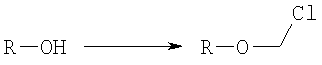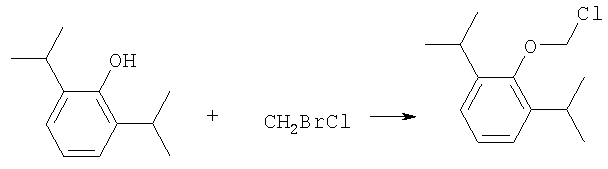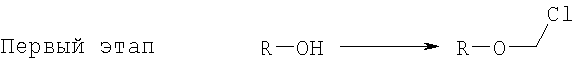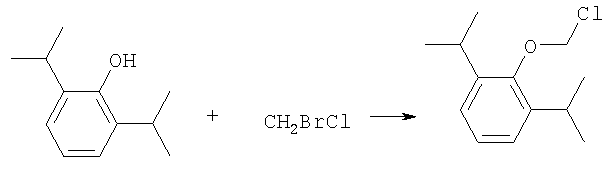Патент на изобретение №2310659
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ФОСФОНООКСИМЕТИЛОВЫХ ПРОИЗВОДНЫХ СПИРТА И ФЕНОЛА
(57) Реферат:
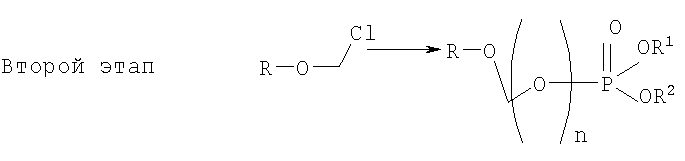
Данное изобретение касается нового способа получения водорастворимых форм фармацевтических препаратов, содержащих блокированную ароматическую гидроксильную группу. Описывается способ получения водорастворимых фосфонооксиметиловых производных, включающий этапы: этап 1:
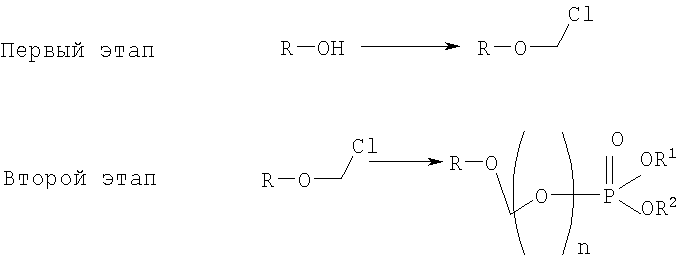
на котором R-OH преобразуется в R-O-CH2Cl и этап 2:
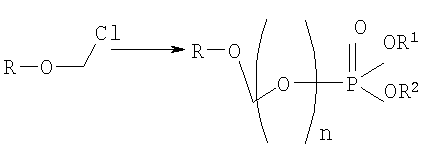
на котором используется продукт, полученный на первом этапе, где R-O-CH2Cl реагирует с молярным избытком фосфорной кислоты и основания в подходящем полярном апротонном растворителе; и где R-OH представляет лекарство, содержащее спирт или фенол, n представляет собой целое число 1 или 2, R1 является водородом, ионом щелочного металла, или лекарственно-приемлемым катионом, и R2 является водородом, ионом щелочного металла или лекарственно-приемлемым катионом. Также описывается способ получения водорастворимого фосфонооксиметилпропофола. Технический результат – упрощение процесса получения водорастворимых форм фармацевтических препаратов. 2 н. и 24 з.п. ф-лы.
