Патент на изобретение №2310848
|
||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБ ПРОГНОЗИРОВАНИЯ РИСКА ВОЗНИКНОВЕНИЯ, КЛИНИЧЕСКОГО ТЕЧЕНИЯ И ИСХОДА ОСТРОГО ИДИОПАТИЧЕСКОГО ПАНКРЕАТИТА
(57) Реферат:
Изобретение относится к медицине, а именно к хирургии, и может быть использовано для прогнозирования риска возникновения, клинического течения и исхода острого идиопатического панкреатита. Задача изобретения: повышение эффективности прогнозирования клинического течения острого идиопатического панкреатита. Поставленную задачу решают за счет того, что из крови пациента с острым идиопатическим панкреатитом получают ДНК и осуществляют генотипирование пациента с помощью полимеразной цепной реакции. Для этого исследуют мутации генов SPINK1, GSTM1, GSTT1, PRSS1 и CFTR, при обнаружении гетерозиготных мутаций в генах SPINK1, PRSS1 и CFTR прогнозируют развитие деструктивной формы идиопатического панкреатита, при обнаружении сочетания не менее чем двух мутировавших генов из SPINK1, GSTM1, GSTT1, PRSS1 и CFTR у гетерозиготных носителей или гомозиготных носителей по мутации N348 гена SPINK1 в сочетании с гетерозиготной мутацией R122H гена PRSS1 прогнозируют формирование панкреонекроза тяжелой степени. Преимущество изобретения в том, что оно позволяет выделить группу больных с высоким риском развития деструктивных осложнений и обоснованно рекомендовать им проведение полномасштабной специализированной интенсивной терапии на ранних сроках, тем самым снизить, или полностью исключить летальность у больных. 3 ил., 4 табл.
(56) (продолжение): CLASS=”b560m”GRIGORERESCU M. Genetic factor in pancreatitis, Romanian Journal of Gastroenterology. 2005, v.14, №1, pp.53-61. LAMPRECHT G. et al. Relapsing pancreatitis due to a novel compound heterozygosity in the CFTR gene involving the second most common mutation in Central and Eastern Europe [CFTRdelete2,3(21kb)], Pancreatology. 2005, v.5, pp.92-96.
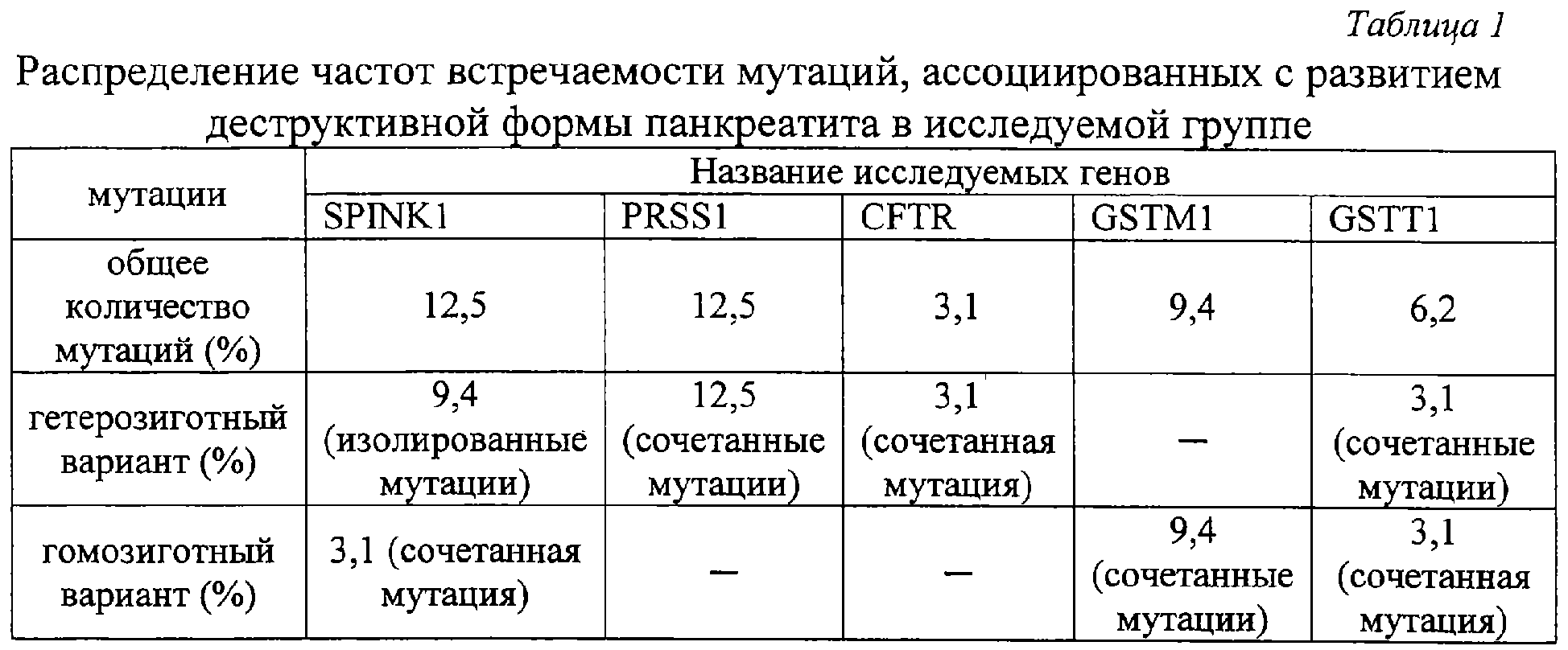
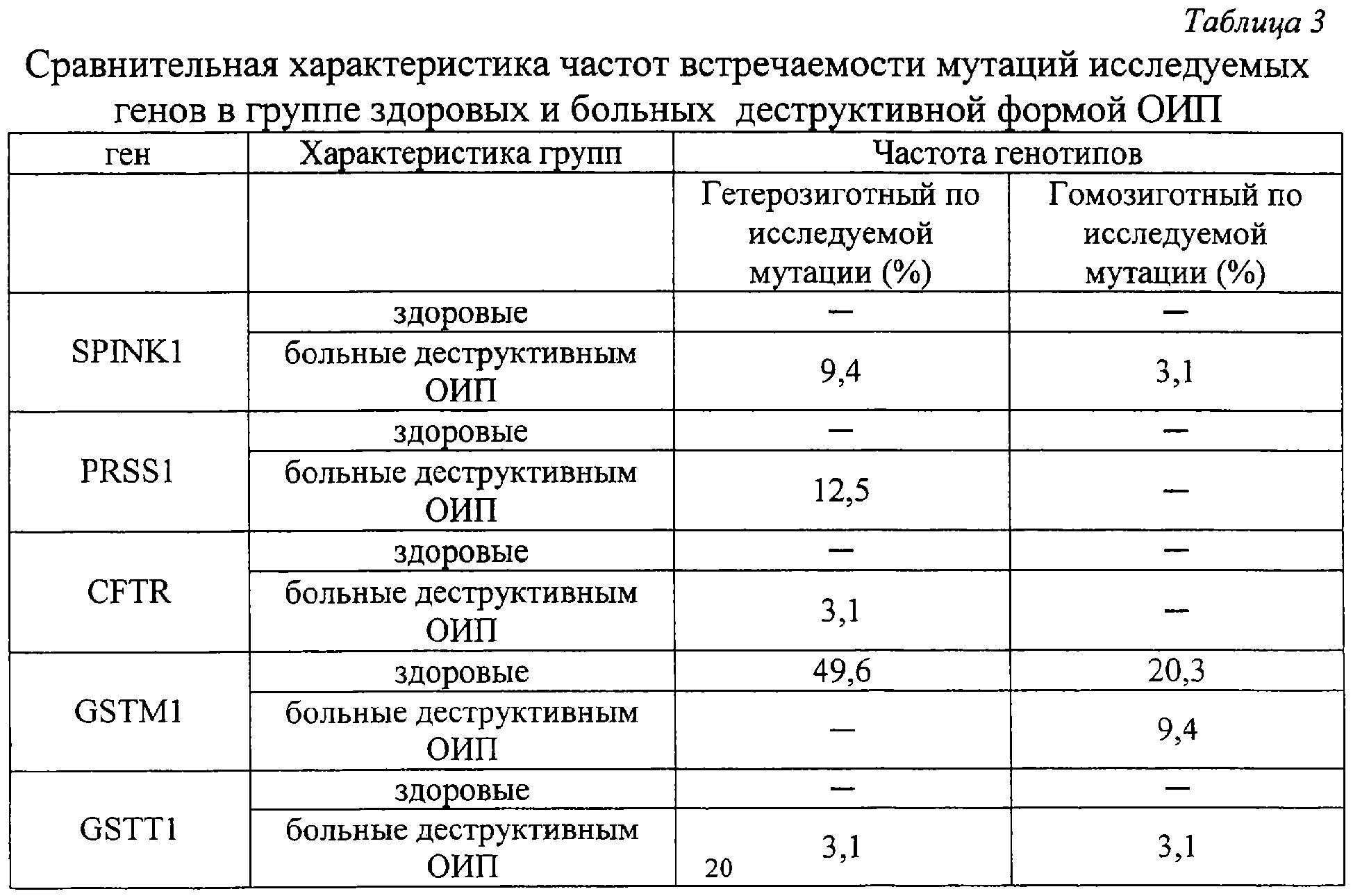
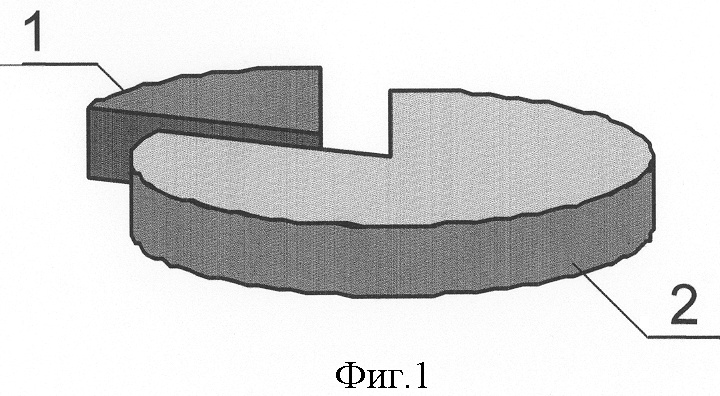
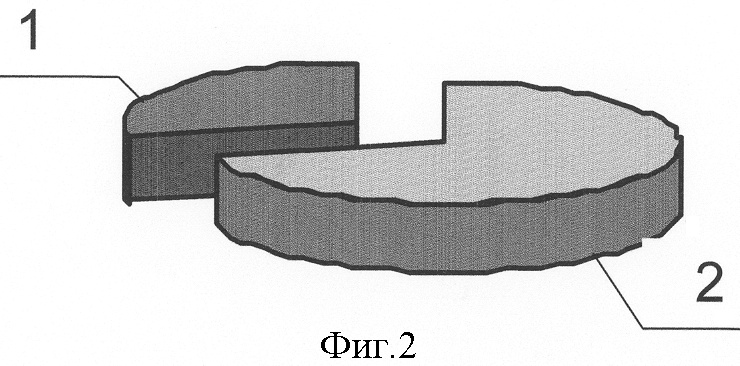
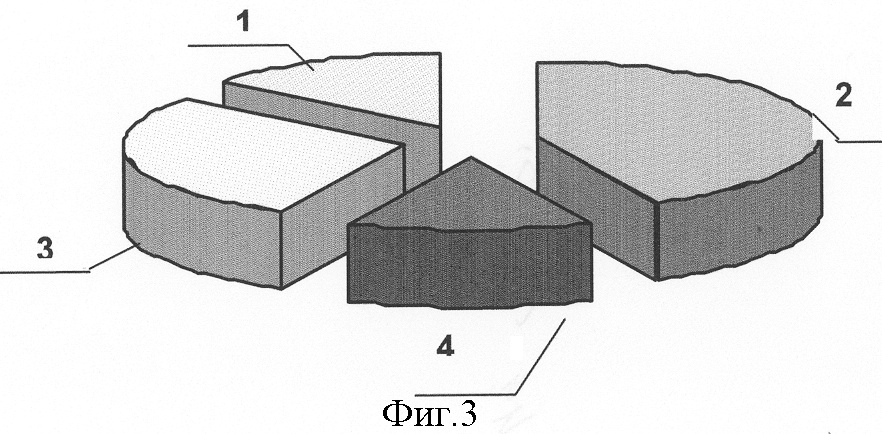
Изобретение относится к медицине, а именно к хирургии, и может быть использовано для прогнозирования риска возникновения, клинического течения и исхода острого идиопатического панкреатита. Острый панкреатит (ОП) – одно из самых сложно диагностируемых и, вместе с тем, широко распространенных заболеваний [1]. В течение нескольких лет он стабильно занимает второе, а по некоторым регионам – первое место в структуре заболеваний «острый живот», опережая острый аппендицит и острый холецистит [2, 3, 4]. Летальность при остром деструктивном панкреатите как в России, так и за рубежом остается неизменно высокой, не опускаясь ниже 20%, достигая при инфицированных формах 60-80% [4, 5, 6, 7, 8, 25]. Примечательным фактом является относительное постоянство на протяжении последних десятилетий величины идиопатического панкреатита в общей структуре заболевания, несмотря на применение современных диагностических методов, на его долю приходится 15-35%, что указывает на несовершенство диагностики. В настоящее время, среди различных критериев прогноза течения острого панкреатита наиболее распространены прогностические системы, основанные на изучении лабораторных параметров, характеризующих состояние пораженного органа и жизненно важных констант организма и предполагающие, в конечном счете, числовое выражение тяжести состояния больного. Достаточную клиническую апробацию прошли: 1. ранние прогностические критерии Ranson; 2. индекс APACHE II; и система стратификации тяжести острого панкреатита на основе компьютерно-томографических критериев [9, 10, 26, 27]. Наиболее известная и повсеместно применяющаяся прогностическая система предложена американским хирургом J.Ranson – она состоит из 11 критериев. Сумма баллов от 0 до 2 указывает на легкое течение панкреатита, от 3 до 5 – на панкреатит средней тяжести, сумма от 6 до 11 баллов соответствует тяжелому течению панкреонекроза. Несколько проще система прогноза Glasgow (Imrie), она состоит из восьми лабораторных показателей, девятым является возраст пациента. Недостатком этих систем