Патент на изобретение №2159243
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТИАТЕРОСКЛЕРОТИЧЕСКОЙ ИЛИ ГИПОХОЛЕСТЕРИНЕМИЧЕСКОЙ АКТИВНОСТЬЮ
(57) Реферат: Изобретение относится к новым производным азетидинона общей формулы (I), в которой R, R1, Ar1 -Ar3, X, Y, m, n, q и r имеют указанные в формуле изобретения значения, и их фармацевтически приемлемым солям, которые представляют собой активное вещество фармацевтической композиции с антиатеросклеротической или гипохолестеринемической активностью. 2 с. и 5 з.п. ф-лы, 2 табл. 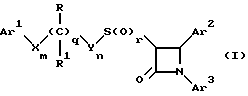
Изобретение относится к новым  -лактамам, обладающим биологической активностью, в частности к производным азетидинона и их фармацевтически приемлемым солям азетидинона, а также к фармацевтической композиции с антиатеросклеротической или гипохолестеринемической активностью.
Известны -лактамы, которые можно применять в качестве гипохолестеринемического средства (см. заявку WO 94/14433, кл. A 61 K 31/395, 07.07.1994 г.).
Задачей изобретения является расширение арсенала производных -лактама, т.е. 2-азетидинона, которые обладают гипохолестеринемической активностью.
Поставленная задача решается предлагаемыми производными азетидинона общей формулы (I) -лактамам, обладающим биологической активностью, в частности к производным азетидинона и их фармацевтически приемлемым солям азетидинона, а также к фармацевтической композиции с антиатеросклеротической или гипохолестеринемической активностью.
Известны -лактамы, которые можно применять в качестве гипохолестеринемического средства (см. заявку WO 94/14433, кл. A 61 K 31/395, 07.07.1994 г.).
Задачей изобретения является расширение арсенала производных -лактама, т.е. 2-азетидинона, которые обладают гипохолестеринемической активностью.
Поставленная задача решается предлагаемыми производными азетидинона общей формулы (I)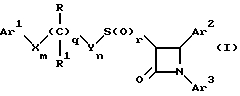 где Ar1 – фенил, незамещенный или замещенный галогеном или 5- или 6-членный гетероароматический радикал, содержащий азот или серу, Ar2 – фенил, замещенный низшим алкилом, группой -OR2 или -O(CO)R2, где R2 означает атом водорода или низший алкил, Ar3 – фенил, незамещенный или замещенный галогеном, X и Y независимо друг от друга означают -CH2-, R – группа -OR3, где R3 означает атом водорода или низший алкил, R1 – атом водорода, или R и R1 вместе означают атом кислорода карбонильной группы (=O), q = 0 или 1, r = 0, 1 или 2, m и n независимо друг от друга имеют значение 0, 1, 2, 3, 4 или 5 при том условии, что m + n + q = 2, 3, 4 или 5. В первую группу предпочтительных производных азетидинона формулы (I) входят соединения, у которых Ar1 – фенил, незамещенный или замещенный галогеном или тиенил; Ar2 – фенил, замещенный гидроксилом, а Ar3 – фенил, замещенный фтором. Во вторую группу предпочтительных производных азетидинона формулы (I) входят соединения, у которых q = 0, Х и Y означают каждый -CH2-, а m + n = 2, 3 или 4. В третью группу предпочтительных производных азетидинона формулы (I) входят соединения, у которых q = 1, Х и Y означают каждый -CH2-, m + n = 1, 2 или 3, R – группа -OR3, где R3 – атом водорода, или R и R1 вместе – атом кислорода карбонильной группы (=O). В частности предпочитаются производные азетидинона формулы (I), выбранные из группы, включающей транс-4-(4-гидроксифенил)-3-[(2-фенилэтил)тио] -1-(4- фторфенил)-2-азетидинон; транс-4-(4-метоксифенил)-1-фенил-3-[(2 фенилэтил)тио]-2-азетидинон; цис-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)тио]-2-азетидинон; транс-4-(4-гидроксифенил)-3-[(2-фенилэтил)сульфинил1-1-(4- фторфенил)] -2-азетидинон: цис-4-(4-гидроксифенил)-3-[(2-фенилэтил)сульфинил]-1-(4- фторфенил)-2-азетидинон; транс-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)сульфинил] – 2-азетидинон; цис-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)сульфинил]- 2-азетидинон; транс-4-[4-кето-3-[(2-фенилэтил)сульфинил] -1-(4-фторфенил)-2- азетидинил]-фенилацетат; цис-4-[4-кето-3-[(2-фенилэтил)сульфинил] -1-(4-фторфенил)-2- азетидинил] -фенилацетат; (+/-)-транс-4-(метоксифенил)-1-фенил-3-[(2-фенилэтил)сульфонил]-2-азетидинон; транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(4- фторфенил)-этил]тио]-2-азетидинон; транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-гидрокси-2- (4-фторфенил)этил]тио]-2-азетидинон; (3R, 4R) 4-(4-гидроксифенил)-1 -(4-фторфенил)-3-[[2-кето-2- (4-фторфенил)-этил]суль-финил]-2-азетидинон; 4(R)-(4-гидроксифенил)-1-(4-фторфенил)-3(R)-[[2(R)-гидрокси- 2-(4-фторфенил)этил]-сульфинил]-2-азетидинон; 4(R)-(4-гидроксифенил)-1-(4-фторфенил)-3(R)-[[2(S)-гидрокси- 2-(4-фторфенил)этил]-сульфинил]-2-азетидинон; (3R,4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето- 2-(2-тиенил)-этил]тио]-2-азетидинон; (3R,4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето- 2-(3-тиенил)-этил]тио]-2-азетидинон; (3R,4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето- 2-(3-пиридинил)этил]-тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2- кето-2-(4-пириди-нил)этил]-тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето- 2-(2-пириди-нил)этил]-тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2- гидрокси-2-(3-тиенил)этил]-тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2- гидрокси-2-(4-пиридинил)-этил]тио]-2-азетидинон; (3S, 4R) цис-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2- (4-фторфенил)этил]-тио]-2-азетидинон: (3S, 4R) цис-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2- гидрокси-2-(4-фторфенил)-этил]тио]-2-азетидинон. Некоторые соединения согласно изобретению являются кислотами (например, соединения, которые содержат карбоксильную группу). Такие соединения образуют фармацевтически приемлемые соли с неорганическими и органическими основаниями. Примерами таких солей являются соли натрия, калия, кальция, алюминия, серебра и золота. Сюда же входят и соли, образованные с фармацевтически приемлемыми аминами, такими как аммиак, алкиламины, гидроксиалкиламины, N-метилглюкамин и тому подобные. Соединения формулы I могут быть получены известными, например, описываемыми далее способами. Способ A: 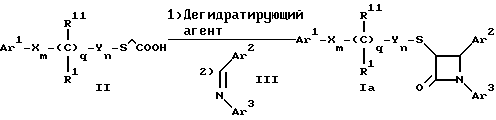 Соединения формулы I, в которой r = 0, R3 означает защищенную гидроксильную группу (варианты таких защищенных гидроксильных групп приведены в таблице 1), а остальные переменные имеют приведенное выше значение, то есть соединения формулы Ia могут быть получены в соответствии с этой схемой реакций в результате взаимодействия карбоновой кислоты формулы II с имином формулы III в присутствии такого основания, как триэтиламин, и соответствующего дегидратирующего агента, например дихлорангидрида диметиламида фосфорной кислоты. Для получения тиосоединения формулы Ia образующееся при этом вещество обрабатывают кислотой, например фтористоводородной кислотой. Если защищенная гидроксильная группа R3 означает алкоксильную группу или бензилоксигруппу, то такая защитная группа не нуждается в снятии для получения соединения формулы I, однако другие защитные группы могут быть сняты с целью получения соединений формулы I, в которой R означает гидроксильную группу, с использованием обычных приемов. Соединения, в которых R означает гидроксильную группу, могут быть превращены хорошо известными способами в другие соединения формулы I, где R означает алкоксил, один из способов сводится к