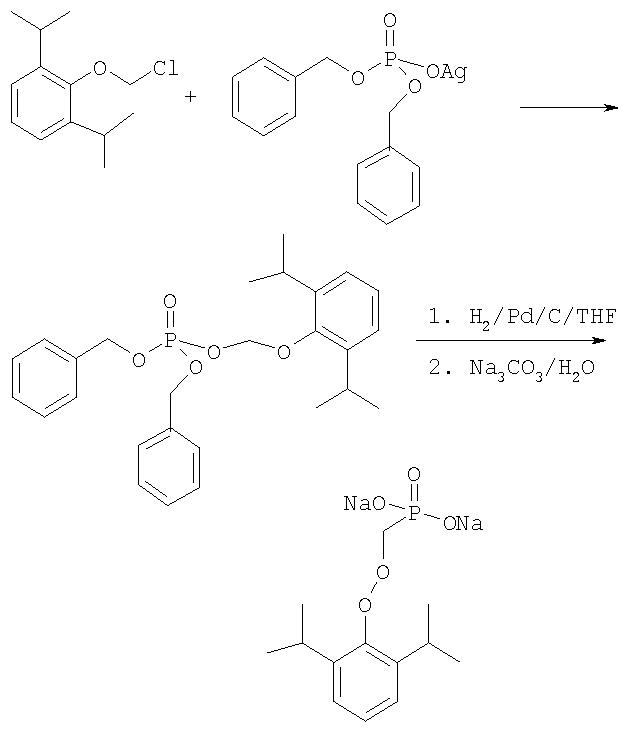
ОБЛАСТЬ ИЗОБРЕТЕНИЯ Данное изобретение касается нового способа получения водорастворимых пролекарств фармацевтических препаратов, содержащих блокированную ароматическую гидроксильную группу. Конкретно, данное изобретение касается способа получения фармацевтических препаратов, содержащих водорастворимые фосфонооксиметиловые эфиры блокированного спирта и фенола типа камтпотецина, пропофола, этопозида, витамина Е и циклоспорина А. ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ Успешная поставка фармацевтической продукции пациенту имеет решающее значение в лечении заболеваний. Однако использование многих клинических лекарств с известными свойствами ограничено из-за их очень низкой растворимости в воде. В результате этого такие лекарства должны быть приготовлены в виде композиций с фармацевтическими носителями, включающими поверхностно-активные вещества. Оказалось, что введенные в организм эти поверхностно-активные вещества приводят к серьезным побочным эффектам, что ограничивает клиническую безопасность этих лекарств и, соответственно, ограничивает их применение для лечения ряда заболеваний. Например, камптотецин является натуральным продуктом, выделенным из коры китайского камптотекового дерева, Camptotheca accuminata. В нескольких модельных экспериментах с животными in vivo было показано, что камптотецин обладает сильной противоопухолевой активностью, включая основные виды опухоли типа рака легкого, груди, яичника, поджелудочной железы, толстой кишки и живота, а также злокачественную меланому. Серьезный недостаток камптотецина – его весьма ограниченная растворимость в воде. Для биологических исследований необходимо растворить соединение в сильном органическом растворителе (DMSO) или приготовить суспензию лекарства в смеси – Tween 80: солевой раствор, который является нежелательной лекарственной композицией для лечения человеческого организма. Недавно два аналога камптотецина с умеренной водорастворимостью были одобрены в Соединенных Штатах для лечения запущенного рака яичника (гикамтин) и рака ободочной и прямой кишки (камптозар). Другие лекарства, подобно камптотецину, которые имеют подобные недостатки – это циклоспорин A (CsA), пропофол, этопозид и витамин Е (альфа токоферол). Подобно камптотецину, структура CsA содержит стерический блокированный спирт, в данном случае – вторичный спирт. CsA сформулирован в виде смеси со CremophorEL/спирт. Примером пространственно блокированного, малорастворимого в воде фенола является анестезирующее средство пропофол. Пропофол сформулирован для клинического использования внутривенно, в виде водной эмульсии. Пропофол не только малорастворим в воде, но также причиняет боль на участке инъекции. Для облегчения боли можно использовать, например, лидокаин. Кроме того, пропофол сформулирован в виде эмульсии, и при добавлении к данной формулировке других лекарств возникают трудности и сомнения, так как увеличение размеров масляных капель может привести к эмболии легкого, и т.д. Патент США 6204257 описывает растворимую в воде форму лекарств, содержащих спирт и фенол, типа камптотецина и пропофола. В случае камптотецина соединениями являются фосфонооксиметиловые эфиры камптотецина в виде свободной кислоты и ее лекарственно-приемлемых солей. Растворимость в воде кислоты и ее солей улучшается при приготовлении фармацевтических формулировок. Однако способы получения растворимых в воде форм лекарств, содержащих спирт и фенол, описанные в патенте США 6204257, сложны и требуют использования дорогих канцерогенных реактивов. Например, синтез О-фосфонооксиметилпропофола требует 6 этапов, как показывает приведенная ниже итоговая схема реакции.
