например, следующие антрациклины известны и могут быть представлены с помощью той же формулы 2: даунорубицин (2a: R1 = R2 = R3 = H, R4 = OCH3, R5 = R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-даунозаминил), доксорубицин (2b: R1 = R2 = R3 = H, R4 = OCH3, R5= R8 = OH, X1 = X2 = CO, R7 = CH2OH, L = O-даунозаминил), 4-деметоксидаунорубицин (2c: R1 = R2 = R3 = R4 = H, R5 = R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-даунозаминил), 11-дезоксидаунорубицин (2d: R1 = R2 = R3 = H, R4 = OCH3, R5 = OH, R8 = H, X1= X2 = CO, R7 = CH3, L = O-даунозаминил), 11-аминодаунорубицин (2e: R1 = R2 = R3 = H, R4 = OCH3, R5 = OH, R8 = NH2, X1= X2 = CO, R7 = CH3, L = O-даунозаминил), 6-дезоксидаунорубицин (2f: R1 = R2 = R3 = H, R4 = OCH3, R5 = H, R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-даунозаминил), 6-аминодаунорубицин (2g: R1 = R2 = R3 = H, R4 = OCH3, R5 = OH, R8 = NH2, X1 = X2 = CO, R7 = CH3, L = O-даунозаминил), 4-аминодаунорубицин (2h: R1 = R2 = R3 = H, R4 = NH2, R5 = R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-даунозаминил), 9-деацитил-9-формил-N-трифторацетилдаунорубицин (2i: R1 = R2 = R3 = H, R4 = OCH3, R5 = R8 = OH, X1 = X2 = CO, R7 = H, L = O-(N-трифторацетил-даунозаминил). Известны также некоторые 7-O-производные формулы 2, например, 7-O-этоксикарбонилдауномицинон (2j; R1 = R2 = R3 = H, R4 = OCH3, R5 = R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-COOC2H5), 7-O-(тетрагидропиранил)-дауномицинон (2k: R1 = R2 = R3 = H, R4 = OCH3, R5 = R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-ТГП), 7-O-ацетилдауномицинон (2l: R1 = R2 = R3 = H, R4 = OCH3, R5 = R8 = OH, X1 = X2 = CO, R7 = CH3, L = O-COCH3). Соединения настоящего изобретения характеризуются высокой ингибиторной активностью в отношении амилоидоза. Настоящее изобретение, таким образом, предлагает применение соединения формулы 1, как отмечалось выше, или его фармацевтически приемлемой соли для лечения амилоидоза. Таким образом, человека или животное, например, млекопитающее, можно лечить способом, который включает введение ему соединения формулы 1 или его фармацевтически приемлемой соли. Термин “амилоидоз” охватывает различные заболевания, общей характеристикой которых является тенденция определенных белков к агрегации и осаждению с образованием нерастворимых фибрилл во внеклеточном пространстве, что вызывает структурные и функциональные повреждения органов и тканей. Классификация амилоидов и амилоидозов недавно была пересмотрена в бюллетене Всемирной организации здравоохранения 71(1): 105(1993). Все различные типы амилоидных участков характеризуются одним и тем же ультраструктурным строением в виде антипараллельных -складчатых слоев, несмотря на тот факт, что они содержат множество совершенно различных белковых субъединиц [смотри: Glenner G.G., New England J. Med. 302(23): 1283 8 1980). AL амилоидоз вызывается особыми моноклональными иммуноглобулиновыми легкими цепями, которые образуют амилоидные фибриллы. Эти моноклональные легкие цепи продуцируются моноклональными плазматическими клетками с низким митотическим индексом, который обусловливает их хорошо известную нечувствительность к химиотерапии. Злокачественное развитие таких клеток состоит в их усиленной белоксинтезирующей активности.
Клиническое течение болезни зависит от селективности вовлечения органов; прогноз может быть крайне неблагоприятным в случае инфильтрации сердца (средняя продолжительность жизни менее 12 месяцев) или более благоприятным в случае вовлечения почек (средняя продолжительность жизни около 5 лет). Принимая во внимание относительную нечувствительность амилоидогенных отложений к протеолитическому гидролизу, видимо, единственной реальной надеждой больных AL амилоидозом может быть молекула, которая может блокировать или замедлять образование амилоида и увеличивать растворимость существующих амилоидных отложений. Более того, как надмолекулярное строение амилоидных фибрилл является одинаковым для всех типов амилоидов, применимость лекарства, которое препятствует образованию амилоида и увеличивает растворимость имеющихся отложений, вызывая их удаление путем обычных механизмов, может иметь существенные преимущества для лечения всех типов амилоидозов и, в особенности, для лечения болезни Альцгеймера.
Действительно, основной характеристикой патологических изменений при болезни Альцгеймера (БА), синдроме Дауна, деменции pugilistic и церебральной амилоидной ангиопатии является отложение амилоида в церебральной паренхиме и сосудистых стенках. Эти маркеры связаны с потерей нервных клеток в коре мозга, лимбических структурах и субкортикальных ядрах. В некоторых исследованиях было показано, что избирательное разрушение различных участков нервной системы и потеря синапсов во фронтальной коре коррелирует со снижением когнитивных функций. Патогенез и молекулярная база нейродегенеративных процессов при БА неизвестны, но роль -амилоида, откладываемого в паренхиме мозга и сосудистых стенках, освещена в недавнем обзоре их нейротоксической активности in vitro и in vivo (Yanker et al., Science, 245:417, 1990, Kowall et al., PNAS, 88:7247, 1991).