обработке спирта алкилгалогенидом в присутствии подходящего основания, такого как гидрид натрия. Соединения формулы Ia, в которых q = 1, а R и R1 вместе образуют карбонильный атом кислорода (=O), могут быть превращены в соответствующие соединения, в которых R1 означает атом водорода, a R означает гидроксильную группу, действием такого восстановительного реагента как борогидрид натрия. Для получения соответствующих сульфинильных соединений, то есть соединений формулы I, в которых r = 1 и остальные радикалы и индексы имеют вышеприведенные значения (то есть соединений формулы Iб) на тиосоединения формулы Ia с защищенной гидроксильной группой действуют одним эквивалентом такого окислителя, как надкислота, например м-хлорнадбензойная кислота или метапериодат натрия: 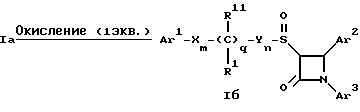 Для получения соответствующих сульфонильных соединений, то есть соединений формулы I, в которых r равно 2 и остальные переменные имеют вышеприведенные значения (то есть соединений формулы Ib) на тиосоединения формулы Ia с защищенной гидроксильной группой действуют двумя эквивалентами вышеприведенного окислителя 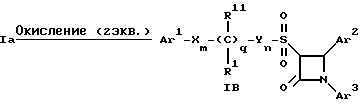 В соединениях Iб и Iв защитная группа в заместителе R11 при необходимости может быть снята с получением соединений формулы I. Способ Б: 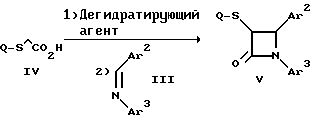 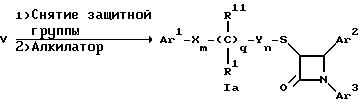 Соединения формулы Ia, в которых переменные имеют определенное выше значение, могут быть получены по реакции защищенной тиогликолевой кислоты формулы IV, где Q означает защитную группу для тиольной функции, например, бензил или замещенный бензил, с имином по описанной в способе А схеме. Защитную группу Q после этого удаляют и алкилируют меркаптогруппу соединением формулы 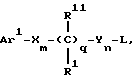 где L означает уходящую группу, такую как атом брома или иода. С использованием описанных в способе А реакций полученные по способу Б соединения формулы Iа могут быть переведены в сульфинильные и сульфонильные соединения, а соединения, в которых R и R1 образуют карбонильный атом кислорода (= O), могут быть превращены в соединения, где R означает атом водорода и R1 означает гидроксильную группу, а соединения, в которых R означает гидроксильную группу, могут быть превращены в соединения, у которых R означает алкоксил. Способ В Соединения формулы I, в которых индекс r = 0 и остальные радикалы и индексы имеют определенное выше значение, могут быть получены энантиоселективным способом по следующей схеме: 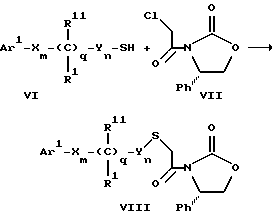 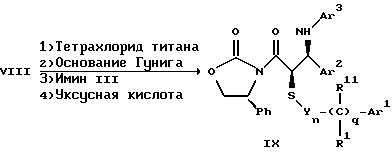 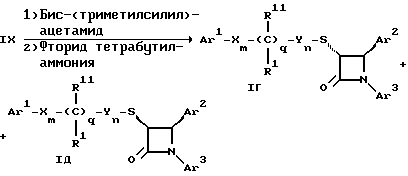 Вспомогательный реагент – хлорацилированный оксазолидинон формулы VII-реагирует с меркаптаном формулы VI, переменные радикалы которого определены ранее, в присутствии такого основания, как триэтиламин, в таком инертном растворителе, как метиленхлорид. Образующееся соединение формулы VIII обрабатывают тетрахлоридом титана в присутствии такого основания, как диизопропилэтиламин (основание Гунига), и вводят в реакцию с имином формулы III, разлагая затем реакционную массу кислотой, например уксусной кислотой. Образующееся соединение формулы IX циклизуют по реакции с таким силилирующим реагентом, как бис(триметилсилил)ацетамид (BSA) и таким фторидным катализатором, как фторид тетрабутиламмония (TBAF). Продукт циклизации разделяют на цис- и транс-изомеры формул Iг и Iд с использованием обычных для этого приемов выделения, например флэш-хроматографии. Соединения формул Iг и Iд могут быть превращены в соответствующие сульфинильные и сульфонильные соединения в результате реакций с надкислотами, как это описано выше, или с таким реагентом, как (R) или (S)(10-камфоросульфонил)- оксазиридин. Например, соединение формулы Iг может быть превращено в соответствующие сульфинильные соединения Iе и Iж по схеме: 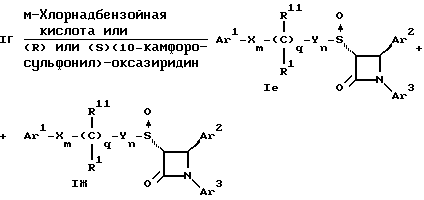 До разделения на цис- и транс-изомеры или после него, как более удобно, в соединениях формул Iг и Iд снимаются защитные группы в радикале R3, если же в соединении R означает ОН, то эта группа может быть функционализирована так, как это описано в способе А. Исходные соединения формул II, III, IV, VI и VII могут быть приобретены по каталогам, они известны специализирующимся в этой области синтетикам или их синтезируют известными способами. Реакционноспособные группы, не участвующие в описанных выше превращениях, могут быть защищены во время реакций обычными защитными группами, которые могут быть удалены после реакции стандартными приемами. Таблица 1 демонстрирует некоторые типичные защитные группы. Как уже указывалось выше, соединения согласно изобретению обладают гипохолестеринемической активностью и поэтому они могут представлять собой активное вещество фармацевтической композиции с антиатеросклеротической и гипохолестеринемической активностью. Данная фармацевтическая композиция, которая помимо эффективного количества активного вещества содержит еще фармацевтически приемлемый носитель, представляет собой дальнейший объект изобретения. Предлагаемая фармацевтическая композиция может также содержать ингибитор биосинтеза холестерина в эффективном количестве. В качестве ингибитора биосинтеза холестерина можно назвать, например, ингибиторы HMG CoA-редуктазы, такие как ловастатин, правастатин, флувастатин, симвастатин и Cl-981; ингибиторы синтетазы гидроксиметилглутарил-кофермента A (HMG CoA-синтетазы), например L-659,699 ((E,E-11-[3’R-(гидроксиметил)-4′-кето-2’R- оксэтанил] -3,5,7R-триметил-2,4-ундекадиеновая кислота); ингибиторы био-синтеза сквалена, например, сквалестатин 1; и ингибиторы сквален-эпоксидазы, например NB-598 (гидрохлорид (Е)-N-этил-N-(6,6-диметил-2-гептен-4-инил)-3-[(3,3′-битиофен-5-ил)метокси] бензол-метанамина), и другие ингибиторы биосинтеза холестерина, такие как DMP-565. Предпочтительными ингибиторами HMG CoA-редуктазы являются ловастатин, правастатин и симвастатин. Активность in vivo соединений формулы I подтверждается следующим опытом: Гиполипидемические действующие начала в опыте in vivo на гиперлипидемических хомячках Хомячков разделяют на группы по шесть животных и дают им корм с контролируемым содержанием холестерина (PurinaChow # 5001, содержание холестерина 0,5%) в течение семи дней. Количество потребляемого корма фиксируют для определения поступления холестерина на фоне действия исследуемых соединений. Животные получают дозы иследуемых соединений один раз в сутки, одновременно с началом кормления вышеуказанным кормом. Пероральная дозировка составляет 0,2 мл растительного масла в чистом виде (для контрольной группы подопытных животных) или раствора (или суспензии) исследуемых соединений в растительном масле. Всех животных в болезненном или в плохом физическом состоянии усыпляют. Через семь дней подопытным животным делают внутримышечную инъекцию кетамина и в наркотическом состоянии забивают их обезглавливанием. Кровь отбирают в вакуумные приемные пробирки, содержащие этилендиаминтетраацетат (ЭДТА), для анализа липидов плазмы, а печень извлекают резекцией для анализа липидов в тканях. Анализ на липиды проводится по опубликованной методике (Schnitzer-Polokoff, R. И др. , Сотр. Biochem. Physiolog., 99А, 4 (1991), стр. 665-670). Данные опытов приводятся в процентах снижения содержания липидов по отношению к контролю. Результаты