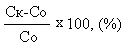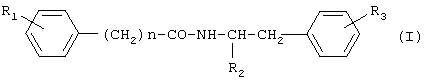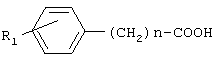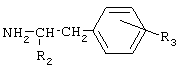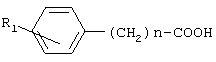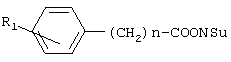Патент на изобретение №2309144
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ФЕНИЛСОДЕРЖАЩИЕ N-АЦИЛЬНЫЕ ПРОИЗВОДНЫЕ АМИНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ И АНАЛЬГЕТИЧЕСКИХ СРЕДСТВ
(57) Реферат:
Изобретение относится к фенилсодержащим N-ацильным производным аминам общей формулы I, где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1; и их фармацевтически приемлемым солям. Изобретение относится к способу получения соединений общей формулы I (где n равно 1 или 2), осуществляемому за счет активации карбоксильной группы п-гидроксифенилуксусной кислоты или фенилуксусной кислоты, взаимодействием с дифенилфосфорилазидом и триэтиламином в органическом растворителе при охлаждении с последующим взаимодействием с аминосоединением. Изобретение относится к способу получения соединений формулы I (где n равно 1), осуществляемому за счет превращения п-гидроксифенилуксусной кислоты или фенилуксусной кислоты в активированный N-оксисукцинимидный эфир N’-дициклогексилкарбодиимидным методом, с последующим взаимодействием активированного N-сукцинимидного эфира с аминопроизводным. Изобретение относится к фармацевтической композиции и средству, обладающим ингибирующей циклооксигеназу активностью, включающим в качестве активного агента эффективное количество соединения общей формулы (I) (где n равно 1 или 2). Также изобретение относится к способу ингибирования циклооксигеназы, осуществляемому путем введения млекопитающему эффективного количества соединения общей формулы (I) (где n равно 1 или 2) или его фармацевтически приемлемой соли. Технический результат – получение фенилсодержащих N-ацильных производных аминов, обладающих ингибирующей циклооксигеназу активностью и применяемых для лечения болевых синдромов, воспалительных и воспалительно-дегенеративных заболеваний суставов и соединительной ткани, а также костно-мышечной системы, других заболеваний, сопровождающихся воспалением. 7 н. и 12 з.п. ф-лы, 9 табл.
(56) (продолжение): CLASS=”b560m”02.07.2002. JP 57-193437 А, 27.11.1982. US 2003/99566 А, 23.10.2003. WO 99/52962 A1, 21.10.1999. RU 2123041 C1, 20.01.1999.
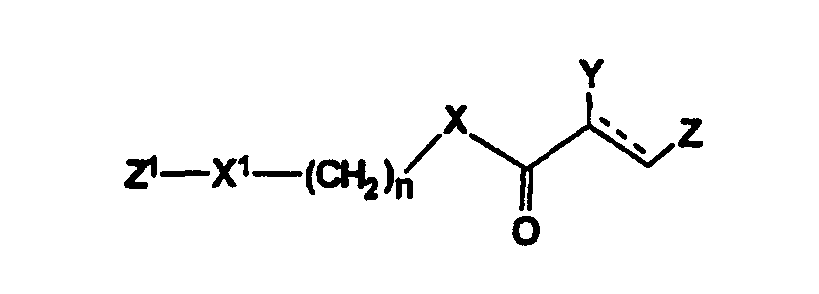
Настоящее изобретение относится к области биоорганической химии и касается новых соединений – фенилсодержащих N-ацильных производных биогенных аминов, а также способа синтеза новых и известных соединений, их применения в медицине в качестве потенциальных анальгетических и противовоспалительных средств. Предшествующий уровень техники В публикации международной заявки WO 97/23202 раскрыты фенилсодержащие N-ацильные производные аминов общей формулы (XV)
включающие, среди прочих, 3-(п-гидроксифенил) пропионилфенилэтиламин, 3-(п-гидроксифенил)пропионилтирамин и 3-фенилпропионилфенилэтиламин (соединения IX, X, XI настоящего изобретения, соответственно), в качестве промежуточных соединений, а также их синтез и применение в качестве селективных лигандов подтипов NMDA рецепторов, используемых для лечения хронической боли, мигреневой головной боли, а также анестетиков. Однако, ни в описании, ни в формуле изобретения указанной публикации не описаны и не охарактеризованы конкретные структуры, соответствующие соединениям X и XI настоящего изобретения, и отсутствуют какие-либо данные, подтверждающие заявленный вид активности, а соединение IX в качестве промежуточного соединения и его синтез раскрыты лишь в способе получения других производных аминов. Соединения IX, X и XI настоящего изобретения также описаны в более ранних публикациях, ставших общедоступными до даты приоритета вышеуказанной международной заявки WO 97/23202, для использования по иному назначению. 