Более того, выявление наследственной формы БА с мутацией гена белка-предшественника амилоида (БПА) повысило интерес к потенциальной патогенетической функции -амилоида при БА [Mullan V. et al., TINS, 16(10): 392 (1993)] . Нейротоксичность -амилоида связана со свойствами белка индуцировать образование фибрилл. Изучение гомологичных синтетических пептидов показывает, что клетки гиппокампа оставались нечувствительными к 1-42 при экспозиции с его свежеприготовленным раствором в течение 24 часов, в то время как их жизнеспособность снижалась при экспозиции нейронов с A1-42 , предварительно выдержанном в солевом растворе в течение 2-4 дней при температуре 37oC, что благоприятствует агрегации пептида. Взаимосвязь между образованием фибрилл и нейротоксичностью дополнительно подтверждается полученными недавно данными, показавшими, что растворимая форма -амилоида вырабатывается in vitro и in vivo в процессе нормального клеточного метаболизма (Hass et al. Nature, 359, 322, 1993) и только их агрегация в виде конгофильных образований связана с дистрофическими нейритами. С другой стороны, неконгофильное “преамилоидное” скопление -амилоида не связано с повреждением нейронов (Tagliavini et al. Neurosci. Lett. 93: 191, 1988). Нейротоксичность -амилоида подтверждена также при использовании пептидного фрагмента 25-35 гомолога -амилоида 25-35), сохраняющего способность полного фрагмента -амилоида – 1-42 к самоагрегации.
Хроническая, но не кратковременная, экспозиция нейронов гиппокампа с микромолярной концентрацией 25-35 индуцировала смерть нейронов путем активации механизма программируемой смерти клеток, известного как апоптоз (Forloni et al. NeuroReport, 4:523, 1993). В этом случае опять нейротоксичность была связана со способностью 25-35 к самоагрегации.
Другие нейродегенеративные нарушения, такие как губчатая энцефалопатия (ГЭ), характеризуются смертью нейронов и внеклеточными отложениями амилоида, в этом случае состоящего из белка Приона (PrP). По аналогии с изучением нейротоксичности -амилоида исследовали действие синтетических пептидов, гомологичных различным фрагментам PrP, на жизнеспособность первичных нейронов гиппокампа крыс. Хроническая аппликация пептида, соответствующего фрагменту 106-126 PrP, индуцировала смерть нейронов путем апоптоза, в то время как при тех же условиях все другие исследуемые пептиды и случайная последовательность PrP 106-126 не снижали жизнеспособности клеток (Forloni et al. Nature, 362:543). PrP обладал высокими способностями к образованию фибрилл in vitro, при окраске Конго красным демонстрировавшими зеленое двойное светопреломление, указывающее на наличие конформации -складок, характерных для амилоида.
Соединения настоящего изобретения могут применяться для изготовления медикаментов, применяемых для предотвращения или приостановки развития болезней, вызываемых амилоидными белками, таких как AL амилоидоз, болезнь Альцгеймера или синдром Дауна.
Соединения настоящего изобретения исследовали на их природную цитотоксичность на культуре клеток PC 12 согласно стандартным процедурам. Было обнаружено, что все соединения нетоксичны в концентрации до 10 мМ.
Настоящее изобретение включает в свою сферу также фармацевтические композиции, состоящие из одного или нескольких компонентов формулы 1 как активных компонентов в комбинации с фармацевтически приемлемыми носителями, наполнителями или, при необходимости, с другими добавками.
Фармацевтические композиции, содержащие соединение формулы 1 или его соли, могут быть приготовлены обычным путем с применением традиционных нетоксичных фармацевтических носителей или разбавляющих средств в виде различных лекарственных форм и с применением разных способов введения.
В частности, соединения формулы 1 могут вводиться:A) Орально, например, в виде таблеток, пастилок, лепешек, водных или масляных суспензий, сыпучих порошков или гранул, эмульсий, твердых или мягких капсул, сиропов или элексиров. Композиции, предназначенные для орального применения, могут быть приготовлены согласно любому известному науке методу для производства фармацевтических композиций, и такие композиции могут содержать один или более агентов, выбранных из группы, включающей подсластители, вкусовые отдушки, подкрашивающие агенты и консерванты, для придания препаратам фармацевтически первоклассных приятных качеств. Таблетки содержат активный компонент с примесью нетоксичных фармацевтически приемлемых наполнителей, которые пригодны для производства таблеток. Этими наполнителями могут быть, например, инертные разбавляющие средства, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; агенты, вызывающие грануляцию или диспергирование, например, маисовый крахмал или альгиновая кислота, связывающие агенты, например, маисовый крахмал, желатин или аравийская камедь, и смазочные агенты, например, стеарат магния или стеариновая кислота, или тальк. Таблетки могут быть без оболочки или они могут покрываться оболочкой с помощью известной технологии для замедления разрушения и улучшения абсорбции в желудочно-кишечном тракте, обеспечивая таким образом, поддержание более продолжительного действия. Например, могут быть применены такие материалы, продлевающие время действия, как моностеарат глицерина или дистеарат глицерина. Состав для орального применения может также быть представлен в виде твердых желатиновых капсул, активный компонент в которых смешан с инертным твердым наполнителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, активный компонент в которых смешан с водой или с масляной средой, например, с арахисовым маслом, жидким парафином или оливковым маслом. Жидкие суспензии содержат активные компоненты с добавлением наполнителей, приемлемых для производства жидких суспензий. Такие наполнители являются суспендирующими агентами, такими как, например, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидрокси-, пропилметилцеллюлоза, альгинат натрия, камедь поливинилпирролидона, трагакант или аравийская камедь; диспергирующие или увлажняющие агенты могут представлять собой природные фосфатиды, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида с алифатическими многоатомными спиртами, например, гептадекаэтиленоксиэтанол, или продукты конденсации этиленоксида с неполными эфирами жирных кислот и ангидридов гекситола, например, полиоксиэтиленосорбитан моноолеат. Указанные водные суспензии могут также содержать один или более консервантов, например, этил- или н-пропил-п-гидроксибензоат, один или более подкрашивающих