опыта сведены в таблице 2. Экспериментальные данные представляют собой процент изменения (то есть процент снижения содержания эфиров холестерина) по отношению к контролю. Из этого следует, что отрицательные значения чисел отражают положительный эффект снижения уровня липидов. В случае рацемических соединений формулы I, а также активных диастереомеров или энантиомеров соединений формулы I, введение их в дозах 0,1-25 мг/кг сопровождается снижением уровня сложных эфиров холестерина в области от -21 до -97% и снижением уровня холестерина в сыворотке от -57 до 0%. Соединения показывают предпочтительно снижение уровня сложных эфиров холестерина в диапазоне от -21 до -97% при дозировке от 0,1 до 3 мг/кг, а более предпочтительно снижение уровня сложных эфиров холестерина в диапазоне от -21 до -97% при дозировке от 0,1 до 1 мг/кг. В рамках вышеописанного опыта исследовалась активность соединения примера N 5 и ловастатина в качестве ингибитора биосинтеза холестерина при раздельной и совместной аппликации. Животным давали имеющийся в торговле корм под названием Purina Chow N 5006, содержащий мальтодекстрин, к которому добавляли либо соединение примера N 5 в дозе 0,01 мг/кг/день, либо ловастатин в дозе 5 мг/кг/день, либо соединение примера N 5 и ловастатин в указанных дозах. Опыты проводились в течение 14 дней, при этом кровь бралась до начала опытов и в 3-й, 11-й и 14-й день. При этом 10 мкл плазмы брали для определения в ней общего содержания холестерина, для чего применяли 1 мл 0,15 М Трис- буфера, pH 7,0, содержащего 0,1% п-хлорфенола, 0,13 ед/мл оксидазы холестерина, 0,13 ед/мл гидролазы сложных эфиров холестерина, 2,4 ед/мл пероксидазы и 0,015% 4-аминоантипиридина. Анализы проводились при 37oC в течение 10 мин. При даче соединения примера N 5 общее содержание холестерина в плазме крови по сравнению с контрольным практически не снизилось, при даче ловастатин наблюдалось снижение примерно на 10%, тогда как при совместной даче соединения примера N 5 и ловастатина снижение общего содержания холестерина в плазме крови составляло порядка 30% по сравнению с контрольным. Суточная гипохолестеринемическая доза соединения формулы I составляет от примерно 0,1 до примерно 30 мг/кг веса тела в день, предпочтительно от примерно 0,1 до примерно 15 мг/кг. Для среднего веса тела 70 кг уровень дозы составляет таким образом от примерно 5 до примерно 1000 мг лекарства в день в виде разовой дозы или разделенных на 2-4 дозы. Однако точная доза определяется наблюдающим клиницистом и зависит от эффективности вводимого соединения, возраста, веса, состояния и ответной реакции пациента. Для комбинаций в рамках настоящего изобретения, когда азетидинон с серусодержащим заместителем вводят в сочетании с ингибитором биосинтеза холестерина, типичная суточная доза ингибитора биосинтеза холестерина составляет от 0,1 до 80 мг/кг веса млекопитающего в день, причем ее вводят в один или в несколько приемов, обычно один или два раза в день: например, в случае ингибиторов редуктазы гидроксиметилглутарилкофермента А от примерно 10 до примерно 40 мг на дозу вводят один или два раза в день, что составляет общую суточную дозу около 10-80 мг в день, а для других ингибиторов биосинтеза холестерина около 1-1000 мг на дозу для приема один-два раза в день, что составляет общую суточную дозу от примерно 1 мг до примерно 2000 мг в день. Точная доза каждого компонента вводимой комбинации определяется наблюдающим клиницистом и зависит от эффективности вводимого соединения, возраста, веса, состояния и ответной реакции пациента. Если компоненты комбинации вводят раздельно, то число доз каждого принимаемого в день компонента не обязательно будет одинаковым, например, когда один компонент имеет более пролонгированное действие, его прием должен быть менее частым. Так как настоящее изобретение относится к снижению уровня холестерина в плазме за счет лечения с помощью комбинации активных ингредиентов и эти активные ингредиенты могут вводиться раздельно, изобретение также относится к сочетанию разделенных фармацевтических композиций в одном комплекте. То есть такой комплект рассматривается как объединение двух отдельных составных частей: фармацевтической композиции ингибитора биосинтеза холестерина и фармацевтической композиции азетидинона с серусодержащим заместителем в качестве ингибитора его абсорбции. Комплект предпочтительно включает указания по приему отдельных компонентов. Такая комбинированная форма особенно предпочтительна в тех случаях, когда отдельные компоненты должны приниматься в разных лекарственных формах, (например, орально и парентерально) или когда дозировки принимаются с различными интервалами. Получение производных азетидинона формулы (I) иллюстрируется следующими примерами. Встречающиеся в них понятия цис и транс представляют собой относительные ориентации в положениях 3 и 4 азетидинона, если не указано ничего другого. Индекс “J” относится к константе спин-спинового взаимодействия в спектре протонного магнитного резонанса в герцах (Гц) для протонов в 3- и 4- положениях замещенного азетидинона. Спектры кругового дихроизма определяли в метанольном растворе. Пример 1 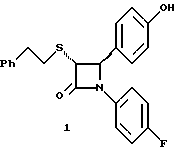 Первая стадия. В 1,2 л толуола кипятят с обратным холодильником, соединенным через ловушку Дина-Старка, смесь 128 мл 4-фторанилина и 290 г 4-трет. -бутилдиметилсилоксибензальдегида. Через 24 часа упаривают в вакууме и растворяют остаток в 0,2 л горячего гексана. Раствор охлаждают до -20oC и отделяют выпавший имин фильтрованием, получают 378 г (94% от теории), т.пл. 51,4- 52,2oC. Вторая стадия. К смеси 0,55 г фенилэтилмеркаптоуксусной кислоты (получена в две стадии: а) взаимодействием фенилэтилмеркаптана с этиловым эфиром бромуксусной кислоты и б) омылением водно-спиртовым раствором гидроксида натрия), полученного на первой стадии имина и 1,2 мл триэтиламина в 20 мл метиленхлорида при температуре 0oC прибавляют дихлорангидрид диметиламидофосфорной кислоты. Оставляют при перемешивании на ночь, позволяя реакционной массе нагреться до комнатной температуры. К смеси добавляют этилацетат и 10%-ный раствор бикарбоната натрия. Промывают водой, сушат над сульфатом магния и упаривают органический слой, затем очищают остаток с помощью флэш- хроматографии на силикагеле смесью гексана и этилацетата (20:1), получают 0,48 г (34% от теории) желтого масла. Составляющие масла разделяют с помощью ВЭЖХ на колонке с хиральной фазой (Chiralcet OD), элюент: гексан/изопропанол (66:1), отбирая второй пик. Третья стадия. К раствору 215 мг полученного на второй стадии вещества в 21 мл ацетонитрила при 0oC добавляют 2,5 мл 48%-ной фтористоводородной кислоты и оставляют при перемешивании на ночь, позволяя реакционной массе нагреться до комнатной температуры. К реакционной массе добавляют серный эфир и холодную воду, промывают органический слой 10%-ным раствором бикарбоната натрия и водой. Органический слой сушат над сульфатом магния и концентрируют. Продукт выделяют флэш-хроматографией на силикагеле, элюируя смесью гексана с этилацетатом (5:1). Целевое соединение 1 получают в виде бесцветного твердого вещества (0,16 г, 96% от теории). Масс-спектр с поверхностной ионизацией 394 (М+Н), 256 (100%). Элементный анализ: вычислено в %: C 69,41, H 5,19, N 3,52. C23H20NO2SF  0,25H2O. Найдено %: C 69,42, H 5,26, N 3,45.