является неспецифичность использованных лабораторных параметров и 48-часовая отсрочка в установлении прогноза течения острого панкреатита [11, 12]. В ведущих отечественных и зарубежных клиниках в последние годы для оценки тяжести состояния больных острым панкреатитом применяется шкала APACHE-2 (Acute physiology and chronic health evaluation) – шкала оценки острых физиологических нарушений и хронических заболеваний. Данная шкала позволяет с высокой точностью определить тяжесть состояния пациента, имеет некоторые преимущества перед системой Рансона и компьютерной томографией, заключающиеся в возможности ежедневной повторной оценки, низкой стоимости скрининга тяжести и незначительным риском, однако большое количество параметров создает неудобства при использовании этой шкалы в условиях ургентной хирургии [28, 29]. Наиболее доступна и удобна в применении прогностическая система, предложенная Санкт-Петербургским НИИ скорой помощи им. И.И.Джанелидзе. Система состоит из 2 разделов, включающих основные и дополнительные признаки тяжести заболевания. Если у пациента имеется минимум 2 основных признака или 1 основной и 2 дополнительных, то с 95% вероятностью может развиться тяжелая форма острого панкреатита (крупноочаговый, либо субтотально-тотальный). Если присутствует один признак, то возможен острый панкреатит средней тяжести. Отсутствие перечисленных признаков характерно для отечной формы острого панкреатита [13, 14]. Необходимость применения балльных систем оценки тяжести состояния больных панкреатитом не вызывает сомнений, они дают возможность унифицировать больных и выработать алгоритм действий. Однако анализ результатов анкетирования, проведенного в рамках подготовки к IX Всероссийскому съезду хирургов, показал, что в 29% случаев используется система АРАСНЕ-П, шкала Ranson применяется у 27% опрошенных, остальные респонденты указали, что не применяют систем оценки тяжести и прогноза заболевания [15]. Возможно этот факт обусловлен отсутствием оценочной системы, удовлетворяющей основным требованиям – простота, доступность и объективность, а также основным недостатком всех существующих на сегодняшний день систем, который заключается в том, что ни одна из них не позволяет прогнозировать риск возникновения, возможного течения и исхода острого панкреатита до его клинико-лабораторной манифестации, то есть не соответствует основным принципам современной предиктивной медицины – медицины предупредительной. Наиболее близким к предлагаемому является способ прогнозирования течения язвенной болезни двенадцатиперстной кишки (ДПК) на основе фармакогенетического анализа метаболизма и клинической эффективности лекарственных препаратов, предназначенных для ее лечения, а именно ингибиторов протонного насоса, разработанный Никоновым Е.Л [16]. Проведенное у больных генотипирование выявило четкую зависимость возникновения осложнений, а также непрерывно-рецидивирующего течения язвенной болезни ДПК от наличия мутаций гена CYPC19 в системе цитохрома Р450. Такие генные мутации обуславливают “быстрый” тип метаболизма ингибиторов протонных помп, что снижает их эффективность и приводит к неудовлетворительным результатам консервативного лечения язвенной болезни ДПК, склонности к ее непрерывно-рецидивирующему течению. Предлагаемый способ прогнозирования играет важную роль: 1) в определениии тактики лечения язвенной болезни ДПК; 2) в выработке показаний к оперативному лечению, выполнению органосохраняющих операций на ДПК и желудке, а также тому или иному способу хирургической коррекции кислотопродуцирующей функции желудка. Основным недостатком, не позволяющим применить его в отношении острого идиопатического панкреатита, является различие в патогенетической сущности этих заболеваний. Для язвенной болезни ДПК ведущим в патогенезе является повышение кислотности желудочного сока, поэтому в основе системы прогноза ее течения и исхода лежит выявление мутаций в системе цитохрома Р450, повышающих активность ферментов, метаболизирующих лекарственные препараты, предназначенные для снижения кислотопродуцирующей функции желудка, что приводит к их клинической неэффективности в связи с быстрым выведением из организма. Для