3-(п-Гидроксифенил)пропионилфенилэтиламин (IX) раскрыт в Jacobson K.A., Kirk K.L. New high-performance liguid chromatographic procedure for the detection and quantification of В публикации международной заявки WO 97/23202 указана возможность использования соединений общей формулы (XV) для предотвращения лишь некоторых специфических видов боли, таких как мигреневая головная боль, хроническая боль, а также применение их для анестезии, обусловленная способностью данных соединений проявлять действие селективных лигандов подтипов NMDA рецепторов. Однако следует отметить, что в описании WO 97/23202 нет подтверждения заявляемой активности и, следовательно, возможности применения соединений по указанному назначению, на конкретных моделях на животных in vivo, и, таким образом, выводы о возможных фармакологических эффектах основаны исключительно на утверждении о том, что вообще все заявленные в патенте соединения являются селективными лигандами подтипов NMDA-рецепторов. В публикации международной заявки WO 97/23202 описан способ синтеза 3-(п-гидроксифенил)пропионилфенилэтиламина (IX) с использованием 1-гидроксибензотриазола в присутствии N,N’- дициклогексилкарбодиимида (DCC). Не описан способ выделения и очистки данного соединения, из физико-химических констант приведены температура плавления и данные 1H-ЯМР-спектроскопии. В статье Jacobson K.A., Kirk K.L. New high-performance liguid chromatographic procedure for the detection and quantification of В статье Herbert R.B., Kattah A.E. The biosynthesis of Sceletium alkaloids in Sceletium subvelutinumSceletium subvelutinum, а также его способ синтеза методом DCC. Недостатком данного способа является необходимость применения для очистки целевого продукта колоночной хроматографии, его сравнительно невысокий выход – около 48%. В статье Maldonado E., Hernandez E., Ortega A. Amides, coumarine and other constituents from simsia cronquistiiSimsia cronquistii и представлены данные масс-спектрометрии, 1Н-ЯМР-спектроскопии, температура плавления. Данных по биологической активности не приведено. Синтез производных аминокислот тирозина и фенилаланина 3-(п-гидроксифенил)пропионилтирозина, фенилпропионилтирозина, фенилацетилтирозина, фенилпропионилфенилаланина и фенилпропионилтирозина метилового эфира (соединения XIV, XV, XVI XVIII и XXI настоящего изобретения, соответственно) и изучение их ингибирующего действия на нейрон TAN, идентифицированный в ганглии улитки Achatina fulica ferussac описаны в статьях Takeuchi H., Ariyoshi Y., Effects of N-beta-phenylpropionyl-L-tyrosine and its derivatives on the excitability of an identifiable giant neuron of Achatina fulica ferussac Фенилпропионилтирозин метиловый эфир (XXI) упоминается в качестве промежуточного соединения в патенте Японии JP 57193437, где его синтез осуществлен методом активированных N-оксисукцинимидных эфиров. 1H-ЯМР- и ИК-спектроскопии, масс-спектрометрии. Было установлено, что фенилацетилфенилаланин (XIX) является индуктором дифференцировки клеток. 3-(п-Гидроксифенил)пропионилтирозина метиловый эфир (XX) упоминается в публикации международной заявки WO 99/52962, однако методика синтеза и физико-химические характеристики не приведены. Соединение (XX) было синтезировано с целью его использования в качестве мономера для получения биоразлагаемых полимеров, совместимых с тканями. Природное соединение, выделенное из симбиотической бактерии Xenorhabdus nematophilus, фенилацетилфенилэтиламин (XXIII) было синтезировано хлорангидридным методом и охарактеризовано физико-химическими данными 1H-ЯМР-, 13 Под общую формулу соединений, раскрытых в публикации международной заявки WO 01/49656, подпадают и другие соединения настоящего изобретения: п-гидроксифенилацетилтирамин, п-гидроксифенилацетилфенилэтиламин и фенилацетилтирамин (соединения VII, VIII и VI настоящего изобретения, соответственно). Однако, ни формул указанных соединений, ни методик синтеза, ни физико-химических констант, ни данных по биологической активности для них не приведено. В статье Peric M., Vercek В., Petric А. В статье Bok S., Lee S., Jeong Т., Phenolic acid