[ 0,25H2O. Найдено %: C 69,42, H 5,26, N 3,45.
[ ]2D5 +44,8o (1,25 мг/мл в метаноле).
1H ЯМР в дейтерохлороформе: 2,95 (м., 4H), 3,93 (д., J=2,4 Гц, 1H), 4,67 (д. , J= 2,4 Гц, 1H), 5,06 (с., 1H), 6,85 (д., 1H), 6,92 (дд. 2H), 7,15-7,3 (9H).
Способ Б ]2D5 +44,8o (1,25 мг/мл в метаноле).
1H ЯМР в дейтерохлороформе: 2,95 (м., 4H), 3,93 (д., J=2,4 Гц, 1H), 4,67 (д. , J= 2,4 Гц, 1H), 5,06 (с., 1H), 6,85 (д., 1H), 6,92 (дд. 2H), 7,15-7,3 (9H).
Способ Б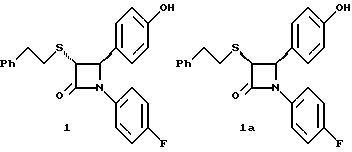 Первая стадия. Раствор 9,76 мл хлорацетилхлорида в 110 мл метиленхлорида при температуре 0oC добавляют к раствору 10,0 г (S)-4-фенилоксазолидинона, 35 мл триэтиламина и 0,5 г диметиламинопиридина в 150 мл метиленхлорида. Постепенно нагревают до комнатной температуры, затем добавляют около 50 г силикагеля и упаривают в вакууме. Остаток очищают флэш-хроматографией на силикагеле, элюируя смесью этилацетат/гексан (1:4). Получают твердое бесцветное вещество, выход 11,3 г (77%). Вторая стадия. К раствору 6,0 г полученного на первой стадии продукта и 5,1 мл триэтиламина в 0,1 л метиленхлорида добавляют фенилэтилмеркаптан. При комнатной температуре перемешивают в течение 16 часов, затем добавляют около 50 г силикагеля и упаривают в вакууме. Остаток очищают флэш-хроматографией на силикагеле, элюируя смесью этилацетат/гексан (1:4). Получают твердое бесцветное вещество, выход 7,81 г (92%), которое может быть перекристаллизовано из смеси этилацетат/гексан (1:4). Третья стадия. К раствору тетрахлорида титана в 200 мл метиленхлорида (получен разбавлением 75 мл 1н раствора тетрахлорида титана в метиленхлориде) при перемешивании добавляют 7,5 мл тетраизопропоксида титана, поддерживая температуру 0oC. Через 15 минут добавляют 34,1 г полученного на второй стадии вещества и еще через 5 минут добавляют 66 г полученного на первой стадии способа А имина. Реакционную массу охлаждают до -40oC, выдерживают 20 минут и добавляют 35 мл диизопропилэтиламина. Выдерживают 15 часов при температуре -40oC, охлаждают реакционную массу до температуры -70oC и добавляют 250 мл изопропилового спирта. Постепенно в течение 6 часов нагревают реакционную массу до комнатной температуры, затем добавляют 500 мл 0,1 н соляной кислоты и серный эфир. Промывают органический слой водой, сушат над сульфатом магния, упаривают и очищают остаток кристаллизацией из метанола. Получают бесцветное твердое вещество, выход 30,9 г (46% от теории). Четвертая стадия. Раствор 10 г полученного на третьей стадии продукта в 0,5 л толуола нагревают до 90oC и добавляют 7,4 мл N,O- бис(триметилсилил)ацетамида. Через 1 час реакционную массу охлаждают до 45oC и добавляют 0,47 г фторида тетрабутиламмония. В течение последующих 18 часов продолжают перемешивание при 45oC, время от времени добавляя еще бис(три-метилсилил)-ацетамид (в сумме 3 молярных эквивалента). Когда общее время реакции составит 24 часа, добавляют 150 мл метанола и перемешивают при комнатной температуре 1 час. Реакционную массу упаривают в вакууме и очищают флэш-хроматографией на силикагеле, элюируя транс-изомер. Продолжая элюирование смесью гексан/этилацетат (5:1) получают цис-изомер. Пятая стадия. Раздельно обрабатывают растворы полученных на четвертой стадии транс- и цис-изомеров в ацетонитриле водной фтористоводородной кислотой в соответствии с операцией на третьей стадии способа А. Получают соответственно транс- и цис-азетидиноны 1 и 1a. 1a: Масс-спектр с химической ионизацией 394 (М+Н), 100%. Элементный анализ: рассчитано для C23H20NO2 SF%: C 70,21, H 5,13, N 3,56, S 8,15; найдено: C 70,33, H 5,43, N 3,71, S 8,20. 1Н ЯМР в дейтерохлороформе: 2,78 (м., 4H), 4,52 (д., J=5 Гц, 1H), 5,23 (д., J=5 Гц, 1H), 6,82-7,3 (13H). При использовании 4-метоксибензилиденанизидина в описанных на третьей и четвертой стадиях способа Б по примеру 1 операциях получают следующие 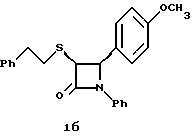 1б: Элементный анализ: рассчитано для C24H23NO2S%: C 74,01, H 5,95, N 3,6, S 8,22; найдено: C 74,19, H 6,0, N 3,73, S 8,03. [  ] 232 нМ = +3,4104, [] 248 нМ = -3,07104.
1H ЯМР в дейтерохлороформе: 2,95 (м., 4H), 3,82 (с., 3H), 3,95 (д., J= 2,2 Гц, 1H), 4,72 (д., J=2,2 Гц, 1H), 6,9-7,3 (14H).
Масс-спектр с поверхностной ионизацией: 390(М+Н), 252 (100%). ] 232 нМ = +3,4104, [] 248 нМ = -3,07104.