создания же прогностической системы в отношении острого идиопатического панкреатита (ОИП) необходимо выявление мутаций в генах, которые способствовали бы снижению эффективности ферментов, участвующих в биотрансформации ксенобиотиков и, соответственно, увеличивающих их токсическое воздействие на организм, а также в генах, вовлеченных в функционирование клеток самой поджелудочной железы. Задача изобретения: повышение эффективности прогнозирования риска возникновения, клинического течения и исхода острого идиопатического панкреатита. Поставленную задачу решают за счет того, что исследуют 5 генов на предмет мутаций, ассоциированных с развитием острого идиопатического панкреатита и влияющих на его течение и исход: ферментов биотрансформации ксенобиотиков – GSTM1 и GSTT1, катионного трипсиногена – PRSS1, ингибитора трипсиногена – SPINK1, трансмембранного регулятора муковисцидоза – CFTR; при наличии мутаций в генах SPINK1, PRSS1, CFTR прогнозируют развитие деструктивной формы острого идиопатического панкреатита с риском в 21,8%; при наличии только изолированной гетерозиготной мутации N34S гена SPINK1 прогнозируют повышение риска возникновения деструктивного идиопатического панкреатита на 9,4%; при сочетании гетерозиготной мутации R122H гена PRSS1 с мутациями в генах SPINK1, CFTR и GSTM1 – на 12,5%; при выявлении “двойных” мутаций в генах, вовлеченных в функционирование клеток поджелудочной железы: SPINK1, PRSS1, CFTR, прогнозируют формировании панкреонекроза тяжелой степени и повышение летального исхода на 44,5%. Оценивали состояние генетических систем: 1. Осуществляющих взаимодействие организма с внешней средой. Исследовали гены ферментов биотрансформации ксенобиотиков – глутатион-S-трансферазы (GSTM1 и GSTT1), где в качестве полиморфных вариантов выявлялись их протяженные делеции. Вследствии таких делеций образуется мутантный “нулевой” аллель генов GSTM1 и GSTT1, у людей гомозиготных по этому аллелю и имеющим, соответственно, “нулевой” генотип (0/0), белковый продукт не синтезируется, то есть ферменты, за образование которых ответственны гены GSTM1 и GSTT1 отсутствуют [21]. 2. Непосредственно вовлеченных в функционирование клеток поджелудочной железы. Исследовали гены: а) катионного трипсиногена PRSS1 и его R122H мутация; б) ингибитора трипсиногена (SPINK1) с мутацией N34S; в) трансмембранного регулятора муковисцидоза (CFTR) с 8 наиболее распространенными мутациями. Выбор исследуемых генов обусловлен тем, что: 1. Ферменты глутатионтрансферазы, экспрессирующиеся с генов GSTM1 и GSTT1 являются ферментами биотрансформации ксенобиотиков, их отсутствие вследствие мутаций этих генов приведет к нарушению их обезвреживания и повышению их токсического воздействия на организм. [17,20]. 2. Фермент с гена GSTM1 экспрессируется в панкреатической паренхиме, поэтому принимает участие в обезвреживании экзогенных токсинов непосредственно в поджелудочной железе, его отсутствие вследствие мутации этого гена приведет к деструкции ее ткани [18]. 3. Фермент с гена GSTM1 входит в состав антиоксидантной системы, при его отсутствии вследствие мутаций нарушится резистентность клеток к перекисному окислению липидов, жиров, повреждающему действию свободных радикалов, что неблагоприятно в отношении тяжести течения и исхода острого панкреатита [19, 30]. 4. Фермент с гена GSTT1 участвует в детоксикации многих поллютантов, его делеция увеличивает вероятность хромосомных повреждений при воздействии оксидов и пероксидов, промежуточных метаболитов [19, 24]. 5. Вследствие мутации гена катионного трипсиногена (PRSS1), трипсиноген становится устойчивым к аутолизу и подверженным более легкой аутоактивации, что может провоцировать развитие генетически обусловленного острого панкреатита [23, 32]. 6. При мутации гена панкреатического ингибитора трипсина (SPINK1) нарушается естественный процесс блокады активированного трипсина в ткани ПЖ, что запускает процесс безудержной активации панкреатических ферментов, вызывая протеолитический некроз ткани поджелудочной железы и лизис стенок венул [33]. 