агентов, одну или более вкусовых отдушек или один или более подсластителей, таких как сахароза или сахарин. Масляные суспензии могут быть составлены путем суспендирования активного компонента в растительном масле, например, арахисовом масле, оливковом масле, sememe масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загущающий агент, например, пчелиный воск, твердый парафин или цетиловый спирт. Могут добавляться подсластители, типа указанных выше, и вкусовые отдушки для создания приятных ощущений при оральном применении препарата. Для лучшего сохранения этих композиций может добавляться антиоксидант, такой как аскорбиновая кислота. Диспергируемые порошки и гранулы, пригодные для приготовления водных суспензий с помощью добавления воды, предлагаются в виде активного компонента с добавлением диспергирующего или увлажняющего агента, суспендирующего агента и одного или более консервантов. Примерами приемлемых диспергирующих или увлажняющих агентов, а также суспендирующих агентов являются те, которые уже были указаны выше. Могут также присутствовать дополнительные наполнители, например, подсластители, вкусовые отдушки. Фармацевтические композиции изобретения могут также быть в форме водно-масляных эмульсий. Масляная фаза может представлять собой растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин, или их смеси. Пригодными эмульгирующими агентами могут быть природные камеди, например, аравийская камедь или трагакант, природные фосфатиды, например, соевые бобы, лецитин и эфиры или неполные эфиры жирных кислот и ангидридов гекситола, например, сорбитан моноолеат, а также продукты конденсации указанных неполных эфиров с этиленоксидом, например, полиоксиэтилен сорбитан моноолеат. Эмульсия может также содержать подсластители и вкусовые отдушки. Сиропы и элексиры могут быть приготовлены с применением подсластителей, например, глицерина, сорбитола или сахарозы. Такие составы могут также содержать смягчающий агент, консервант, вкусовую отдушку и подкрашивающий агент. В) Парэнтерально либо подкожно, либо внутривенно, либо внутримышечно, либо внутригрудинно (интрастериально), либо с помощью техники инфузии, в форме стерильной водной или масляной суспензии для инъекции. Фармацевтические композиции могут быть в форме стерильной водной или масляной суспензии для инъекции. Эта суспензия может быть составлена в соответствии с известными в науке способами, с применением тех подходящих диспергирующих или увлажняющих агентов, которые упоминались выше. Стерильный препарат для инъекций может также быть стерильным раствором или суспензией в нетоксичном, приемлемом для парэнтерального введения разбавителе или растворителе, например, применяться растворенным в 1,3-бутандиоле. В качестве приемлемых носителей и растворителей может применяться вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла обычно применяются как растворители или суспензионные среды. Для этой цели обычно могут использоваться любые мягкие нелетучие масла, включая синтетические моно- или диглицериды. Кроме того, показана применимость в препаратах для инъекций жирных кислот, таких как олеиновая кислота. Настоящее изобретение предлагает также способ контроля над болезнями, связанными с амилоидозом, и/или способ предотвращения или торможения развития болезней, вызванных амилоидными белками, данный способ включает введение терапевтически эффективного количества одного или более компонентов формулы 1 человеку или животному, например, млекопитающему, нуждающемуся в таком лечении. Суточные дозы колеблются в интервале от около 0,1 до 50 мг на кг веса тела, в соответствии с активностью специфического компонента, с возрастом, весом и состоянием субъекта, подвергающегося лечению, с типом и тяжестью заболевания, частотой и схемой введения препарата, предпочтительно, чтобы количество суточной дозы было в пределах от 5 мг до 2 г. Количество активного компонента, который может вводиться в сочетании с носителями в качестве единой лекарственной формы, будет варьироваться в зависимости от субъекта, подвергаемого лечению, и особенностями способа введения препарата. Например, состав, предназначенный для орального введения, может содержать от 5 мг до 2 г активного компонента, дополненного соответствующим и удобным количеством носителя, содержание которого может варьироваться от 5 до 95 процентов от суммарного состава композиции. Единица лекарственных форм обычно должна содержать от 5 до 500 мг активного компонента. Последующие примеры иллюстрируют изобретение, не ограничивая его возможностей. Пример 1 Получение 8-N-(3,4-диметоксибензил)-антразалона (1a) 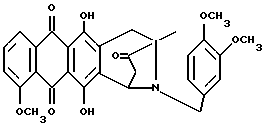 Даунорубицин (2a, 1.58 г, 3 ммоль) растворяли в сухом пиридине (20 мл) с добавлением 3,4-диметоксибензиламина (2 г, 12 ммоль) и выдерживали при комнатной температуре в течение 16 часов. После этого к реакционной смеси добавляли водный раствор 1N HCl (400 мл) и экстрагировали метиленхлоридом (200 мл). Органическую фазу промывали водой (2х200 мл), высушивали над безводным сульфатом натрия, концентрировали до небольшого объема в условиях пониженного давления и подвергали флэш-хроматографии на силикагеле, применяя в качестве элюирующей системы смесь толуол – ацетон (9:1 объем), получая соединение 1a, (1 г), указанное в заголовке. Тонкослойная хроматография (ТСХ) на пластине кизельгеля F254 (Merk), элюирующая система – метиленхлорид – ацетон (95 : 5 объем), Rf = 0.56 FAB-MS (+): m/z 530 [MH]+; 380 [M-CH2(C6H3)(OCH3)2+2H]+;  1H ЯМР (400 МГц CDCl3)  : 1.43 (s, 3H, CH3); 2.34 (d, J = 17.5 Гц, 1H, CH(H)-12); 2.66, 2.77 (два дублета, J = 19.4 Гц, 2H, CH2-10); 2.81 (dd, J = 7.3, 17.5 Гц, 1H, CH(H)-12); 3.24, 3.79 (два дублета, J = 12.8 Гц, 2H, N-CH2-Ph); 3.85, 3.86 (2xs, 6H, 2xOCH3); 4.08 (s, 3H, 4-OCH3); 4.77 (d, J = 7.3 Гц, 1H, H-7); 6.6-6.8 (m, 3H, водороды ароматического кольца); 7.38 (d, J = 7.6 Гц, 1H, H-3); 7.77 (dd, J = 7.8 Гц, 1H, H-2); 8.03 (d, J = 7.8 Гц, 1H, H-1); 13.22 (s, 1H, OH-11); 13.50 (s, 1H, OH-6).
Пример 2 : 1.43 (s, 3H, CH3); 2.34 (d, J = 17.5 Гц, 1H, CH(H)-12); 2.66, 2.77 (два дублета, J = 19.4 Гц, 2H, CH2-10); 2.81 (dd, J = 7.3, 17.5 Гц, 1H, CH(H)-12); 3.24, 3.79 (два дублета, J = 12.8 Гц, 2H, N-CH2-Ph); 3.85, 3.86 (2xs, 6H, 2xOCH3); 4.08 (s, 3H, 4-OCH3); 4.77 (d, J = 7.3 Гц, 1H, H-7); 6.6-6.8 (m, 3H, водороды ароматического кольца); 7.38 (d, J = 7.6 Гц, 1H, H-3); 7.77 (dd, J = 7.8 Гц, 1H, H-2); 8.03 (d, J = 7.8 Гц, 1H, H-1); 13.22 (s, 1H, OH-11); 13.50 (s, 1H, OH-6).