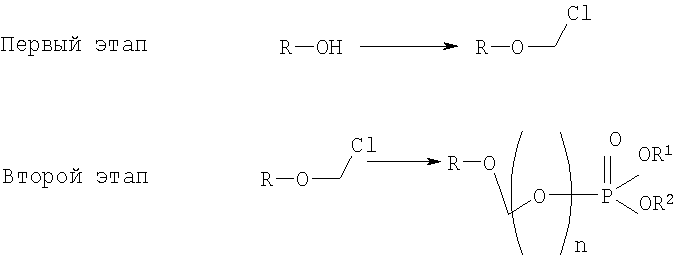
Желательно иметь более короткий и не требующий использования дорогих канцерогенных реактивов способ. КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Данное изобретение касается нового способа получения растворимых в воде лекарств, содержащих фосфонооксиметиловые производные спирта и фенола, в частности, динатриевой соли фосфоно-О-метил-2,6-диизопропилфенола. Данное изобретение конкретно направлено на получение растворимых в воде фосфонооксиметиловых производных согласно этапам:
где R-ОН представляет собой лекарство, содержащее спирт или фенол, n представляет собой целое число 1 или 2, R1 является водородом, ионом щелочного металла или лекарственно-приемлемым катионом, R2 является водородом, ионом щелочного металла или лекарственно-приемлемым катионом. В предпочтительном осуществлении 2,6-диизопропилфенол реагирует с бромхлорметаном с образованием O-хлорметил-2,6-диизопропилфенола.
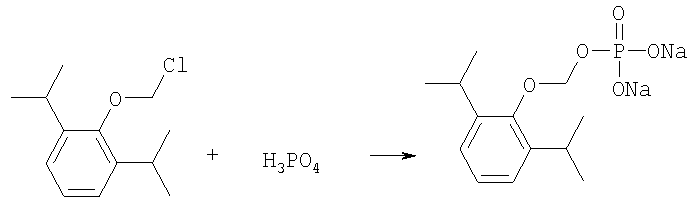
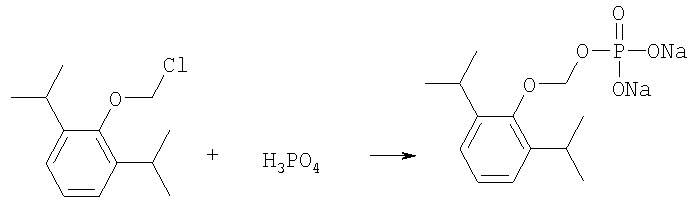
O-хлорметил-2,6-диизопропилфенол реагирует с фосфорной кислотой с образованием динатриевой соли фосфоно-O-метил-2,6-диизопропилфенола.
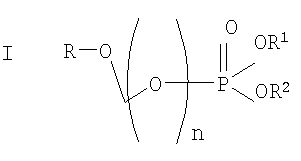
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Данное изобретение касается нового способа получения растворимых в воде лекарств, содержащих фосфонооксиметиловые производные спирта и фенола, конкретно, динатриевой соли фосфоно-O-метил-2,6-диизопропилфенола. Такие фосфонооксиэтиловые производные описаны в патенте США 6204257, который полностью включен сюда в виде ссылки. Способ данного изобретения требует только двух этапов и не требует канцерогенного и дорогого сырья, которое необходимо для осуществления способов-прототипов. Кроме того, не требуется хроматография. Процесс кончается высокими выходами продукта, обычно, до 85% и приблизительно 40-85%. Описанное здесь изобретение использует новый способ для получения водорастворимых фармацевтических препаратов, содержащих фосфонооксиметиловые производные спирта и фенола, представленные формулой I:
Формула I является производным R-ОН, где R-ОН представляет лекарство, содержащее спирт или фенол, типа камптотецина, пропофола, этопозида, витамина Е и циклоспорина А. Предпочтительно, R-ОН является фенолсодержащим фармацевтическим препаратом типа пропофола. Включены также некоторые лекарства, инъекционные формы которых невозможны из-за свойственной им низкой растворимости в воде. Они включают, без ограничения, даназол, метилтестостерон, йодохинол, атовахинол и флуконал, Термин n представляет собой целое число 1 или 2, предпочтительно 1. R1 – водород или ион щелочного металла, включая натрий, калий, литий, протонированный амин, протонированную аминокислоту или любой другой лекарственно-приемлемый катион. R2 – водород или ион щелочного металла, включая натрий, калий, литий, протонированный амин, протонированная аминокислота или любой другой лекарственно-приемлемый катион. Производные формулы I могут быть получены согласно следующей реакционной схеме:
где R-ОН представляет лекарство, содержащее спирт или фенол, n представляет целое число 1 или 2, R1 является водородом, ионом щелочного металла или лекарственно-приемлемым катионом, R2 является водородом, ионом щелочного металла или лекарственно-приемлемым катионом. На первом этапе R-OH реагирует с большим избытком бромхлорметана в присутствии основания и тетрагидрофурана (THF). После этого образующийся продукт реагирует с избытком фосфорной кислоты и основания в подходящем растворителе. Пример вышеприведенной схемы можно иллюстрировать с использованием в качестве исходного материала 2,6-диизопропилфенола. На первом этапе, 2,6-диизопропилфенол реагирует с бромхлорметаном с образованием О-хлорметил-2,6-диизопропилфенола.