7. Мутации гена трансмембранного регулятора муковисцидоза (CFTR) реализуются на клеточном уровне недостаточной гидратацией и защелачиванием первичного секрета экзокринных желез и увеличением его вязкости [22, 31]. Увеличение концентрации гликопротеидов в панкреатическом секрете приводит к затруднению его экскреции по протокам и нарушению транспорта зимогенных гранул через апикальную мембрану, способствующих интрапанкреатической активации энзимов и аутолизу ткани железы [19, 31]. Для осуществления предлагаемого способа проводят молекулярно-генетический анализ образцов ДНК, полученных из 4 мл крови обследуемых методом полимеразной цепной реакции (ПЦР). Экстракцию ДНК осуществляют по стандартной методике с обработкой протеинкиназой К (фирма “СибЭнзим”) и фенолхлороформной смесью. Детекцию делеционных полиморфизмов GSTM1 и GSTT1 генов проводили методом ПЦР. Для генов PRSS1 и SPINK1 использовали ПЦР-ПДРФ-анализ с рестрикторами BstFNl (фирма «Сибэнзим») и TspRI (фирма «New England BioLab»). Анализ мутаций CFTR гена проводят с использованием диагностического набора фирмы «ДНК-лаборатория» на 8 наиболее распространенных мутаций: del21kb, delF508, dell507, 1677delTA, 2143delT, 2184insA, 394delTT, 3821delT. Визуализацию результатов осуществляют электрофоретически в 3% агарозном геле (для генов глутатион-S-трансферазы, PRSS1 и SPINK1) и в 7% полиакриламидном геле для гена CFTR с окраской бромистым этидием. Статистическую обработку данных проводят с помощью закона Харди-Вайнберга для аутосомных признаков, критерия За период с 2003 по 2005 годы был обследован 101 человек. В группу сравнения вошли 69 здоровых донора, из них 42 мужчин и 27 женщин, средний возраст составил 30±10,4 лет. Исследуемые группы составили 32 пациента с острым панкреатитом идиопатической этиологии, из которых было 19 мужчин и 13 женщин, средний возраст больных составил 50,2±7,8 лет. Диагноз острого панкреатита устанавливали на основании анамнеза, данных объективного обследования, лабораторных и инструментальных методов. Исследование, проведенное в группе больных острым идиопатическим панкреатитом (ОИП), выявило, что из 32 пациентов этой группы деструктивная форма заболевания зарегистрирована у 9, что составило 28,1%, из которых у 7 больных (21,8%), были выявлены мутации генов, непосредственно участвующих в регуляции ферментативной функции поджелудочной железы (SPINK1, CFTR и PRSS1), что доказывает значимость этих мутаций в развитии деструктивных форм ОИП. У 23 (71,9%) больных, с отечной формой идиопатического панкреатита, ни одной мутации генов SPINK1, CFTR и PRSS1 обнаружено не было. Распределение клинических форм ОИП представлено на фиг.1, где 1 – 28,1% больные с деструктивной формой заболевания, 2 – 71,9% больные с отечной формой заболевания. Значение мутаций генов SPINK1, CFTR и PRSS1 в развитии его деструктивных форм представлено на фиг.2, где 1 – 21,8% больные с деструктивной формой ОИП, обусловленной мутациями генов SPINK1, CFTR и PRSS1, 2 – общее количество больных острым идиопатическим панкреатитом. В результате исследования полиморфного варианта гена ингибитора трипсина среди больных острым панкреатитом идиопатической этиологии мутантный аллель был зафиксирован у 4 больных, что составило 12,5%. Трое больных (9,4%) являлись гетерозиготами, так как у них только один аллель содержал замену N34S, мутаций других генов у них не было выявлено. Один пациент (3,1%) оказался гомозиготным в отношении данной мутации, причем именно у этого больного была выявлена еще и R122H мутация в гене катионного трипсиногена [табл.1, табл.2]. Учитывая, что среди здоровых обследованных не было выявлено ни одного случая полиморфизма гена SPIHK1, участвующего в инактивации внутрипанкреатического активного трипсина, можно предположить, что обнаруженная мутация N34S гена SPIHK1 является одним из предрасполагающих факторов к развитию идиопатического панкреатита [Табл.3]. Проведенный анализ клинического течения острого панкреатита показал, что у больных с генетически детерминированной слабостью механизма ингибирования трипсина патологический процесс протекает по более тяжелому сценарию и практически во всех случаях сопровождается развитием деструкции поджелудочной железы. У трех больных (9,4%) острым панкреатитом, у которых был выявлен мутантный аллель