derivatives and composition for preventing or treating blood lipid level-related diseases comprising the same. Pat. US. US 2003199566 описан синтез 3-(п-гидроксифенил)пропионилфенилаланина (XVII) и 3-(п-гидроксифенил)пропионилфенилаланина метилового эфира (XIII) с использованием 1-гидроксибензотриазола и 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида в присутствии триэтиламина. В случае 3-(п-гидроксифенил)пропионилфенилаланина (XVII) далее проводили омыление с выходом 78%. Для обоих соединений приведены данные 1H-ЯМР- и 13С-ЯМР- спектроскопии. Соединения XVII и XIII предлагается использовать для предупреждения и лечения заболеваний, связанных с уровнем липидов в крови. Известно, что анальгетическое действие может осуществляться в соответствии с различными механизмами, в частности, путем ингибирования фермента циклооксигеназы в каскаде арахидоновой кислоты [Машковский М.Д. Лекарственные средства. / М., Медицина, 1993, т.2]. Наиболее выраженным обезболивающим эффектом среди препаратов, снижающих синтез альгогенов, обладают ненаркотические анальгетики и нестероидные противовоспалительные средства. Ненаркотические анальгетики представлены салицилатами (аспирин), производными пиразолона (амидопирин, анальгин) и пара-аминофенола (парацетамол). К нестероидным противовоспалительным средствам относятся производные салициловой, уксусной, пропионовой и антраниловой кислот. Ненаркотические анальгетики и нестероидные противовоспалительные средства, наряду с болеутоляющим эффектом, обладают противовоспалительным и жаропонижающим действием [Кукушкин М.Л., Хитров Н.К. Общая патология боли. / Москва, Медицина, 2004, 142 с.]. Основным побочным эффектом нестероидных противовоспалительных средств является ульцерогенность. Целью настоящего изобретения является синтез и применение новых и известных фенилсодержащих N-ацильных производных биогенных аминов и аминокислот в качестве нетоксичных, более эффективных анальгетиков и потивовоспалительных средств, без побочных эффектов, в частности ульцерогенности. Краткое описание изобретения Настоящее изобретение относится к новым фенилсодержащим N-ацильным производным аминам общей формулы I:
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -COOH, -COOR4, где R4 представляет С1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; при условии, что когда n равно 1, R1 представляет водород и R2 представляет -COOH, -COOR4, где R4 представляет метил, то R3 не является гидроксигруппой; когда n равно 1, R1 и R3 одновременно представляют водород, то R2 не является -COOH, -COOR4, где R4 представляет метил; и n не равно 2, и их фармацевтически приемлемым солям, обладающим ингибирующей циклооксигеназу активностью, противовоспалительным и анальгетическим действием, не проявляющим побочного ульцерогенного эффекта. Настоящее изобретение также относится к применению соединений общей формулы I:
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; и их фармацевтически приемлемых солей в качестве ингибиторов циклооксигеназы, анальгетических и противовоспалительных средств. Далее, настоящее изобретение относится к фармацевтической композиции или средству, обладающим ингибирующей циклооксигеназу активностью, противовоспалительным и анальгетическим действием, не проявляющим побочного ульцерогенного эффекта, содержащим эффективное количество соединения общей формулы I или его фармацевтически приемлемой соли, а также, если требуется фармацевтически приемлемый носитель. Еще одним объектом изобретения является способ лечения болевых синдромов различного генеза, а также воспалительных заболеваний, включающий введение эффективного количества соединения общей формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также относится к новым способам получения соединений общей формулы I. Детальное описание изобретения Предпочтительными соединениями формулы I являются соединения, в которых R2 представляет -СООН, -СООСН3. Новые предпочтительные соединения общей формулы I представлены в Таблице 1.