1H ЯМР в дейтерохлороформе: 2,95 (м., 4H), 3,82 (с., 3H), 3,95 (д., J= 2,2 Гц, 1H), 4,72 (д., J=2,2 Гц, 1H), 6,9-7,3 (14H).
Масс-спектр с поверхностной ионизацией: 390(М+Н), 252 (100%).
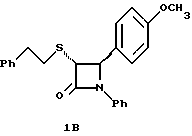 1H ЯМР в дейтерохлороформе: 2,78 (м., 4H), 3,8 (с., 3H), 4,53 (д., J=5,5 Гц, 1H), 5,27 (д., J=5,5 Гц, 1H), 6,9-7,3 (14H). Пример 2 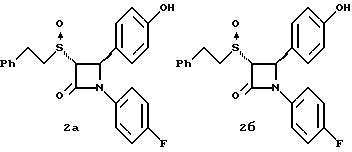 Раствор 2,3 г полученного по примеру 1 соединения 1 и 1,48 г (1s)-(+)-(10-камфоросульфонил)оксазиридина в 40 мл тетрагидрофурана кипятят с обратным холодильником. Через 14 часов реакционную массу упаривают и очищают остаток с помощью флэш-хроматографии на силикагеле, используя для элюирования первого диастереомера 2a смесь метиленхлорид/изопропиловый спирт (100: 1), выход 0,6 г (25%). Элементный анализ: рассчитано для C23H20NO3SF%: C 67,47, H 4,92, N 3,42; найдено: C 67,12, H 5,02, N 3,43. [ ] 219 нМ = -5,49104, [] 254 нМ = +5,2 104[ ]2D5 +214,4o (1,25 мг/мл в метаноле).
1H ЯМР в дейтерохлороформе: 3,15 (м., 3H), 3,92 (м., 2H), 5,25 (д., J= 2,5 Гц, 1H), 6,0 (ш., 1H), 6,8-6,9 (4H), 7,15-7,35 (8H).
Масс-спектр с химической ионизацией 410 (М+Н).
Затем элюируют диастереомер 2б и кристаллизуют диастереомер 2б из изопропилового спирта. Получают 1,48 г (62%) бесцветного твердого вещества.
Элементный анализ: рассчитано для C23H20NO3SF%: C 67,47, H 4,92, N 3,42;найдено: C 67,28, H 4,89, N 3,45. [ ] 233 нМ = +5,56104, [] 251 нМ = -2,79 104.
[]2D5 -16o (1,25 мг/мл в метаноле).
1H ЯМР в дейтерохлроформе: 3,1-3,14 (м., 4h), 4,2 (д., J=2 Гц, 1H), 5,39 (д., J=2 Гц, 1H), 6,7 (д., 2H), 6,95 (м., 2H), 7,15- 7,35 (8H).
Масс-спектр с химической ионизацией 410 (М+Н).
Применив методику способа по примеру 2 на полученное в примере 1 соединение 1a получают следующие соединения: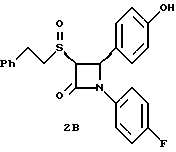 2в: Элементный анализ: рассчитано для C23H20 NO3SF%: C 67,47 H 4,92, N 3,42 S 7,83; найдено: C 67,21, H 5,0, N 3,5, S 7,48. 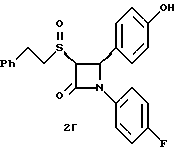 2г: Элементный анализ: рассчитано для C23H20NO3SF%: C 67,47, H 4,92, N 3,42 S 7,83; найдено: C 67,5, H 5,11, N 3,6, S 7,71. 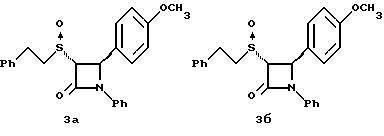 Раствор 0,36 г полученного в примере 1 соединения 1б, в 15 мл метиленхлорида при 0oC обрабатывают м-хлорнадбензойной кислотой (0,16 г) при -78oC. Через 2 часа добавляют разбавленный бисульфит натрия и нагревают реакционную массу до комнатной температуры. Добавляют этилацетат и последовательно промывают органический слой 10%-ным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, затем сушат над сульфатом магния и упаривают в вакууме. Остаток очищают с помощью ВЭЖХ на силикагеле, элюируя смесью этилацетат/гексан (1:2) соединения 3а, выход 0,185 г, и 3б, выход 0,10 г. 3а: Элементный анализ: рассчитано для C24H23NO3S%: C 71,09, H 5,72, N 3,45; найдено: C 70,87, H 5,55, N 3,52. [ ] 220 нМ = -5,36104, [] 257 нМ = +5,46104.
1H ЯМР в дейтерохлороформе: 3,15 (м., 3H), 3,8 (с., 3H), 3,9 (м., 1H), 3,94 (д., J=2,5 Гц, 1H), 5,33 (д., J=2,5 Гц, 1H), 6,9-7,35 (14H).
3б: Элементный анализ: рассчитано для C24H23NO3S%: C 71,09, H 5,72, N 3,45, S 7,83; найдено: C 70,90, H 5,72, N 3,55.
[] 220 нМ = -4,8103, [] 233 нМ = +7,44104, [] 250 нМ = -4,0104.
1H ЯМР в дейтерохлороформе: 3,18 (м., 4H), 3,8 (с., 3H), 4,12 (д., J=2 Гц, 1H), 5,5 (д., J=2 Гц, 1H), 6,9-7,35 (14H).
Применив методику способа по примеру 3 на соединение 1в получают следующие соединения: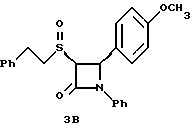 3в: Элементный анализ: рассчитано для C24H23NO3S 0,2 H2O в %: C 70,46, H 5,77, N 3,42: найдено: C 70,49, H 5,78, N 3,52.
1H ЯМР в дейтерохлороформе: 2,85 (м., 1H), 2,95 (м., 1H), 3,12 (м., 1H), 3,62 (м. , 1H), 3,8 (с., 3H), 4,4 (д., J=5,6 Гц, 1H), 5,35 (д., J=5,6 Гц, 1H), 6,9-7,35 (14H).
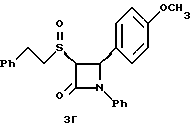 3г: Элементный анализ: рассчитано для C24H23NO3S 0,2 H2O в %: C 70,46, H 5,77, N 3,42; найдено: C 70,32, H 5,78, N 3,46.
1H ЯМР в дейтерохлороформе: 3,17 (м., 3H), 3,4 (м., 1H), 3,83 (с., 3H), 4,69 (д., J=5,2 Гц, 1H), 5,55 (д., J=5,2 Гц, 1H), 6,95-7,4 (14H).