Пример 2Получение антразалона (1b) 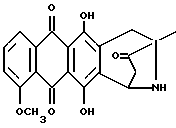 8-N-(3,4-диметоксибензил)-антразалон (1a, 0,5 г, 1 ммоль) растворяли в смеси метиленхлорида (20 мл) и воды (1 мл) и обрабатывали 2,3-дихлоро-5,6-дициано-1,4-бензохиноном (ДДХ, 0.25 г, 1 ммоль) при комнатной температуре. Спустя 4 часа реакционную смесь промывали 5% водным раствором бикарбоната натрия (3х100 мл) и затем – водой. Органическую фазу сушили над безводным сульфатом натрия, растворитель удаляли при пониженном давлении, что позволяло получить соединение 1b, указанное в заголовке (0.35 г), которое превращали в соответствующую хлористоводородную соль обработкой безводной соляной кислотой в метаноле. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид-ацетон (90:10, объем) Rf = 0,26. FD-MS: 380 [MH]+; 362 [M-NH3]+ 1H ЯМР (400 МГц, CDCl3) :1.45 (s, 3H, CH3); 2.43 (d, J = 17.5 Гц, 1H, CH(H)-12); 2.76, 2.84 (два дублета, J = 19.2 Гц, 2H, CH2-10); 2.86 (dd, J = 7.3, 17.5 Гц, 1H, CH(H)-12); 4.08 (s, 3H, OCH3); 5.14 (d, J = 7.3 Гц 1H, H-7); 7.37 (d, J = 8.5 Гц, 1H, H-3); 7.76 (dd, J = 7.7, 8.5 Гц, 1H, H-2); 8.01 (d, J = 7.7 Гц, 1H, H-1); 13.14 (s, 1H, OH-11); 13.60 (s, 1H, OH-6). Пример 3 Получение 8-N-(пиридинметил)-антразалона (1c) 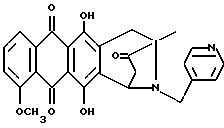 Указанное в заголовке соединение 1c получали из даунорубицина (2a, 1.58 г, 3 ммоля) и 4-аминометилпиридина (1.2 г, 12 ммолей) в соответствии с процедурой, описанной в примере 1. Выход составил 0.95 г. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид – ацетон (80:20, объем) Rf = 0.4. FAB-MS (+): m/z 471 [MH]+; 380 [M-CH2(C5H4N)+2H]+; 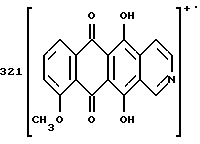 1H ЯМР (400 МГц, CDCl3) :1.39 (s, 3H, CH3); 2.50 (d, J = 17.9 Гц, 1H, CH(H)-12); 2.78 (s, 2H, CH2-10); 2.96 (dd, J = 7.3, 17.9 Гц, 1H, CH(H)-12); 3.70, 4.07 (два дублета, J = 16.7 Гц, 2H, N+-CH2-Пиридин); 4.07 (s, 3H, OCH3); 4.76 (d, J = 7.3 Гц, 1H, H-7); 7.40 (d, J = 7.3 Гц, 1H, H-3); 7.79 (dd, J = 7.3 Гц, 1H, H-2); 7.89 (d, J = 6.0 Гц, 2H, водороды пиридина); 8.02 (d, J = 7.7 Гц, 1H, H-1); 8.70 (d, J = 6.0 Гц, 2H, водороды пиридина); 13.14 (s, 1H, OH-11); 13.45 (s, 1H, OH-6). Пример 4 Получение 4-деметокси-8-N-(пиридинметил)-антразалона (1d) 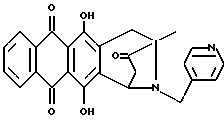 Указанное в заголовке соединение 1d получали из 4-деметоксидаунорубицина (23, 1.38 г, 3 ммоля) и 4-аминометилпиридина (1.2 г, 12 ммолей) в соответствии с процедурой, описанной в примере 1. Выход составил 0.87 г. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид – ацетон (80:20, объем) Rf = 0.46. FAB-MS (+): m/z 441 [MH]+; 350 [M-CH2(C5H4H)+2H]+; 1H ЯМР (200 МГц CDCl3) :1.41 (s, 3H, CH3); 2.46 (d, J = 17.6 Гц, 1H, CH(H)-12); 2.73 (m, 2H, CH2-10); 2.89 (dd, J = 7.0, 17.6 Гц, 1H, CH(H)-12); 3.37, 3.85 (два дублета, J = 14.6 Гц, 2H, N-CH2-Пиридин); 2.73 (d, J = 7.0 Гц, 1H, H-7); 7.24 (m, 2H, водороды пиридина); 7.80 (m, 2H, H-2 + H-3); 8.28 (m, 2H, H-1 + H-4); 8.54 (2H, водороды пиридина); 13.05, 13.16 (2xs, 2H, OH-6 + OH-11). Пример 5 Получение 8-N-бензил-антразалона (1e) 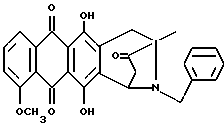 Указанное в заголовке соединение 1e получали из даунорубицина (2a, 1.58 г, 3 ммоля) и бензиламина (1.2 г, 12 ммолей) в соответствии с процедурой, описанной в примере 1. Выход составил 1 г. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид – ацетон (90:10, объем) Rf = 0.7. FAB-MS (+): m/z 470 [MH]+; 320 [M-CH2(C6H5)+2H]+; 1H ЯМР (200 МГц CDCl3) :1.42 (s, 3H, CH3); 2.37 (d, J = 17.4 Гц, 1H, CH(H)-12); 2.68, 2.76 (два дублета, J = 19.6 Гц, 2H, CH2-10); 2.81 (dd, J = 7.0, 17.4 Гц, 1H, CH(H)-12); 3.30, 3.84 (два дублета, J = 13.2 Гц, 2H, N-CH2-Ph); 4.07 (s, 3H, 4-OCH3); 4.74 (d, J = 7.0 Гц, 1H, H-7); 7.2-7.3 (m, 5H, водороды ароматического кольца); 7.38 (dd, J = 1.0, 8.4 Гц, 1H, H-3); 7.77 (dd, J = 7,7, 8.4 Гц, 1H, H-2); 8.02 (dd, J = 1.0, 7,7 Гц, 1H, H-1); 13.22, 13.42 (2xs, 2H, OH-6 + OH-11). Пример 6 Получение 4-деметокси-8-N-бензил-антразалона (1f) 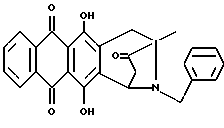 Указанное в заголовке соединение 1f получали из 4-деметоксидаунорубицина (2c, 1.38 г, 3 ммоля) и бензиламина (1.2 г, 12 ммолей) в соответствии с процедурой, описанной в примере 1. Выход составил 0.9 г. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид – ацетон (80:20, объем) Rf = 0.84. FAB-MS (+): m/z 440 [MH]+; 290 [M-CH2(C6H5)+2H]+; 1H ЯМР (200 МГц CDCl3) :1.44 (s, 3H, CH3); 2.38 (d, J = 17.4 Гц, 1H, CH(H)-12); 2.70, 2.78 (два дублета, J = 19.7 Гц, 2H, CH2-10); 2.85 (dd, J = 7.2, 17.4 Гц, 1H, CH(H)-12); 3.31, 3.87 (два дублета, J = 13.0 Гц, 2H, N-CH2-Ph); 4.74 (d, J = 7.2 Гц, 1H, H-7); 7.2-7.3 (m, 5H, водороды фенила); 7.83 (m, 2H, H-2 + H-3); 8.33 (m, 2H, H-1 + H-4); 13.1, 13.2 (2xs, 2H, OH-6 + OH-11). Пример 7 Получение 4-деметокси-8-N-(3,4-диметоксибензил)-антразалона (1g) 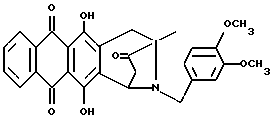 Указанное в заголовке соединение 1g получали взаимодействием 4-деметоксидаунорубицина (2c, 1.38 г, 3 ммоля) с 3,4-диметоксибензиламином (2 г, 12 ммолей) в соответствии с процедурой, описанной в примере 1. Выход составил 0.9 г. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид – ацетон (95:5, объем) Rf = 0.65. FAB-MS (+): m/z 500 [MH]+; 350 [M-CH2(C6H3)(OCH3)2+2H]+. Пример 8 Получение 4-деметокси-антразалона (1h) 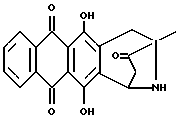 4-деметокси-8-N-(3,4-диметоксибензил)-антразалон (1g, 0.5 г, 1 ммоль) превращали в указанное в заголовке соединение 1h в присутствии ДДХ, как описано в примере 2. Выход составил 0.4 г. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид – ацетон (95:5, объем) Rf = 0.34. FD-MS: 350 [MH]+ 1H ЯМР (200 МГц CDCl3) :1.46 (s, 3H, CH3); 2.45 (d, J = 17.7 Гц, 1H, CH(H)-12); 2.81, 2.86 (два дублета, J = 19.4 Гц, 2H, CH2-10); 2.87 (dd, J = 7.0, 17.7 Гц, 1H, CH(H)-12); 5.14 (d, J = 7.0 Гц, 1H, H-7); 7.83 (m, 2H, H-2 + H-3); 8.33 (m, 2H, H-1 + H-4); 13.18, 13.25 (2xs, 2H, OH-6 + OH-11). Пример 9 Получение 8-N-аллил-антразалона (1i) 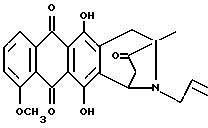 Указанное в заголовке соединение 1i получали взаимодействием даунорубицина (2a, 1.58 г, 3 ммоля) с аллиламином (0.9 г, 12 ммолей) как описано в примере 1. Сырой материал подвергали флэш-хроматографии на силикагеле с использованием смеси метиленхлорид-ацетон (98:2) в качестве элюента, и получали чистый 1i (0.85 г). При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck), элюируя метиленхлоридом, Rf = 0.1. 1H ЯМР (200 МГц CDCl3) :1.37 (s, 3H, CH3); 2.41 (d, J = 17.6 Гц, 1H, CH(H)-12); 2.64 (m, 2H, CH2-10); 2.88 (dd, J = 7.2, 17.6 Гц, 1H, CH(H)-12); 2.8-3.4 (m, 2H, CH2CH= CH2); 4.04 (s, 3H, 4-OCH3); 5.0-5.2 (m, 2H, CH2CH=CH2); 5.90 (m, 1H, CH2CH= CH2); 7.37 (d, J = 8.4 Гц, 1H, H-3); 7.75 (dd, J = 7.6, 8.4 Гц, 1H, H-2); 8.00 (d, J = 7.6 Гц, 1H, H-1); 13.0, 13.5 (2xs, 2H, OH-6 + OH-11). Пример 10 Получение 8-N-бензил-13-дигидро-антразалона (1j) 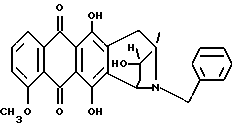 8-N-бензилантразалон (1e, 0.75 г, 1.5 ммоля), полученный как описано в примере 5, растворяли в безводном метиленхлориде (209 мл) и обрабатывали борогидридом тетрабутиламмония (1.6 г) при комнатной температуре в течение 5 мин. Реакционную смесь затем выливали в водный раствор 1 N соляной кислоты и экстрагировали метиленхлоридом. Органическую фазу отделяли, промывали водой и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, а сырой материал подвергали флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси толуол – ацетон (9:1, объем/объем) для выделения указанного в заголовке соединения 1j (0.65 г). При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид-ацетон (90:10, объем) Rf = 0.4. 1H ЯМР (200 МГц CDCl3) :1.42 (s, 3H, CH3); 1.51 (m, 1H, CH(H)-12); 2.6 (m, 2H, CH(H)-12 + CH(H)-10); 3.06 (d, J = 19.6 Гц, 1H, CH(H)-10); 3.21, 3.79 (два дублета, J = 12.9 Гц, 2H, N-CH2-Ph); 4.08 (s, 3H, OCH3); 4.20 (m, 1H, H-9); 4.34 (d, J = 7.2 Гц, 1H, H-7); 7.1-7.3 (m, 5H, водороды фенила); 7.37 (dd, J = 1.0, 8.8 Гц, 1H, H-3); 7.76 (dd, J = 7.7, 8.8 Гц, 1H, H-2); 8.02, (dd, J = 1.0, 7.7 Гц, 1H, H-1); 13.24, 13.51 (2xs, 2H, OH-6 + OH-11). Пример 11 Получение 8-N-(3,4-диметоксибензил)-13-дигидро-антразалона (1k) 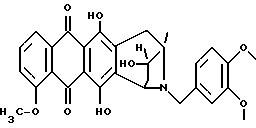 8-N-(3,4-диметоксибензил)-антразалон (500 мг, 1.1 ммоль) растворяли под аргоном в тетрагидрофуране (20 мл) и добавляли MgBr2  OEt2 (1.13 г, 4.4 ммоля) при перемешивании. Смесь охлаждали до -50oC и добавляли NaBH4 (84 мг, 2.2 ммоля) небольшими порциями в течение 10 мин. Затем добавляли метанол (2 мл) и реакционную смесь перемешивали в течение еще одного часа. После этого добавляли ацетон (2 мл) и смесь выливали в охлажденный водный раствор оксаловой кислоты (100 мг в 100 мл воды) и экстрагировали метиленхлоридом. Органическую фазу отделяли, промывали водой и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, а неочищенный материал подвергали флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорид – метанол – уксусная кислота (30:2:1, объем), что дало 320 мг единственного изомера продукта. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид-метанол-уксусная кислота (30:2:1, объем) Rf = 0.5 OEt2 (1.13 г, 4.4 ммоля) при перемешивании. Смесь охлаждали до -50oC и добавляли NaBH4 (84 мг, 2.2 ммоля) небольшими порциями в течение 10 мин. Затем добавляли метанол (2 мл) и реакционную смесь перемешивали в течение еще одного часа. После этого добавляли ацетон (2 мл) и смесь выливали в охлажденный водный раствор оксаловой кислоты (100 мг в 100 мл воды) и экстрагировали метиленхлоридом. Органическую фазу отделяли, промывали водой и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, а неочищенный материал подвергали флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорид – метанол – уксусная кислота (30:2:1, объем), что дало 320 мг единственного изомера продукта. При тонкослойной хроматографии на пластинах Кизельгеля F254 (Merck) в системе элюирования метиленхлорид-метанол-уксусная кислота (30:2:1, объем) Rf = 0.5FAB-MS: m/z 532 [M+H]+; m/z 382 [M+2H-CH2C6H3(OCH3)2]+. 1H ЯМР (600 МГц (DMSO-d6) d: 1.57 (m, 1H, H-8); 1.70 (s, 3H, CH3); 2.74 (m, 1H, H-8); 2.98 (d, 1H, J = 19.0 Гц, H-10); 3.40 (d, 1H, J = 19.0 Гц, H-10); 3.64 (s, 3H, OCH3); 3.74 (s, 3H, OCH3); 3.84 (m, 1H, CH(H)-Ph); 3.99 (s, 3H, OCH3); 4.33 (m, 1H, H-9); 4.42 (m, 1H, CH(H)-Ph); 4.53 (m, 1H, H-7); 5.95 (s, 1H, OH-9); 6.77 (m, 1H, водород ароматического кольца); 6.92 (m, 1H, водород ароматического кольца); 6.94 (m, 1H, водород ароматического кольца); 7.69 (m, 1H, водород ароматического кольца); 7.94 (m, 2H, водороды ароматического кольца); 11.09 (размытый сигнал, 1H, NH+), 13.03 (s, 1H, OH), 13.56 (s, 1H, OH). Пример 12 Получение 8-N-(пиридинметил)-13-антразалона оксима (1l) 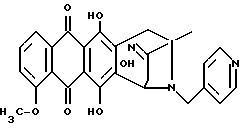 8-N-(пиридинметил)-13-антразалон (1c, 210 мг, 0.5 ммоля) растворяли в EtOH (10 мл) и обрабатывали гидроксиламингидрохлоридом (59.5 мг, 0.85 ммоля) и тригидратом ацетата натрия (66 мг, 0.5 ммоля), растворенным в 0.25 мл воды. Реакционную смесь нагревали при перемешивании с обратным холодильником 2 часа, выливали в воду и экстрагировали метиленхлоридом. Органическую фазу отделяли, промывали водой и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, а сырой продукт подвергали флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорид – ацетон (8:2, объем), что дало 120 мг смеси оксимов. При тонкослойной хроматографии на пластинах Кизельгеля F234 (Merck) в системе элюирования метиленхлорид – ацетон (8:2, объем) Rf = 0.44 и 0.36. FAB-MS (+): m/z 486 [M+H]+; m/z 470 {M+2H-O]+; m/z 468 [M+H-H2O]+ 1H-ЯМР (400 МГц DMSO-d6) d: 1.40 (s, 3H, CH3); 2.52 (d, 1H, J = 17.2 Гц, H-8a); 2.57 (d, 1H, J = 18.8 Гц, H-10); 2.89 (dd, 1H, J = 17.2 и 6.8 Гц, H-8b); 2.97 (d, 1H, J = 18.8 Гц, H-10); 3.63 (d, 1H, J = 18.0 Гц, CH(H)-Ph); 3.96 (s, 3H, OCH3); 4.22 (d, 1H, J = 18.0 Гц, CH(H)-Ph); 4.52 (d, 1H, J = 6.8 Гц, H-7); 7.65 (dd, 1H, J = 6.8 и 2.9 Гц, H-3); 7.91 (m, 2H, H-l + H-2): 7.95 (m, 2H, H-3′ + H-5′); 8.78 (m, 2H, H-2′ + H-6′); 10.77 (s, 1H, =N-OH), 13.09 (s, 1H, OH), 13.53 (s, 1H, OH). Пример 13 Получение 8-N-(4-метоксибензил)-антразалона (соединение 1m) 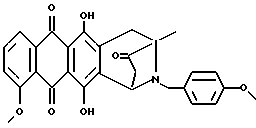 Искомое соединение 1m получали путем взаимодействия даунорубицина (2a, 1,58 г, 3 ммоль) с 4-метоксибензиламином (1,65 г, 12 ммоль), как описано в примере 1. Неочищенный продукт подвергли флэш-хроматографии на силикагеле с использованием смеси этилацетата-циклогексана (3:1 объем) в качестве элюирующей системы, до получения искомого соединения 1m (0,25 г). Соединение растворяли в дихлорметане, добавляли раствор газообразной соляной кислоты в метаноле и соответствующий гидрохлорид осаждали диэтиловым эфиром: температура плавления 225-227oC. Тонкослойная хроматография на пластине кизельгеля F254 (Merck), элюирующая система – хлороформ-метанол (9:1 объем), Rf = 0,74. Пример 14 Получение 8-N-метил-антразалона (соединение 1n) 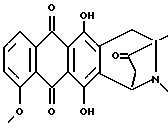 Искомое соединение 1n получали из антразалона 1b. В раствор антразалона 1b (0,4 г, 0,79 ммоль) в 8 мл безводного диметилформамида добавляли метилиодид (0,136 мл, 1,58 ммоль) и N-этил-N-диизопропиламин (0,36 мл, 1,58 ммоль). После перемешивания при комнатной температуре в течение 24 часов реакционную смесь выливали в 100 мл воды и экстрагировали этилацетатом. Органическую фазу отделяли, промывали водой, высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле, используя смесь хлороформа и метанола (50:1 объем) в качестве элюирующей системы, получая 0,141 г искомого соединения 1n, температура плавления 244-246oC (разл.). Тонкослойная хроматография на пластине кизельгеля