гена SPINK1, наблюдалось субтотальное поражение паренхимы поджелудочной железы. У одного пациента (3,1%), являющегося гомозиготой в отношении мутации гена SPINK1 и гетерозиготой по R122H мутации в гене катионного трипсиногена, зона деструкции паренхимы органа характеризовалась тотальным распространением с обширным поражением забрюшинной клетчатки. Масштаб поражения поджелудочной железы при изолированной и сочетанной мутации гена SPINK1 фиг.3, где 1 – 3,1% тотальное поражение при сочетании мутации R122H гена PRSS1 и delF508 CFTR, 2 – 9,4% субтотальное поражение при изолированной мутации N34S гена SPINK1, 3 – 6,2% субтотальное поражение при мутации R122H гена PRSS1 и GSTM1 0/0, 4 – 3,1% тотальное парожение с вовлечением забрюшинной клетчатки при сочетании мутаций N34S гена SPINK1 и R122H гена PRSS1. Таким образом, полученные результаты позволяют утверждать, что наличие мутации гена ингибитора трипсина является патогномоничным фактором для развития ОИП, протекающего по преимущественно деструктивному типу, так как она ассоциирована с ним в 12,5% случаев, из них 9,4% обусловлены изолированными мутациями SPINK1 и 3,1% сочетанием мутации SPINK1 и R122H мутации гена катионного трипсиногена [табл.1, табл.2]. Кроме того, необходимо отметить, что для больных острым панкреатитом, детерминированного мутацией гена SPINK1, формирование зон некроза поджелудочной железы происходит в сравнительно короткие сроки и характеризуется быстрым прогрессированием объема девитализированных тканей. Вероятно, такая клиническая особенность течения генетического панкреатита является причиной выраженной панкреатогенной токсемии в ранние сроки заболевания, развития панкреатогенного шока и полиорганной недостаточности – основной причины смерти больных в первую неделю заболевания. К неблагоприятному исходу ОИП приводит сочетание гомозиготной мутации N34S гена SPINK1 и гетерозиготной R122H мутации гена катионного трипсиногена, так как больной с таким генотипом умер в ранние сроки после госпитализации. По-видимому, сочетание нескольких мутантных генов увеличивает не только риск возникновения деструктивного панкреатита, но и тяжесть патологического процесса. Поэтому выявление в генотипе больного мутаций нескольких генов, участвующих в функционировании клеток поджелудочной железы, можно рассматривать как летальный вариант панкреонекроза, характеризующегося короткой стадией отека и быстрым формированием некротических изменений паренхимы органа. Анализ полиморфизма гена катионного трипсиногена в группе больных идиопатическим панкреатитом показал наличие мутации R122H у четырех больных, что составило 12,5%. Причем у всех больных выявленные мутации гена PRSS1 были амплифицированы только в одном аллеле (все больные оказались гетерозиготами по мутации R122H). Наличие мутантного аллеля гена PRSS1 во всех случаях сопровождалось развитием деструктивного панкреатита [табл.1]. Однако необходимо отметить, что у всех этих больных имелись двойные мутации, то есть помимо мутации гена PRSS1 у них были выявлены: две гомозиготные мутации гена глутатион-S-трансферазы Ml (GSTM1 0/0); гомозиготная мутация N34S гена ингибитора трипсина (SPINK1); гетерозиготная мутация delF508 гена регулятора трансмембранного муковисцидоза (CFTR) [табл.2]. Анализируя особенности клинического течения острого панкреатита, детерминированного полиморфным вариантом гена катионного трипсиногена, были отмечены тенденции, сходные с панкреатитом, обусловленным мутацией гена ингибитора трипсина. У двух больных (6,2%), гетерозиготных по замене R122H в сочетании с “нулевыми генотипами” GSTM1 0/0 (гомозиготный вариант гена), панкреонекроз характеризовался субтотальным поражением поджелудочной железы с вовлечением в воспалительный процесс забрюшинной клетчатки. У двух других больных (6,2%) с “двойными” мутациями, такими как, сочетание гетерозиготной R122H мутации гена PRSS1 с гомозиготной мутацией N34S гена SPINK1 и гетерозиготной R122H мутации гена PRSS1 в комбинации с мажорной гетерозиготной мутацией delF508 гена CFTR, был выявлен тотальный жировой панкреонекроз с поражением ретродуоденальной, параколической и паранефральной клетчатки. Масштаб поражения поджелудочной