На втором этапе, O-хлорметил-2,6-диизопропилфенол реагирует с фосфорной кислотой с образованием динатриевой соли фосфоно-О-метил-2,6-диизопропилфенола.
Более конкретно, на первом этапе 2,6-диизопропилфенол реагирует с большим молярным избытком бромхлорметана в присутствии основания и соответствующего растворителя, предпочтительно, тетрагидрофурана (THF), с образованием O-хлорметил-2,6-диизопропилфенола. Температура реакции может быть в пределах приблизительно 20-100°С, и предпочтительно в пределах приблизительно 25-65°С. В качестве основания предпочтительно применяют гидроокись щелочного металла или гидрид щелочного металла. Подходящие гидроокиси щелочных металлов и гидриды щелочных металлов включают, без ограничения, гидрид натрия и гидроокись натрия. Количество основания составляет, по крайней мере, приблизительно 1,5 моля основания на 1 моль 2,6-диизопропилфенола. Количество бромхлорметана составляет, по крайней мере, приблизительно 10 молей, предпочтительно приблизительно 10-30 молей бромхлорметана на 1 моль 2,6-диизопропилфенола. Предполагается, что бромхлорметан можно заменить йодхлорметаном. THF может быть заменен другими соответствующими растворителями, такими как апротонные, кислородсодержащие растворители, с сильной растворяющей способностью, типа гликолевых эфиров. На втором этапе O-хлорметил-2,6-диизопропилфенол реагирует с молярным избытком фосфорной кислоты и основания в соответствующем растворителе. К 1 молю O-хлорметил-2,6-диизопропилфенола добавляется по крайней мере 3, предпочтительно около 3-10, конкретно около 6 молей фосфорной кислоты и основания. Температура реакции ниже 100°С, обычно около 25-80°С. Пригодные растворители включают полярные апротонные растворители, такие как ацетонитрил, диметилформамид (DMF), диметилсульфоксид (DMSO) или N-метилпирролидон (NMP). Растворитель должен обладать способностью растворять триэтиламмонийную соль продукта. Предпочтительными растворителями являются алкиламины, пиридины или замещенные производные пиридина. Наиболее предпочтительным растворителем является триэтиламин. После этого растворитель удаляют, остаток растворяют в воде и подкисляют, например, соляной кислотой до рН приблизительно 1-2, предпочтительно приблизительно 1,5. Продукт в форме свободной кислоты экстрагируют с помощью МТВЕ. После этого растворитель удаляют под вакуумом. Остаток растворяют в воде соответствующим реагентом, например гидроокисью натрия, рН доводят до приблизительно 8-11,5 и раствор экстрагируют подходящим растворителем, например толуолом. Далее раствор концентрируют, прибавляют изопропанол или другой подходящий растворитель и продукт осаждают и перекристаллизуют. Пригодные для выделения динатриевой соли фосфоно-О-метил-2,6-диизопропилфенола растворители включают полярные, смешивающиеся с водой, органические растворители, такие как ацетон, ацетонитрил, спирт, THF, или диоксан. Пример 1 Этап 1: Четырехгорлая круглодонная колба на 500 мл была снабжена механической мешалкой, трубкой для входа азота, обратным холодильником и термометром. В колбу загружали 17,8 г (0,10 моля) 2,6-диизопропилфенола, 200 мл THF, 8,0 г (0,2 моля) гранулированной гидроокиси натрия и 387 г (3,0 моля, 194 мл) бромхлорметана. Реакционную смесь нагревали до 64°С и выдерживали в течение 2-3 часов до тех пор, пока анализ методом ГЖХ не показал отсутствие в реакционной смеси 2,6-диизопропилфенола. После охлаждения до 25°С суспензию отфильтровывали и осадок на фильтре промывали THF. THF удаляли на ротационном испарителе и полученное масло перегоняли под вакуумом (0-1 торр, температура кипения 