F254 (Merck), элюирующая система – хлороформ-метанол (49:1 объем), Rf = 0,35. Пример 15 Получение 8-N-пропил-антразалона (соединение 1o) 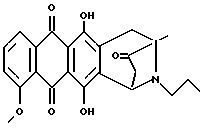 Искомое соединение 1o получали, используя в качестве исходных веществ антразалон 1b (0,4 г, 0,79 ммоль), иодопропан (0,30 мл, 3,16 ммоль), и N-этил-N-диизопропиламин (0,54 мл 3,16 ммоль) в 8 мл безводного диметилформамида. Реакционную смесь нагревали до 80oC в течение 16 часов, выливали в 100 мл воды и экстрагировали этилацетатом. Органическую фазу отделяли, промывали водой, высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле, используя смесь хлороформа и метанола (100:1 объем) в качестве элюирующей системы и получая 0,230 г искомого соединения 1o, температура плавления 188-189oC (разл. ). Тонкослойная хроматография на пластине кизельгеля F254 (Merck), элюирующая система – хлороформ-метанол (100:1 объем), Rf = 0,41. Пример 16 Получение 8-N-фенэтил-антразалона (соединение 1p) 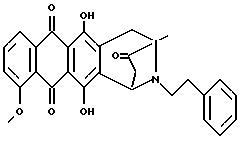 Искомое соединение 1p получали, используя в качестве исходных веществ антразалон 1b (0,4 г, 0,79 ммоль), 2-бромо-1-фенилэтан (0,56 мл, 4,2 ммоль), N-этил-N-диизопропиламин (0,71 мл, 4,2 ммоль) и иодид калия (0,697 г, 4,2 ммоль) в 8 мл безводного диметилформамида. Реакционную смесь нагревали до 80oC в течение 24 часов, выливали в 100 мл воды и экстрагировали этилацетатом. Органическую фазу отделяли, промывали водой, высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле, используя смесь хлороформа и метанола (100:1 объем) в качестве элюирующей системы, и получая 0,220 г неочищенного продукта, который очищали с применением колоночной хроматографии, используя смесь гексан-этилацетат (6:4 объем) в качестве элюирующей системы. Искомое соединение 1p получали в виде твердого вещества красного цвета, температура плавления 118-120oC. Тонкослойная хроматография на пластине кизельгеля F254 (Merck), элюирующая система – хлороформ-метанол (100:1 объем), Rf = 0,26. Пример 17 Получение 8-N-никотиноил-антразалона (соединение 1q) 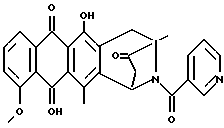 Искомое соединение 1q получали, используя в качестве исходных веществ антразалон 1b (0,4 г, 0,79 ммоль), никотиноилхлорида гидрохлорид (0,56 мл, 3,16 ммоль), и N-этил-N-диизопропиламин (0,54 мл, 3,16 ммоль) в 20 мл безводного дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, выливали в 100 мл воды и экстрагировали этилацетатом. Органическую фазу отделяли, промывали водой, высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле, используя смесь хлороформа и метанола (49:1 объем) в качестве элюирующей системы, получая 0,222 г искомого соединения 1q, температура плавления 173-175oC (разл.). Тонкослойная хроматография на пластине кизельгеля F254 (Merck), элюирующая система – хлороформ-метанол (49:1 объем), Rf = 0,22. Пример 18 Получение 8-N-изоникотиноил-антразалона (соединение 1r) 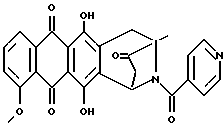 Искомое соединение 1r получали, используя в качестве исходных веществ антразалон 1b (0,5 г, 1,31 ммоль) изоникотиноилхлорида гидрохлорид (0,34 мл, 1,9 ммоль), и N-этил-N-диизопропиламин (0,65 мл, 3,8 ммоль) в 20 мл безводного дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, выливали в 100 мл воды и экстрагировали этилацетатом. Органическую фазу отделяли, промывали водой, высушивали над безводным сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле, используя смесь этилацетата, гексана и метанола (80: 20: 2 объем) в качестве элюирующей системы, и получая 0,146 г искомого соединения 1r, температура плавления 175-177oC (разл.). Тонкослойная хроматография на пластине кизельгеля F254 (Merck), элюирующая система – этилацетат и гексан (9:1 объем), Rf = 0,13. Пример 19 (см. табл. 1) Процедура изготовления капсул: Этап 1: Смешать ингредиенты N 1 и N 2 в подходящем смесителе. Этап 2: Пропустить смесь, полученную на этапе 1, через сетчатый фильтр N 10 меш (0,59 мм). Этап 3: Поместить смесь, пропущенную через сетчатый фильтр, в подходящий смеситель с ингредиентом N 3 и перемешивать до тех пор, пока смесь не пластифицируется. Этап 4: Заполнить смесью твердые желатиновые капсулы N 1 в машине для изготовления лекарств в капсулах. Пример 20 (см. табл. 2) Процедура изготовления таблеток: Этап 1: Смешать ингредиенты N 1, N 2, N 3 и N 4 в подходящем смесителе. Этап 2: Порциями добавить достаточное количество воды в смесь, полученную на этапе 1, тщательно перемешивая после добавки каждой порции. Повторять добавку воды и перемешивание до тех пор, пока не будет получена масса такой консистенции, чтобы из нее можно было получить влажные гранулы. Этап 3: Из влажной массы получают гранулы, пропуская ее через вибрирующий гранулятор, используя сетчатый фильтр N 8 меш (2,38). Этап 4: Затем влажные гранулы досуха высушивают в печи при 140oF (60oC). Этап 5: Сухие гранулы смазывают ингредиентом N 5. Этап 6: Смазанные гранулы прессуют в таблетки в подходящем прессе для изготовления таблеток. Биологический тест Производные антразалинона формулы 1 препятствуют способности фрагмента 25-35 -амилоида и фрагмента 106-126 PrP к самоагрегации, что показано анализом светорассеяния. 