железы у больных с деструктивной формой ОИП, обусловленной вышеперечисленными мутациями, представлен на фиг.3, где 1 – 3,1% тотальное поражение при сочетании мутации R122H гена PRSS1 и delF508 CFTR, 2 – 9,4% субтотальное поражение при изолированной мутации N34S гена SPINK1, 3 – 6,2% субтотальное поражение при мутации R122H гена PRSS1 и GSTM1 0/0, 4 – 3,1% тотальное парожение с вовлечением забрюшинной клетчатки при сочетании мутаций N34S гена SPINK1 и R122H гена PRSS1. При этом патологический процесс развивался в короткие сроки и отличался низкой управляемостью, несмотря на применение интенсивной дезинтоксикационной и противошоковой терапии, что привело к гибели больных в течение первых 3-х суток госпитализации. Таким образом, полученные результаты исследования свидетельствуют о том, что в ряде случаев развитие идиопатического панкреатита обусловлено мутациями в гене, кодирующем катионный трипсиноген. Однако, учитывая то, что во всех случаях данная мутация сочеталась с полиморфными вариантами других генов, можно предположить, что особое значение для развития деструктивного панкреатита генетической этиологии принадлежит именно двойным мутациям [табл.2]. Частота встречаемости мутации delF508 гена трансмембранного регулятора муковисцидоза (CFTR) в группе больных идиопатическим панкреатитом составила 3,1% (гетерозиготный вариант) и зафиксирована только у одного больного с тотальным поражением поджелудочной железы, содержащего также мутантный аллель гена катионного трипсиногена [табл.1, табл.2]. Таким образом, согласно нашим исследованиям, патогенетическое значение гетерозиготной мутации delF508 для острого панкреатита идиопатической этиологии проявляется только в сочетании с полиморфными вариантами других генов и ассоциировано с формированием острого деструктивного панкреатита тяжелой степени тяжести. Проведенный сравнительный анализ распределения частот аллелей и генотипов гена биотрансформации ксенобиотикой GSTM1 и GSTT1 в группе больных идиопатическим панкреатитом и здоровых обследованных выявил следующие закономерности. В группе больных идиопатическим панкреатитом гомозиготный делеционный вариант гена GSTM1 был выявлен у 3 больных (9,4%), что более чем в 2 раза реже по сравнению с частотой встречаемости данной протяженной делеции среди соматически здоровых обследованных, что свидетельствует об отсутствии взаимосвязи полиморфизма гена глутатион-S-трансферазы M1 с развитием ОИП [табл.4]. При исследовании распределения частот полиморфного варианта гена GSTT1 и генотипа GSTT1 О/О среди здоровых лиц и больных исследуемой группы мутантные аллели были обнаружены только у трех больных, что составило 9,4%. Нулевой генотип был выявлен у одного больного, остальные два пациента являлись гетерозиготами [табл.4]. Клинико-генетический анализ ассоциаций полифорфизма генов биотрансформации ксенобиотиков с риском развития идиопатического острого панкреатита выявил следующие закономерности. Полиморфные варианты гена GSTM1 были зафиксированы у 10 больных отечным панкреатитом (31,25%) и у 3 больных панкреонекрозом (9,4%) [табл.1]. В одном случае отмечено сочетание гомозиготной мутации гена GSTM1 и гетерозиготной мутации GSTT1 [табл.2]. Распределение частот делеции гена GSTT1 среди больных ОИП составило: один человек (3,1%) с отечным панкреатитом и 2 человека (6,2%,) с деструктивным, [табл.1]. Полученные результаты указывают на отсутствие ассоциации мутаций в генах биотрансформации ксенобиотиков и тяжелым течением ОИП. Таким образом, проведенный молекулярно-генетический анализ в группе больных идиопатическим панкреатитом позволяет утверждать, что развитие заболевания у данной категории больных связано с определенными генетическими механизмами. Согласно нашим исследованиям, установлено: – воспалительный процесс в ткани поджелудочной железы детерминирован мутациями в генах, непосредственно участвующих в регуляции ферментной активности железы, а не в генах, осуществляющих взаимодействие организма с внешней средой. – главным геном, детерминирующим развитие деструктивного панкреатита в группе больных ОИП, является ген, кодирующий ингибитор трипсина – SPINK1. Мутация N34S явилась причиной развития