Известные предпочтительные соединения общей формулы I представлены в Таблице 2.
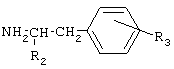
Соединения общей формулы I получают активацией карбоксильной группы п-гидроксифенилуксусной кислоты или фенилуксусной кислоты взаимодействием с дифенилфосфорилазидом (DPPA) и триэтиламином (TEA) в органическом растворителе при охлаждении, с последующим осуществлением взаимодействия с аминопроизводным. Предпочтительно активацию карбоксильной группы осуществляют с использованием 1-1,2 эквивалентов DPPA и TEA. В качестве аминопроизводного могут быть использованы эфиры тирозина и фенилаланина. Для получения соединений II и III, в качестве исходного аминопроизводного используют бензиловые эфиры тирозина и фенилаланина, соответственно, с последующим удалением бензильной группы путем каталитического гидрогенолиза. В отличие от ранее используемых способов синтеза известных соединений общей формулы I, применение дифенилфосфорилазидного способа позволило уменьшить число стадий, а именно, исключить стадию выделения активированного производного карбоксильного компонента, ограничиться экстракцией для выделения целевых веществ и повысить выходы ( Общая схема синтеза дифенилфосфорилазным методом представлена на Схеме 1. Схема 1
Новые соединения II, III, IV, V, VII, VIII, в том числе, содержащие фенольные гидроксильные группы, могут быть получены также методом активированных N-оксисукцинимидных эфиров, преимуществами которого являются доступность реагентов, водорастворимость выделяющегося N-гидроксисукцинимида, быстрота протекания как реакции получения N-оксисукцинимидных эфиров карбоксильных компонентов, так и реакции образования амидной связи, и возможность достижения высоких выходов целевых продуктов (70-80%), несмотря на наличие в них фенольного гидроксила. В соответствии с предлагаемым способом синтез N-оксисукцинимидных эфиров карбоксильных компонентов осуществляется превращением п-гидроксифенилуксусной кислоты или фенилуксусной кислоты в активированный N-оксисукцинимидный эфир N,N’-дициклогексилкарбодиимидным методом (DCC-методом) с высоким выходом (около 90%), с последующим образованием амидной связи реакцией N-оксисукцинимидных эфиров с аминопроизводным, также с высокими выходами (70-80%), за короткое время (1-2 часа) и без применения хроматографической очистки. В качестве аминопроизводного могут быть использованы эфиры тирозина и фенилаланина. Аналогично могут быть получены известные соединения X, XI, XII, XIII, XV, XVII, XIX, XX, XXII, XXIII, XXIV, синтез которых методом активированных N-оксисукцинимидных эфиров не описан в литературе. Общая схема синтеза соединений общей формулы I методом активированных N-оксисукцинимидных эфиров представлена на Схеме 2. Схема 2
п-гидроксифенил)пропионовой кислоты проходит практически полностью и быстро (за 2 часа). После выделения экстракцией без применения хроматографической очистки, выход целевого (XIV) продукта составил около 63%. Соединения общей формулы I также могут быть получены в виде фармацевтически приемлемых аддитивных солей с нетоксичными кислотами, такими как фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, лимонная кислота, винная кислота, оксалиновая кислота и подобные, и солей с основаниями, такими как гидроксид натрия, гидроксид калия, карбонат натрия и подобные. Соединения общей формулы I обладают ингибирующей циклооксигеназу активностью и могут быть использованы для лечения болевых синдромов различного генеза, воспалительных и воспалительно-дегенеративных заболеваний суставов и соединительной ткани, а также костно-мышечной системы, других заболеваний, сопровождающихся воспалением. В частности, соединения настоящего изобретения могут быть использованы для лечения послеоперационной боли, посттравматической боли, а также болевых синдромов гинекологической, неврологической, онкологической, стоматологической