[]2D5 -136o (в метаноле);Пример 4 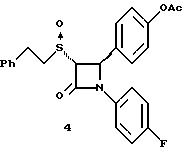 К 60 мг соединения 2б в 5 мл метиленхлорида добавляют 0,025 мл триэтиламина и 0,017 мл уксусного ангидрида. Через 2 часа реакционную массу упаривают и очищают остаток с помощью флэш- хроматографии на силикагеле, элюируя смесью этилацетат/гексан (1:1). Получают белое твердое вещество. Элементный анализ: рассчитано для C25H22NO4SF%: C 66,5, H 4,91, N 3,1, S 7,1; найдено: C 66,28, H 5,10, N 3,29, S 6,99. Применив приведенный выше способ получения соединения 4 на исходные продукты 2в и 2г получают соответственно: 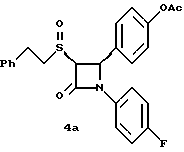 4а: Элементный анализ: рассчитано для C25H22NO4SF%: C 66,5, H 4,91, N 3,1, S 7,1; найдено: C 66,36, H 5,13, N 3,23, S 7,02. 1H ЯМР в дейтерохлороформе: 2,32 (с., 3H), 2,92 (м., 2H), 3,14 (м., 1H), 3,7 (м., 1H) 4,42 (д., J=5,7 Гц, 1H), 5,38 (д., J=5,7 Гц, 1H), 7,0 (м. 2H), 7,12-7,35 (9H), 7,44 (д., 2H). 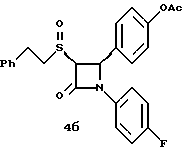 1H ЯМР в дейтерохлороформе: 2,32 (с., 3H), 3,15 (м., 3H), 3,38 (м., 1H), 4,72 (д. , J= 5,3 Гц, 1H), 5,58 (д., J=5,2 Гц, 1H), 7,0 (м. 2H), 7,15-7,35 (9H), 7,40 (д., 2H). Пример 5 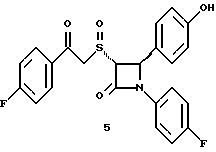 Первая стадия. К смеси 13 мл 4-метоксибезилхлорида и 10 мл этил-2-меркапто-ацетата в 0,2 л метиленхлорида в атмосфере азота прибавляют 14 мл триэтил-амина. Через 48 часов разбавляют реакционную массу 0,5 л серного эфира и последовательно промывают органическую фазу 0,3 н соляной кислотой (трижды) и 10%-ным бикарбонатом натрия. После высушивания и упаривания органического слоя получают 22 г масла. Порцию масла (5 г) растворяют в 75 мл тетрагидрофурана и 75 мл воды и добавляют 1 г гидроксида лития. После перемешивания в течение 72 часов добавляют 0,15л воды и экстрагируют 0,2 л гексана. Водную фазу подкисляют 1 н соляной кислотой и экстрагируют этилацетатом. Органический слой промывают водой, сушат над сульфатом магния и упаривают. Получают 4,25 г (96%) твердого вещества желтого цвета. Вторая стадия. При температуре 0oC к смеси 1 г полученного на первой стадии продукта и 1,55 г полученного на первой стадии примера 1 по способу А имина в 40 мл метиленхлорида прибавляют 0,56 мл диметиламинофосфорилдихлорида. Смесь нагревают до комнатной температуры и перемешивают 16 часов. Реакционную массу разбавляют 100 мл серного эфира и последовательно промывают 1 н соляной кислотой, 10%-ным бикарбонатом натрия и насыщенным раствором хлорида натрия. Органическую фазу высушивают над сульфатом магния, упаривают и очищают образующийся остаток с помощью флэш-хроматографии на силикагеле смесью гексан/этилацетат (20:1). Получают 0,75 г (30%) масла. Третья стадия. К раствору 0,2 г полученного на второй стадии продукта в 5 мл трифторуксусной кислоты при температуре 0oC добавляют 121 мг ацетата ртути. Через 15 минут добавляют воду и серный эфир, промывают, сушат и упаривают органический слой. Остаток очищают с помощью флэш-хроматографии на силикагеле смесью гексан/этилацетат (10:1). Получают 0,15 г масла. Четвертая стадия. К смеси 0,15г полученного на третьей стадии продукта и 0,06 мл триэтиламина в 5 мл метиленхлорида в атмосфере азота прибавляют 86 мг 2-бром-4′-фторацетофенона. Через 5 часов реакционную массу разбавляют серным эфиром и промывают последовательно 1н соляной кислотой, 10%-ным бикарбонатом натрия и насыщенным раствором хлорида натрия. Органический слой сушат, упаривают и очищают остаток с помощью флэш-хроматографии на силикагеле смесью гексан/этилацетат (9:1). Полученное масло разделяют методом ВЭЖХ на колонке с хиральным сорбентом (Chirlacel AS), элюируя смесью гексан/изопропиловый спирт (85:15) энатиомер 5(1) ([ ] 228 нМ = -3,77103, [] 244 нМ = +3,34103) и затем энантиомер 5(2)([ ] 228 нМ = +3,65103, [] 244 нМ = -3,24103).
Пятая стадия. При обработке энантиомера 5(2) фтористоводородной кислотой аналогично третьей стадии примера 1 по способу А получают соединение 5. Элементный анализ: рассчитано для C23H17NO3SF2%: C 64,93, H 4,03, N 3,29, S 7,52; найдено: C 64,87, H 4,39, N 3,31, S 7,25.
Пример 6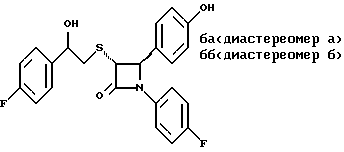 Первая стадия. К раствору 0,4 г полученного на четвертой стадии примера 5 энантиомера 5(2) в 20 мл метанола прибавляют 28 мг борогидрида натрия. Через 2 часа добавляют серный эфир и воду, органические слои сушат и упаривают, при очистке остатка с помощью флэш-хроматографии смесью этилацетат/гексан (1:6) элюируют диастереомеры 6(1) и 6(2). Вторая стадия. Полученные на первой стадии диастереомеры 6(1) и 6(2) раздельно обрабатывают фтористоводородной кислотой по аналогии с третьей стадией примера 1 по способу А. Получают 6(а) и 6(б). 6а: 1H ЯМР в дейтерохлороформе: 2,85 (дд., J=6,12 Гц, 1H), 3,04 (дд., J= 3,12 Гц, 1H), 4,06 (д., J=2,4 Гц, 1H), 4,7 (д., J=2,4 Гц, 1H), 4,9 (д., J= 3,9 Гц, 1H), 6,85-7,35 (12H). 6б: 1H ЯМР в дейтерохлороформе: 3,01 (м., 2H), 3,97 (д., J=2,2 Гц, 1H), 4,7 (д., J=2,2 Гц, 1H), 4,92 (д., J=4,8 Гц, 1H), 6,85-7,36 (12H). Пример 7 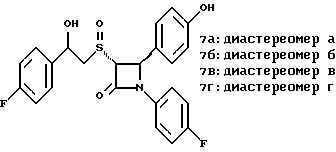 Первая стадия. Полученный по примеру 6 на первой стадии диастереомер 6(1) обрабатывают м-хлорнадбензойной кислотой аналогично примеру 3. Продукты реакции очищают с помощью ВЭЖХ на силикагеле, элюируя смесью этилацетат/гексан (1:2). Получают диастереомеры 7(1) и 7(2). Вторая стадия. Полученные на первой стадии диастереомеры 7(1) и 7(2) раздельно обрабатывают фтористоводородной кислотой по аналогии с третьей стадией примера 1 по способу А. Получают 7a и 7б. 1H ЯМР в дейтерохлороформе с 10% дейтерометанола: 7a: 3,35 (д., 1H), 3,75 (дд. , 1H), 4,22 (с., 1H), 5,20 (м., 2H), 6,80 (д., 2H), 6,9 (м., 2H), 7,04 (м. , 2H), 7,24 (м., 4H), 7,38 (м., 2H). 