80°С); в результате получали 16,9 г (0,074 моля, выход 75%) O-хлорметил-2,6-диизопропилфенола. Этап 2: Четырехгорлая круглодонная колба на 1 л, которая была снабжена термопарой для контроля температуры, была помещена в нагревательный кожух. В колбу загружали 300 мл ацетонитрила, а далее триэтиламин (48,7 мл, 0,349 моль) и 85% фосфорную кислоту (18,7 мл, 0,318 моля). После этого добавляли O-хлорметил-2,6-диизопропилфенол (12 г, 0,0529 моля) и реакционный раствор нагревали до 65°С в течение 2 часов. Реакцию считали законченной при исчезновении исходного материала, который определяли методами TLC (тонкослойная хроматография) и HPLC (жидкостная хроматография высокого давления). Раствору позволяли охладиться и смесь концентрировали под уменьшенным давлением. Остаток растворяли в 500 мл воды и подкисляли 8N HCl до рН 1,5. Этот раствор экстрагировали три раза с 500 мл МТВЕ. Объединенные органические экстракты промывали один раз раствором соли и органический слой фильтровали через целит. К остатку добавляли 60 мл воды и 20%-ный раствор NaOH, до рН 8,6. Этот раствор промывали дважды 50 мл толуола. Водный раствор был сконцентрирован под уменьшенным давлением до половины первоначального объема, после чего к нему добавляли 315 мл изопропанола. Для растворения продукта смесь была нагрета до 70°С и далее была охлаждена до 0°С. Фильтрацией путем отсасывания выделяли белое кристаллическое вещество, которое промывали один раз 45 мл изопропанола и сушили в вакуумной сушилке (30 дюймов Hg, 45°С) в течение 48 часов; в результате получали 13.1 грамм (0.039 моля, выход 75%) белого твердого вещества. Пример 2 Этап 1: 2,6-диизопропилфенол (20 кг, FW (молекулярная масса) = 178, 112 моля, 1 эквивалент) реагирует с бромхлорметаном (347 кг, FW=129, 2.682 моля, 24 эквивалента) и гидроокисью натрия (11 кг, FW=40, 280 моля, 2,5 эквивалента) в тетрагидрофуране (108 кг, FW=72, 1.498 моля, 13,3 эквивалента) при кипячении в течение приблизительно 1,5 часа. После охлаждения до 20°С реакционную смесь разлагают водой (87 кг). Органический слой отделяют и дважды промывают 15%-ным водным раствором хлорида натрия (78 кг). После расслоения смеси органический слой снова отделяют и из него отгоняют растворитель; в результате получается сырое масло, которое дистиллируют обычным способом и получают очищенный хлорметил-2,6-диизопропилфениловый эфир (FW=227) в виде масла светло-желтого цвета. Хлорметил-2,6-диизопропилфениловый эфир (20 кг, FW=227, 88,20 моль, 1 эквивалент) реагирует с фосфорной кислотой (81 кг, 85%, FW=98,705 моль, 8 эквивалент) и триэтиламином (89 кг, FW=101,883 моля, 10 эквивалентов) в ацетонитриле (200 кг, FW=41, 4,872 моля, 55 эквивалентов) при приблизительно 75°С в течение 3 часов. Реакционную смесь охлаждают и концентрируют под уменьшенным давлением. Полученную кашеобразную массу растворяют в воде, рН смеси доводят до 1,5 концентрированной соляной кислотой. Подкисленную смесь дважды экстрагируют толуолом. Экстракты объединяют и промывают водой один раз. Органический раствор концентрируют под вакуумом. Полученное масло смешивают с очищенной водой, USP. рН смеси доводят до 11 50%-ным водным раствором гидроокиси натрия. Водный раствор дважды промывают толуолом. Далее водный раствор частично концентрируют под вакуумом до приблизительно 40-50% первоначального объема. К концентрированному водному раствору при 70°С прибавляют изопропиловый спирт (525 кг) и полученную смесь охлаждают до 0°С с целью кристаллизации продукта. Твердое вещество выделяют путем фильтрации и сушат под вакуумом, в результате чего получают динатриевую