деструктивной формы острого панкреатита у 4-х больных (12,5%), при этом изолированные гетерозиготные мутации N34S гена SPINK1 ассоциировались с тяжелым течением в 9,4%, и только в одном случае (3,1%) этому способствовала гомозиготная мутация N34S гена SPINK1 в сочетании с заменой R122H гена PRSS1 [табл.1, табл.2]. – особое значение имеет тот факт, что наличие мутаций в генах SPINK1, PRSS1 и CFTR сопровождается развитием панкреонекроза в 21,8% случаев, полиморфизм гена катионного трипсиногена в комбинации с “нулевым” генотипом GSTM1, мутацией N34S гена SPINK1, мажорной мутацией delF508 гена CFTR ассоциирован с развитием деструктивной формой ОИП в 12,5%, сочетание мутаций в генах, вовлеченных в функционирование клеток поджелудочной железы: SPINK1, PRSS1 и CFTR, приводит к формированию панкреонекроза тяжелой степени тяжести, сопровождается тотальным поражением ткани поджелудочной железы и забрюшинной клетчатки, повышая риск развития осложнений и летальных исходов [табл.1]. Масштаб поражения поджелудочной железы при наличии сочетанных мутаций представлен на фиг.3, где 1 – 3,1% тотальное поражение при сочетании мутации R122H гена PRSS1 и delF508 CFTR, 2 – 9,4% субтотальное поражение при изолированной мутации N34S гена SPINK1, 3 – 6,2% субтотальное поражение при мутации R122H гена PRSS1 и GSTM1 0/0, 4 – 3,1% тотальное парожение с вовлечением забрюшинной клетчатки при сочетании мутаций N34S гена SPINK1 и R122H гена PRSS1. Это подтверждает тот факт, что из 9 больных с деструктивной формой ОИП «двойные» мутации были зафиксированы у пяти больных, четверо из них погибли в течение первых 2-3 суток с момента госпитализации, что составило 44,5% от общего количества больных панкреонекрозом идиопатической этиологии. У всех больных наблюдалось тотальное некротическое поражение паренхимы поджелудочной железы и обширный воспалительный процесс забрюшинной клетчатки. Основной причиной смерти у этих больных явилась полиорганная недостаточность. Пример 1: Больной Ш., 34 года, 8.09.05 г доставлен бригадой скорой помощи в панкреатологический центр г.Красноярска с диагнозом: острый панкреатит. Жалобы при поступлении: боли в эпигастрии и левом подреберье опоясывающего характера, тошнота, рвота. Из анамнеза известно, что заболел около 6 суток назад, без видимой причины. Объективно: состояние тяжелое, кожные покровы бледные, с «мраморным» оттенком. Дыхание ослаблено, хрипов нет. ЧДД = 18 в минуту. Тоны сердца приглушены, ритмичные. Пульс = 92 уд. в мин, АД=90/60 мм рт.ст. Язык сухой, обложен белым налетом. Живот в акте дыхания не участвует, при пальпации напряжен и резко болезненный в верхних отделах. Симптомы Воскресенского, Мейо-Робсона положительные. Симптомы раздражения брюшины положительные. Диурез 1200 мл/сутки. УЗИ от 8.09.05: неоднородность эхоструктуры, контуры поджелудочной железы размыты, снижена эхонегативность парапанкреатической клетчатки. R-граммы органов грудной клетки и живота от 8.09.05 без особенностей. В анализах крови: Hb = 152 г/л, Эр. = 4,3×1012/л, L = 22×10%; билирубин общий = 24 мкмоль/л, прямой = 11,8 мкмоль/л, непрямой = 12,2 мкмоль/л. Амилаза крови 31 мг/с×л, мочевина 9,2 ммоль/л. Сахар крови = 5,8 ммоль/л. АлАТ=0,9 ммоль/л, АсАТ=0,8 ммоль/л. Параметры гемокоагуляции в пределах нормы. Диастаза в моче: 150 ед. Диагноз при поступлении: острый панкреатит средней степени тяжести. При генотипировании: выявлено сочетание гомозиготного варианта мутации N34S гена SPINK1 и гетерозиготного варианта R122H мутации в гене катионного трипсиногена, что говорит о высоком риске тяжелого течения заболевания у данного пациента и возможном неблагоприятном исходе. Учитывая полученные результаты с 8.09.05 для дальнейшего лечения больной переведен в отделение реанимации и интенсивной терапии. Лечение: проведена консервативная терапия, включающая обезболивание анальгином, назначение сандостатина 100 мг/сут, контрикала 40-60 т.ед./сут, спазмолитиков и холинолитиков, кваматела 40 мг/сут, цефабола 3 г/сут, инфузионная терапия в объеме до 7,5 л/сут, под контролем ЦВД и диуреза. Мониторное наблюдение, лабораторный мониториг. Несмотря на проводимое лечение состояние больного ухудшилось. Усилились боли в животе, снизилось артериальное давление, принято решение об оперативном лечении. 