7б: 3,02 (дд., 1H), 3,26 (м., 1H), 4,21 (д., J=2,1 Гц, 1H), 5,14 (дд., 1H), 5,41 (д., J=2,1 Гц, 1H), 6,78 (д., 2H), 6,9 (м., 2H), 6,98 (м., 2H), 7,18 (м., 4H), 7,28 (м., 2H). Температуры плавления: 7a: 207-211oC; 7б: 110oC (разл.). Аналогично методикам первой и второй стадий из диастереомера 6(2), полученного на первой стадии примера 6, получают 7в и 7г. 1H ЯМР в дейтерохлороформе с 10% дейтерометанола: 7в: 3,12 (дд., 1H), 3,30 (м., 1H), 4,45 (д., J=2,2 Гц, 1H), 5,04 (дд., 1H), 5,39 (д., J=2,2 Гц, 1H), 6,78 (д., 2H), 6,88 (м., 2H), 6,94 (м., 2H), 7,20 (м., 6H). 7г: 3,10 (дд., 1H), 3,72 (м., 1H), 4,07 (д., J=2,5 Гц, 1H), 5,09 (дд., J= 2,3 Гц, 1H), 5,17 (д., J=2,5 Гц, 1H), 6,78 (д., 2h), 6,85 (м., 2H), 6,98 (м., 2H), 7,18 (м., 4H), 7,30 (м., 2H). Температуры плавления: 7в: 98oC (разл.); 7г: 106,5oC (разл.). Пример 8 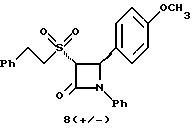 К раствору 0,185 г рацемического продукта, полученного на второй стадии примера 1 по способу А, в 20 мл метиленхлорида прибавляют м-хлорнадбензойную кислоту. Через 3 часа добавляют бисульфит натрия и бикарбонат натрия и перемешивают в течение 10 минут, затем экстрагируют этилацетатом. Органическую фракцию очищают с помощью флэш-хроматографии на силикагеле, элюируя смесью гексан/этилацетат (4: 1). Получают соединение 8 в виде белого твердого вещества, выход 0,15 г (76%). Элементный анализ: рассчитано для C24Н23NO4S%: C 68,39, H 5,5, N 3,32; найдено: C 68,12, H 5,49, N 3,37. Масс-спектр с ионизацией при электронном ударе 421 (М+). 1H ЯМР: 3,2 (м., 2H), 3,55 (м., 2H), 3,80 (с., 3H), 4,23 (д., J=2,4 Гц, 1H), 5,53 (д., J=2,4 Гц, 1H), 6,9 (д., 2H), 7,1 (м., 1H), 7,28 (11H). Пример 9 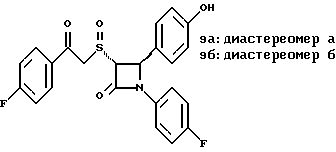 Первая стадия. Энантиомер 5(2), полученный на четвертой стадии примера 5 обрабатывают по аналогии с примером 3. Продукт реакции очищают с помощью флэш-хроматографии, элюируя смесью этилацетат/гексан (1:3) диастереомер 9(1) и диастереомер 9(2). Вторая стадия. Полученные на первой стадии диастереомеры 9(1) и 9(2) обрабатывают фтористоводородной кислотой по аналогии с третьей стадией примера 1 по способу А. Получают 9а и 96. 9а: Т.пл. 112,5-117oC. 1H ЯМР в дейтерохлороформе: 4,39 (д., J=2,4 Гц, 1H), 4,93 (д., J=16 Гц, 1H), 5,25 (д. , J= 16 Гц, 1H), 5,32 (д., J=2,4 Гц, 1H), 5,55 (ш.с. 1H), 6,85-6,95 (м., 4H), 6,88 (м., 2H), 7,18-7,30 (м., 6H), 8,05-8,09 (м., 2H). 9б: Т.пл. 188-195oC. 1H ЯМР в дейтерохлороформе с 5% дейтерометанола: 4,39 (д., J=2,1 Гц, 1H), 4,46 (д., J=15 Гц, 1H), 4,62 (д., J=15 Гц, 1H), 5,42 (д. J=2,1 Гц, 1H), 6,75 (д., 2H), 6,9 (д.д., 2H), 7,08-7,20 (м., 6H), 7,90 (м., 2H). Пример 10 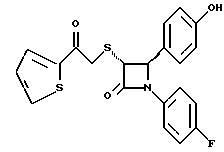 Первая стадия. Повторяют синтезы по примеру 1, способ Б, стадии с первой до четвертой, заменив на второй стадии п- метоксибензилмеркаптан на фенилэтилмеркаптан. Вторая стадия. Полученный на первой стадии транс-изомер обрабатывают ацетатом ртути. Получают продукт третьей стадии примера 5 в оптически чистом виде. Третья стадия. По аналогии с четвертой и пятой стадиями примера 5 взаимодействием полученного на второй стадии продукта с 1′-бромо-2-ацетил-тиофеном получают целевой продукт в виде твердого вещества с т.пл. 148-150oC. Пример 11 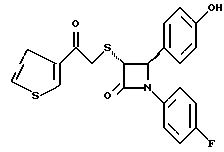 Повторяют операции примера 10, используя на третьей стадии 1′-бромо-3-ацетилтиофен. Получают целевое соединение в виде твердого вещества с т.пл. 176-178oC. Элементный анализ: рассчитано для C21H16NO3S2F%: C 61,01, H 3,90, N 3,39, S 15,48; найдено: C 61,33, H 4,12, N 3,51, S 15,37. Пример 12 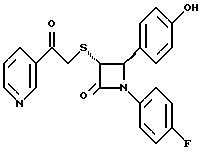 Повторяют операции примера 10, используя на третьей стадии 1′-бромо-3-ацетилпиридин. Получают целевое соединение в виде твердого вещества с т.пл. 74-90oC. Масс-спектр с ионизацией бомбардировкой быстрыми атомами 409(М+H). Пример 13 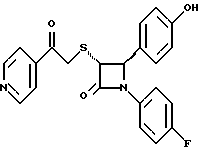 Повторяют операции примера 10, используя на третьей стадии 1′-бромо-4-ацетилпиридин. Получают целевое соединение в виде твердого вещества с т.пл. 65-69oC. Элементный анализ: рассчитано для C22H17N2SF%: C 64,69, H 4,20, N 6,86, S 7,85; найдено: C 65,00, H 4,43, N 6,77, S 7,65. Пример 14 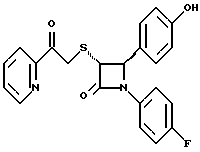 Повторяют операции примера 10, используя на третьей стадии 1′-бромо-2-ацетилпиридин. Получают целевое соединение в виде твердого вещества с т.пл. 59-64oC. Пример 15 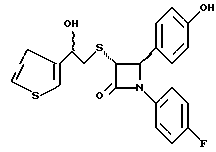 Полученное в примере 11 соединение в метаноле обрабатывают борогидридом натрия, получают смесь диастереомеров в виде твердого вещества с т.пл. 65-70oC. Элементный анализ: рассчитано для C21H18NO3S2F%: C 60,71, H 4,37, N 3,37, S 15,4; найдено: C 60,67, H 4,48, N 3,40, S 15,87. Пример 16 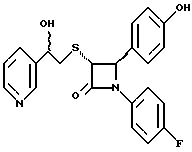 Полученное по примеру 12 соединение в метаноле обрабатывают борогидридом натрия при температуре 0oC. Через 30 минут выливают в смесь метиленхлорида и воды, отделяют раствор в метиленхлориде и очищают продукт с помощью флэш-хроматографии на силикагеле, элюируя смесью метиленхлорид/метанол (95:5). Получают целевое соединение в виде твердого вещества с т.пл. 85-90oC. Пример 17 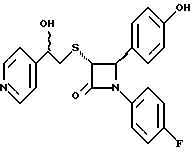 По аналогии с примером 16 из полученного по примеру 13 соединения синтезируют целевой продукт с т.пл. 95-98oC Элементный анализ: рассчитано для C22H19N2O3SF%: C 64,38, H 4,67, N 6,82, S 7,81; найдено: C 64,09, H 4,95, N 6,67, S 8,06. Пример 18 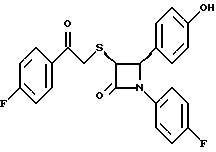 Первая стадия. Полученный на первой стадии примера 10 цис-изомер обрабатывают по аналогии со второй стадией примера 10. Получают твердое вещество. Вторая стадия. Проводят взаимодействие полученного на первой стадии соединения по аналогии с четвертой и пятой стадиям примера 5. Получают целевой продукт в виде твердого вещества с т.