соль фосфоно-O-метил-2,6-дизопропилфенола (C13H19Na2O5P, FW=332). Пример 3 Этап 1: Четырехгорлая круглодонная колба на 500 мл была снабжена механической мешалкой, трубкой для входа азота, обратным холодильником и термометром. В колбу загружали 8,9 г (0,05 моля) 2,6-диизопропилфенола, 100 мл THF и 4,0 г (0,10 моля) гранулированной гидроокиси натрия. Полученную зеленую суспензию нагревали при 60-65°С и выдерживали в течение 1 часа. Далее реакционную смесь охлаждали до 30°С, добавляли 100 мл (199 г, 1,54 моль) бромхлорметана и снова нагревали. Реакционную смесь выдерживали при 64°С в течение 2-3 часов до тех пор, пока анализ методом ГЖХ не показал отсутствие в реакционной смеси 2,6-диизопропилфенола. После охлаждения до 25°С суспензию отфильтровывали через целитовый фильтр и осадок на фильтре промывали THF. THF удаляли на ротационном испарителе и полученное масло перегоняли под вакуумом (0-1 торр, т.кип.=80°С), в результате получали 9,6 г (0,042 моля, выход 85%) O-хлорметил-2,6-диизопропилфенола. Этап 2: Четырехгорлая круглодонная колба на 1 л, которая была снабжена термопарой для контроля температуры, была помещена в нагревательный кожух. В колбу загружали 300 мл ацетонитрила, а далее триэтиламин (48,7 мл, 0,349 моля) и 85% фосфорную кислоту (18,7 мл, 0,318 моля). После этого добавляли O-хлорметил-2,6-диизопропилфенол (12 г, 0,0529 моля) и реакционный раствор нагревали до 65°С в течение 2 часов. Реакцию считали законченной при исчезновении исходного материала, который определяли методами TLC (тонкослойная хроматография) и HPLC (жидкостная хроматография высокого давления). Раствору позволяли охладиться и смесь концентрировали под уменьшенным давлением. Остаток растворяли в 500 мл воды и подкисляли 8N НС1 до рН 1,5. Этот раствор экстрагировали три раза с 500 мл МТВЕ. Объединенные органические экстракты промывали один раз раствором соли и органический слой фильтровали через целит. К остатку добавляли один эквивалент TEA и раствор концентрировали под уменьшенным давлением. К полученному остатку добавляли 60 мл воды и 20%-ный раствор NaOH до рН 8,6. Этот раствор промывали дважды 50 мл толуола. Водный раствор был сконцентрирован под уменьшенным давлением, после чего к нему была добавлена вода (30 мл) и раствор был охлажден в ледяной ванне. Далее к нему прибавляли по каплям ацетонитрил (300 мл). Полученная смесь была охлаждена в холодильнике в течение ночи. Смесь снова охлаждали в ледяной ванне в течение 1 часа, а далее твердое вещество отделяли фильтрованием. Полученное твердое вещество белого цвета сушили в вакуумной сушилке (30 дюймов Hg, 45°С) в течение 48 часов, в результате получали 7,50 г (0.023 моля, выход 43%) белого твердого вещества. Пример 4 2,6-Диизопропилфенол был обработан бромхлорметаном в присутствии годроокиси натрия и THF; в результате с 85%-ным выходом был получен О-хлорметил-2,6-диизопропилфенол, который был очищен путем вакуумной дистилляции. Обработка O-хлорметил-2,6-диизопропилфенола фосфорной кислотой и триэтиламином в ацетонитриле и последующее удаление растворителя, растворение полученного продукта в метаноле, регулирование рН и осаждение ацетоном дает с 85%-ным выходом динатриевую соль фосфоно-O-метил-2,6-диизопропилфенола. В то время как изобретение было описано относительно определенных примеров, включая, предпочтительные в настоящее время способы осуществления изобретения, специалисту должно быть понятно, что существуют многочисленные разновидности и перестановки вышеупомянутых описанных систем и способов, которые совпадают с духом и возможностями данного изобретения.