10.09.05 операция: Верхнесрединная лапоротомия, абдоминизация поджелудочной железы, вскрытие парапанкреатической и параколической забрюшинной клетчатки. Дренирование и марсупиализация сальниковой сумки. Санация и дренирование брюшной полости, забрюшинного пространства справа. DS: Тотальный жировой панкреонекроз. Разлитой ферментативный перитонит. Парапанкреатит. Забрюшинный целлюлит с поражением ретродуоденальной, паранефральной и пароколической клетчатки справа. В послеоперационном периоде проводилась традиционная терапия, включающая коррекцию гемодинамических, водно-электролитных нарушений, спазмолитическую, антисекреторную, цитостатическую терапию, назначение блокаторов H2-рецепторов, антибиотикотерапию, санация гнойных затеков через установленные дренажи. Несмотря на интенсивную полномасштабную терапию, проводимую в послеоперационном периоде, состояние пациента прогрессивно ухудшалось. 11.09.05 констатирована смерть. Приведенный пример доказывает диагностическую и прогностическую значимость генотипирования у больных острым панкреатитом. Своевременное использование предлагаемого способа, разработка в сооответствии с полученными результатами профилактических мер до клинико-лабораторной манифестации этого заболевания позволила бы снизить риск возникновения острого идиопатического панкреатита у данного пациента. Выявление сочетания гомозиготного варианта мутации N34S гена SPINK1 и гетерозиготного варианта R122H мутации в гене катионного трипсиногена дало возможность прогнозировать высокий риск тяжелого течения заболевания у данного пациента и, учитывая позднее обращение пациента, его неблагоприятный исход, несмотря на интенсивную терапию. Предлагаемый способ позволяет: 1. Прогнозировать риск развития острого идиопатического панкреатита до его клинико-лабораторной манифестации, что дает возможность своевременно разработать меры по его профилактике. 2. Выделить группу больных с высоким риском развития осложнений и обоснованно рекомендовать им проведение полномасштабной специализированной терапии на ранних сроках. 3. В будущем определить оптимальную стратегию при лечении заболевания с точки зрения предиктивной медицины и снизить или полностью ликвидировать летальность у больных.
Примечания: «+» – нормальный аллель гена; «0» – делеционный («нулевой») аллель гена Список литературы: 10. Черданцев Д.В. Коррекция синдрома системной воспалительной реакции при остром панкреатите (экспериментально-клиническое исследование): автореф. дис. … д-ра мед. наук / Д.В.Черданцев. – Красноярск, 2002. – 59 с. 14. Багненко С.Ф., Рухляда Н.В., Толстой А.Д. и др. Лечение острого панкреатита на ранней стадии заболевания: НИИ Скорой помощи им. И.И.Джанелидзе. – СПб, 2002. – 24 с. 16. Никонов, Е.Л. Клинико-патогенетические особенности различных видов антисекреторной терапии у больных кислото-зависимыми заболеваниями: автореф.дис. … д-ра мед. наук./ Е.Л Никонов. – Москва, 2004. – 43 с. 17. Геном человека и гены «предрасположенности» (введение в предиктивную медицину) / А.С.Баранов, Е.В.Баранова, Т.Э.Иващенко, М.В.Асеев. – СПб.: Интермедика, 2000. – 271 с. Формула изобретения
Способ прогнозирования клинического течения острого идиопатического панкреатита, включающий генотипирование пациента с помощью полимеразной цепной реакции, отличающийся тем, что генотипирование осуществляют по ДНК, полученной из крови пациента с острым идиопатическим панкреатитом, исследуют мутации генов SPINK1, GSTM1, GSTT1, PRSS1 и CFTR, при обнаружении гетерозиготных мутаций в генах SPINK1, PRSS1 и CFTR прогнозируют развитие деструктивной формы идиопатического панкреатита, при обнаружении сочетания не менее чем двух мутировавших генов из SPINK1, GSTM1, GSTT1, PRSS1 и CFTR у гетерозиготных носителей или гомозиготных носителей по мутации N34S гена SPINK1 в сочетании с гетерозиготной мутацией R122H гена PRSS1, прогнозируют формирование панкреонекроза тяжелой степени.
РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 12.04.2008
Извещение опубликовано: 20.09.2009 БИ: 26/2009
|
||||||||||||||||||||||||||||||||||||||||
 7. Пирсона.
7. Пирсона.