пл. 180-185oC. Элементный анализ: рассчитано для C23H17NO2SF2%: C 64,93, H 4,03, N 3,29, S 7,54; найдено: C 65,13, H 4,16, N 3,43, S 7,70. Пример 19 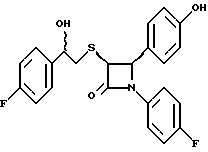 Полученное на первой стадии примера 18 соединение обрабатывают борогидридом натрия по аналогии с примером 16 и затем действуют на продукт реакции фтористоводородной кислотой по аналогии с третьей стадией примера 1 по способу А. Получают целевое соединение с т.пл. 95-105oC. Предлагаемая фармацевтическая композиция иллюстрируется следующими примерами некоторых конкретных лекарственных препаратов (см. в конце описания). В каждом составе термин “активное начало” означает соединение формулы I. Способ приготовления Смешивают ингредиенты N 1 и N 2 в подходящем смесителе в течение 10-15 минут. Добавляют ингредиент N 3 и гранулируют смесь. Влажные гранулы протирают, если это необходимо, через сетку с крупной ячейкой (например, 0,25 дюйма, 0,63 см). Влажные гранулы высушивают и, если это необходимо, просеивают, затем смешивают с ингредиентом N 4 и перемешивают 10-15 минут. Добавляют ингредиент N 5 и перемешивают 1-3 минуты. Смесь прессуют до требуемого размера и веса в устройстве для таблетирования. Способ приготовления Смешивают ингредиенты N 1, N 2 и N 3 в подходящем для этого смесителе в течение 10-15 минут. Добавляют ингредиент N 4 и перемешивают 1-3 минуты. Смесью заполняют предназначенные для этого капсулы из твердого желатина, состоящие из двух частей, в устройстве для капсулирования. При желании в вышеприведенные препараты можно включить еще ингибитор биосинтеза холестерина в нужном количестве. Формула изобретения
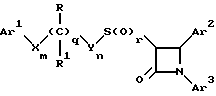 где Ar1 – фенил, незамещенный или замещенный галогеном, или 5- или 6-членный гетероароматический радикал, содержащий азот или серу; Ar2 – фенил, замещенный низшим алкилом, группой -OR2 или -O(CO)R2, где R2 – атом водорода или низший алкил; Ar3 – фенил, незамещенный или замещенный галогеном; X и Y независимо друг от друга означают -CH2-; R – группа -OR3, где R3 – атом водорода или низший алкил; R1 – атом водорода или R или R1 вместе – атом кислорода карбонильной группы (=О), q = 0 или 1; r – 0, 1 или 2, m и n независимо друг от друга имеют значение 0, 1, 2, 3, 4 или 5 при том условии, что m + n + q = 2, 3, 4 или 5, и их фармацевтически приемлемые соли. 2. Производные азетидинона формулы (I) по п.1, где Ar1 – фенил, незамещенный или замещенный галогеном, или тиенил, Ar2 – фенил, замещенный гидроксилом, а Ar3 – фенил, замещенный фтором. 3. Производные азетидинона формулы (I) по п.1 или 2, где q = 0, X и Y означают каждый -CH2-, а m + n = 2, 3 или 4. 4. Производные азетидинона формулы (I) по п.1 или 2, где q = 1, X и Y означают каждый -CH2-, m + n = 2 или 3, R – группа -OR3, где R3 – атом водорода, или R и R1 вместе – атом кислорода карбонильной группы (=О). 5. Производные азетидинона формулы (I) по п.1, выбранные из группы, включающей транс-4-(4-гидроксифенил)-3-[(2-фенилэтил)тио] -1-(4-фторфенил)-2-азетидинон; транс-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)тио]-2-азетидинон; цис-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)тио]-2-азетидинон; транс-4-(4-гидроксифенил)-3-[(2-фенилэтил)сульфинил] -1-(4-фторфенил)-2-азетидинон; цис-4-(4-гидроксифенил)-3-[(2-фенилэтил)сульфинил] -1-(4-фторфенил)-2-азетидинон; транс-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)сульфинил] -2-азетидинон; цис-4-(4-метоксифенил)-1-фенил-3-[(2-фенилэтил)сульфинил]-2-азетидинон; транс-4-[4-кето-3-[(2-фенилэтил)сульфинил]-1-(4-фторфенил)-2-азетидинил] -фенилацетат; цис-4-[4-кето-3-[(2-фенилэтил)сульфинил] -1-(4-фторфенил)-2-азетидинил] -фенилацетат; (+/-)-транс-4-(метоксифенил)-1-фенил-3-[(2-фенилэтил)сульфонил]-2-азетидинон; транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(4-фторфенил)-этил]тио]-2-азетидинон; транс-4-[4-гидроксифенил)-1-(4-фторфенил)-3-[[2-гидрокси-2-(4-фторфенил)этил]тио]-2-азетидинон; (3R, 4R) 4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(4-фторфенил)-этил]сульфинил]-2-азетидинон; 4(R)-(4-гидроксифенил)-1-(4-фторфенил)-3(R)-[[2(R)-гидрокси-2-(4-фторфенил)этил]сульфинил]-2-азетидинон; 4(R)-(4-гидроксифенил)-1-(4-фторфенил)-3(R)-[[2(S)-гидрокси-2-(4-фторфенил)этил]сульфинил]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(2-тиенил)-этил]тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(3-тиенил)-этил]тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(3-пиридинил)этил]тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(4-пиридинил)этил]тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(2-пиридинил)этил]тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-гидрокси-2-(3-тиенил)этил]тио]-2-азетидинон; (3R, 4R) транс-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-гидрокси-2-(4-пиридинил)этил]тио]-2-азетидинон; (3S, 4R) цис-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-кето-2-(4-фтор-фенил)этил]тио]-2-азетидинон; (3S, 4R) цис-4-(4-гидроксифенил)-1-(4-фторфенил)-3-[[2-гидрокси-2-(4-фтор-фенил)этил]тио]-2-азетидинон. 6. Фармацевтическая композиция с антиатеросклеротической или гипохолестеринемической активностью, содержащая активное вещество на основе производных азетидинона и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве производных азетидинона содержит соединения общей формулы (I) по любому из пп.1 – 5 или их фармацевтически приемлемые соли в эффективном количестве. 7. Фармацевтическая композиция по п.6, отличающаяся тем, что она дополнительно содержит ингибитор биосинтеза холестерина в эффективном количестве. Приоритет по пунктам и признакам: 18.11.94 по пп.1, 2, 5 – 7, где Ar1 – фенил, незамещенный или замещенный галогеном; по пп.3 и 4; 05.06.95 по пп.1, 5 – 7, где Ar1 – 5- или 6-членный гетероароматический радикал, содержащий азот или серу. РИСУНКИ
|
||||||||||||||||||||||||||