Формула изобретения
1. Способ получения водорастворимых фосфонооксиметиловых производных, включающий этапы: этап 1:
на котором R-OH преобразуется в R-O-CH2Cl и этап 2:
на котором используется продукт, полученный на первом этапе, где R-O-CH2Cl реагирует с молярным избытком фосфорной кислоты и основания в подходящем полярном апротонном растворителе; и где R-OH представляет лекарство, содержащее спирт или фенол, n представляет собой целое число 1 или 2, R1 является водородом, ионом щелочного металла, или лекарственно-приемлемым катионом, и R2 является водородом, ионом щелочного металла или лекарственно-приемлемым катионом. 2. Способ согласно п.1, где R-OH представляет камптотецин, пропофол, этопозид, витамин Е или циклоспорин А. 3. Способ согласно п.1, где n равно 1. 4. Способ согласно п.1, где R1 и R2 независимо отобраны из группы, состоящей из водорода, натрия, калия, лития, протонированного амина и протонированной аминокислоты. 5. Способ согласно п.1, где на первом этапе R-OH реагирует с молярным избытком бромхлорметана в присутствии основания и подходящего растворителя. 6. Способ согласно п.1, где на первом этапе R-OH реагирует с молярным избытком йодхлорметана в присутствии основания и подходящего растворителя. 7. Способ согласно п.5, где основанием является гидроокись щелочного металла или гидрид щелочного металла. 8. Способ согласно п.7, где основанием является гидроокись натрия или гидрид натрия. 9. Способ согласно п.5, где растворителем является апротонный растворитель или апротонный кислородсодержащий растворитель. 10. Способ согласно п.9, где растворителем является тетрагидрофуран. 11. Способ согласно п.5, где на каждый один моль R-OH приходится по крайней мере приблизительно 1,5 моля основания. 12. Способ согласно п.5, где на каждый один моль R-OH приходится по крайней мере приблизительно 10 молей бромхлорметана. 13. Способ согласно п.12, где на каждый один моль 2,6-диизопропилфенола приходится по крайней мере приблизительно 10-30 молей бромхлорметана. 14. Способ согласно п.1, где R-OH представляет 2,6-диизопропилфенол. 15. Способ согласно п.1, где температура реакции на первом этапе составляет приблизительно 25-65°С. 16. Способ согласно п.1, где на каждый один моль R-O-CH2Cl приходится по крайней мере приблизительно 3 моля фосфорной кислоты. 17. Способ согласно п.16, где на каждый один моль R-O-CH2Cl приходится по крайней мере приблизительно 3-10 молей фосфорной кислоты. 18. Способ согласно п.1, где температура реакции на втором этапе ниже 100°С. 19. Способ согласно п.18, где температура реакции составляет приблизительно 25-65°С. 20. Способ согласно п.1, где полярный апротонный растворитель отобран из группы, включающей ацетонитрил, диметилформамид, диметилсульфоксид, или N-метил пирролидон. 21. Способ согласно п.1, где основанием является алкиламин, пиридин, или замещенное производное пиридина. 22. Способ согласно п.21, где основанием является триэтиламин. 23. Способ согласно п.1, где на втором этапе рН сначала выдерживают в пределах приблизительно 1-2, а далее доводят до значения приблизительно 8-11,5. 24. Способ получения водорастворимого фосфонооксиметил-пропофола, включающий реакцию 2,6-диизопропилфенола с бромхлорметаном с образованием О-хлорметил-2,6-диизопропилфенола:
и далее, реакцию O-хлорметил-2,6-диизопропилфенола с фосфорной кислотой с образованием динатриевой соли фосфоно-О-метил-2,6-диизопропилфенола.
25. Способ согласно п.6, где на каждый один моль R-O-H приходится по крайней мере приблизительно 10 молей йодохлорметана. 26. Способ согласно п.6, где на каждый один моль 2,6-диизопропилфенола приходится по крайней мере приблизительно 10-30 молей йодохлорметана.
Прежний патентообладатель:
(73) Патентообладатель:
Договор № РП0000853 зарегистрирован 25.06.2010
Извещение опубликовано: 10.08.2010 БИ: 22/2010
PC4A – Регистрация договора об уступке патента СССР или патента Российской Федерации на изобретение
Прежний патентообладатель:
(73) Патентообладатель:
Договор № РД0068249 зарегистрирован 10.08.2010
Извещение опубликовано: 20.09.2010 БИ: 26/2010
|
||||||||||||||||||||||||||