25-35 (GSNKGAIIGLH) и PrP 106-126 (KTNMKHMAGAAAAGAVVGGLG) получали химическим синтезом на твердофазном носителе с применением 430A Applied Biosystems Instruments и очищали высокоэффективной жидкостной хроматографией (ВЭЖХ) с обращенной фазой (Beckman Inst. mod. 243) по Forloni et al., Nature 362: 543, 1993.
Светорассеяние пептидных растворов оценивали спектрофлуорометрически (Perkin Elmer LS 50B), возбуждение и эмиссию определяли при 600 нм.
Фрагмент 25-35 -амилоида и фрагмент 106-126 PrP, разведенные до концентрации от 0.5 до 1.0 мг/мл (0.4-0.8 мМ и 0.2-0.4 мМ соответственно) в 10 мМ фосфатном буфере, pH 5, спонтанно агрегировали в течение часа.
8-N-пиридинметилен-антразалон (1c), разведенный до концентрации в интервале от 0.2 до 2 мМ в 5 мМ Трис-буфере, pH 7.4, добавляли к пептидным растворам в момент их приготовления, чтобы оценить развитие процесса образования фибрилл.
Соединение 1c, добавленное в эквимолярной концентрации по отношению к фрагменту 25-35 -амилоида и фрагменту 106-126 PrP, вызывало полное предотвращение агрегации.
Определение тиофлавина TМаточный раствор A 25-35 пептида готовили растворением лиофилизированного пептида в диметилсульфоксиде (ДМСО) до концентрации 7.07 мг/мл.
Аликвоты данного раствора растворяли в 50 мМ фосфатном буфере, pH 5 до получения конечной концентрации пептида 100 мМ и инкубировали в течение 24 часов при 25oC с или без тестируемого соединения в концентрации 30 мМ и конечном объеме 113 мл. Соединения предварительно растворяли в ДМСО до концентрации 3.39 мМ, конечный % объем ДМСО (объем/объем) в инкубационных смесях составлял менее 3%.
Измерения флуоресценции выполняли по Naiki et al., Anal. Biochem. 177, 244, 1989 и по H.LeVine III, Protein Sci. 2, 404, 1993. В кратком виде образцы инкубата разводили до концентрации пептида 8 мг/мл в 50 мМ натрийцитратном буфере, pH 5, содержащем 47 мМ тиофлавина T (ThT) до конечного объема 1.5 мл. Флуоресценцию измеряли при длине волны возбуждающего излучения 420 нм и при длине волны светоэмиссии 490 нм в Kontron флуоресцентном спектрофотометре и полученные величины усредняли после вычитания фоновой флуоресценции 47 мМ ТФТ.
Результаты выражали в виде относительной флуоресценции, то есть в процентах от флуоресценции, определяемой при инкубации одного A 25-35 пептида (контроль).
В табл. 3 представлены результаты, полученные для некоторых соединений.
Формула изобретения
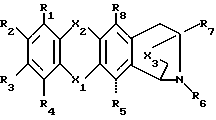 где Х1 и Х2 означают С=О; Х3 выбран из С=О, СНОН или С=N(R9), где R9 представляет собой гидрокси; R1, R2 и R3 означают водород; R4 означает водород или С1-6 алкоксил; R5 и R8 означают гидроксил; R6 выбран из водорода, Rв-СН2-, где Rв означает фенил, незамещенный или замещенный С1-6 алкоксилом, пиридинил или группу формулы Rc-CH=CH-, где Rс означает водород; С1-12 алкила, ацила формулы -С(R11)=О, где R11 – пиридил; R7 означает метил, или их фармацевтически пригодные соли. 2. Соединение по п.1, в котором R4 означает водород или С1-4 алкоксил, или его фармацевтически приемлемая соль. 3. Соединение по п.1, в котором Х3 выбран из С=О или СНОН; R4 означает водород или метоксигруппу; R6 выбран из водорода, бензила, аллила, 3,4-диметоксибензила, пиридинметила и никотила, или его фармацевтически приемлемая соль. 4. Соединение по п.1, в котором Х3 является С=О; R4 означает водород или метоксигруппу; R6 выбран из водорода, пиридинметила и никотила, или его фармацевтически приемлемая соль. 5. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении амилоидоза, отличающаяся тем, что включает в качестве активного компонента эффективное количество соединения формулы 1 или его фармацевтически приемлемой соли, как указано в любом из пп.1 – 4, в смеси с фармацевтически приемлемым носителем или разбавителем. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 24.07.2007
Извещение опубликовано: 10.08.2010 БИ: 22/2010
|
||||||||||||||||||||||||||