природы, ревматоидного артрита, артропатии, болезни Бехтерева, неспецифических спондилоартритов, подагрического артрита, остеоартроза, внесуставного ревматизма и тромбофлебита. Соединения настоящего изобретения вводятся в эффективном количестве, которое обеспечивает желаемый терапевтический результат. Для лечения болевых синдромов различного генеза, таких как послеоперационные боли, посттравматические боли, а также болевых синдромов гинекологической, неврологической, онкологической, стоматологической природы, воспалительных и воспалительно-дегенеративных заболеваний суставов и соединительной ткани, а также костно-мышечной системы, таких как ревматоидный артрит, артропатия, болезнь Бехтерева, неспецифические спондилоартриты, подагрический артрит, остеоартроз, внесуставный ревматизм и тромбофлебит, других заболеваний, сопровождающихся воспалением, соединения формулы (I) могут быть введены перорально, местно, парентерально, путем ингаляций и ректально в виде стандартных лекарственных форм, содержащих нетоксичные фармацевтически приемлемые носители. Используемый в настоящем описании термин «парентеральное введение» означает подкожные, внутривенные, внутримышечные или внутригрудные инъекции или вливания. Соединения настоящего изобретения могут быть введены пациенту в дозах, составляющих от 0,1 до 10 мг/кг веса тела в день, предпочтительно в дозах от 0,5 до 5 мг/кг один или более раз в день. При этом следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от многих факторов, включая активность данного используемого соединения, возраст, вес тела, пол, общее состояние здоровья и режим питания пациента, время и способ введения лекарственного средства, скорость его выведения из организма, конкретно используемую комбинацию лекарственных средств, а также тяжесть заболевания, подвергаемого лечению. Фармацевтические композиции по настоящему изобретению содержат соединение по настоящему изобретению в количестве, эффективном для достижения желаемого результата, и могут быть введены в виде стандартных лекарственных форм (например, в твердой, полутвердой или жидкой формах), содержащих соединения настоящего изобретения в качестве активного ингредиента в смеси с носителем или наполнителем, пригодным для внутримышечного, внутривенного, перорального, сублингвального, ингаляционного и интраректального введения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, драже, суппозиториев, эмульсий, суспензий, мазей, гелей и любых других лекарственных форм. В качестве наполнителей могут быть использованы различные вещества, такие как сахариды, например глюкоза, лактоза или сахароза, манит или сорбит, производные целлюлозы и/или фосфаты кальция, например, трикальций фосфат или кислый фосфат кальция, в качестве связующего компонента могут быть использованы такие, как крахмальная паста, например кукурузный, пшеничный, рисовый, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон. При необходимости могут быть использованы разрыхляющие агенты, такие как вышеупомянутые крахмалы и карбоксиметилкрахмал, поперечно сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия. Могут быть использованы необязательные добавки, такие как агенты, регулирующие текучесть, и смазывающие агенты, такие как диоксид кремния, тальк, стеариновая кислота и ее соли, такие как стеарат магния или стеарат кальция и/или пропиленгликоль. Ядро драже обычно покрывают слоем, который устойчив к действию желудочного сока. Для этой цели могут быть использованы концентрированные растворы сахаридов, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, и подходящие органические растворители или их смеси. В качестве добавок могут быть также использованы стабилизаторы, загустители, красители и отдушки. В качестве мазевой основы могут быть использованы углеводородные мазевые основы, такие как вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции, такие как твердый парафин и воск; абсорбтивные мазевые основы, такие как гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens); мазевые основы, смываемые водой, такие как гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы, такие как полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие. В качестве основы для гелей могут быть использованы метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, оксипропилцеллюлоза, полиэтиленгликоль или полиэтиленоксид, карбопол. В качестве основы для суппозитория могут быть использованы основы, нерастворимые в воде, такие как масло какао; основы, растворимые в воде или смешиваемые с водой, такие как желатино-глицериновые или полиэтиленоксидные; комбинированные основы – мыльно-глицериновые. При приготовлении стандартной лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьироваться в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства. Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций, содержание активного агента в них составляет 0,01-5%. В качестве разбавителей могут быть использованы 0,9% раствор хлорида натрия, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы, специфические добавки для растворения. При введении в организм соединений настоящего изобретения в виде таблеток и суппозиториев, их количество составляет 5,0-500 мг на стандартную лекарственную форму. Лекарственные формы настоящего изобретения получают по стандартным методикам, таким как, например, процессы смешивания, гранулирования, формирование драже, растворение и лиофилизация. Следует отметить, что соединения настоящего изобретения проявляют биологическую активность в дозах на два-три порядка ниже по сравнению с известными препаратами, использованными для сравнения, при практически одинаковой эффективности, и для них не выявлено отрицательных побочных действий и не обнаружено противопоказаний к применению. При этом, при исследовании токсичности соединений настоящего изобретения в дозе 1000 мкг/кг, перорально, не зарегистрировали гибели экспериментальных животных. Детальное описание соединений настоящего изобретения, их получения и исследования фармакологической активности представлено в нижеследующих примерах, предназначенных для иллюстрации предпочтительных вариантов изобретения и не ограничивающих его объем. Примеры синтеза соединений настоящего изобретения В качестве исходных соединений в синтезе использовали п-гидроксифенилуксусную кислоту, 3-(п-гидроксифенил)пропионовую кислоту, 3-фенилпропионовую кислоту (“Sigma”, США), фенилэтиламин, тирамин (“Fluka”, Швейцария). Индивидуальность полученных соединений проверяли методом ТСХ на пластинках “Kieselgel 60 F254” (“Merck”, Германия) в системе растворителей: хлороформ – метанол 9:1 (1). Хроматограммы проявляли хлор-толидиновым реактивом, нингидрином, йодом и по свечению в УФ-свете. 1H-ЯМР регистрировали на приборе “АМХ-400 Bruker” (Германия). ИК-Фурье спектры снимали в таблетках KBr на приборе “Magna 750” (“Nicolet” США). Температуру плавления определяли на приборе “Boetius” (Германия). Масс-спектры высокого разрешения получали на времяпролетном масс-спектрометре методом матриксной лазернодесорбционной ионизации, с использованием в качестве матрицы 2,5-дигидроксибензойной кислоты, на приборах и REFLEX Аналитическую обращенно-фазовую ВЭЖХ проводили на приборах: – хроматограф “Breeze”, детектор “Waters” (США), детекция при 214 нм, скорость элюирования 1 мл/мин, в условиях (1): колонка Symmetry 300 C18, 4,6×250 мм, 20 мкм, элюция 0,1%-ной TFA с градиентом 0,09% TFA в смеси 60:40 ацетонитрил-вода от 0% до 100% за 15 мин; в условиях (2): колонка Symmetry 300 C18, 3,9×150 мм, 5 мкм, элюция 0,1%-ной водной TFA с градиентом ацетонитрила от 0% до 60% за 18 мин; – хроматографах “System Gold” (“Beckman”, США), скорость элюирования 0,25 мл/мин, детекция при 220 нм, в условиях (3): колонка “Phenomenex” (США) C18, 2×250 мм, 5 мкм, элюция 0,1%-ной TFA с градиентом 0,08% TFA в 100% MeCN от 0% до 100% за 50 мин. Пример 1 п-Гидроксифенилацетилтирамин (VII) Методика А. При перемешивании к раствору 0,40 г (2,63 ммоль) п-гидроксифенилуксусной кислоты в 3,5 мл DMF прибавляли 0,35 г (2,63 ммоль) тирамина. Раствор охлаждали до -10°С и прибавляли 0,68 мл (3,16 ммоль) дифенилфосфорилазида и 0,44 мл (3,16 ммоль) триэтиламина. Перемешивали 2 ч при -10°С и оставляли при 20°С на 15 ч. К реакционной массе прибавляли 35 мл воды, экстрагировали 20 мл этилацетата. Этилацетатный слой промывали 10 мл 5% раствора Na2CO3, водой до pH 7, 10 мл 5% раствора HCl, водой до pH 7. Этилацетатный слой сушили над Na2SO4, отфильтровывали Na2SO4, этилацетат удаляли в вакууме. Маслообразный остаток растирали со смесью эфир-гексан (1:1). Образующийся белый осадок отфильтровывали и сушили в вакууме над CaCl2. Выход 0,68 г (95%). Rf 0,7 (1). Тпл= 147-149°С. [М]+ 271,6. 1H-ЯМР, CD3OD, ИК-Фурье, см-1: 3276 (вал. OH); 3108 (вал., =C-H, аром.); 1612 (амид I); 1591 (амид II); 1515 (аром. -C-C-); 1226 (вал., -C-O, фенольный). Найдено, %: С 70,57; H 6,43; N 5,50 C16H17NO3. Вычислено, %: С 70,83; H 6,32; N 5,16. ВЭЖХ в условиях (2): индивидуальный пик, время удерживания 8,71 мин. Методика Б К раствору 0,70 г (4,60 ммоль) п-гидроксифенилуксусной кислоты в 17 мл этилацетата при перемешивании прибавляли 0,53 г (4,60 ммоль) N-гидроксисукцинимида, раствор охлаждали до 0°С и прибавляли 0,95 г (4,60 ммоль) N,N’- дициклогексилкарбодиимид (DCC). Перемешивали 2 часа при 0°С и оставили на 20 часов при 4°С. Осадок N,N’- дициклогексилмочевины (DCU) отфильтровали. Растворитель удалили в вакууме. Маслообразный остаток растирали с гексаном. Образовавшийся белый твердый осадок отфильтровывали, промывали гексаном и сушили в вакууме над CaCl2. Получили 1,08 г (94,6%). Rf 0,58 (1). При перемешивании к раствору 0,30 г (1,2 ммоль) N-оксисукцинимидного эфира п-гидроксифенилуксусной кислоты в 8 мл N,N-диметилформамида (DMF) прибавляли 0,16 г (1,2 ммоль) тирамина. Реакционную смесь перемешивали 2 часа при 20°С, оставляли при 4°С на 20 часов. DMF удаляли в вакууме. Маслообразный остаток растирали с водой. Образующийся белый осадок отфильтровывали, промывали водой. Выход 0,26 г (80%). Rf 0,68 (1). Тпл= 146-148°С. [М+H]+ 272,3. Найдено, %: С 71,05; H 6,10; N 5,25 C16H17NO3. Вычислено, %: С 70,83; H 6,32; N 5,16. Пример 2 п-Гидроксифенилацетилфенилэтиламин (VIII) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,57 г (90,5%). Rf 0,82 (1). Тпл= 69-70°С. [М]+ 255,5. 1Н-ЯМР DMSO-d6, ИК-Фурье, см-1: 3332 (вал. OH); 3087 (вал., =C-H, аром.); 1626 (амид I); 1558 (амид II); 1515 (аром. -C-C-); 1249 (вал., -C-O, фенольный). Найдено, %: С 75,57; H 6,80; N 5,77 C16H17NO2. Вычислено, %: С 75,27; H 6,71; N 5,49. ВЭЖХ в условиях (2): индивидуальный пик, время удерживания 11,17 мин. Синтез проводили в соответствии с методикой Б, приведенной для соединения VII. Выход 0,50 г (79,4%). Rf 0,85 (1). Тпл= 68-70°С. [М]+ 255,7. Найдено, %: С 75,17; H 6,87; N 5,75 C16H17NO2. Вычислено, %: С 75,27; H 6,71; N 5,49. Пример 3 3-(п-Гидроксифенил)пропионилтирамин (X) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,41 г (95%). Rf 0,38 (1). Тпл 174-176°С. 1H-ЯМР, DMSO-d6, ИК-Фурье, см-1: 3249 (вал. OH), 1621 (амид I), 1515 (аром.), 1541 (амид II). Найдено %: С 71,56; H 6,78; N 4,97. Вычислено %: С 71,56; H 6,71; N 4,91, C17H19NO3. ВЭЖХ в условиях (3): индивидуальный пик, время удерживания 25,62 мин. Синтез проводили в соответствии с методикой Б, приведенной для соединения VII. Выход 0,37 г (85%). Rf 0,35 (1). Тпл 172-174°С. [M]+ 285,3. Пример 4 3-Фенилпропионилфенилэтиламин (XI) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,26 г (97%). Rf 0,78 (1). Тпл 94-96°С. 1H-ЯМР, DMSO-d6, ИК-Фурье, см-1: 1637 (амид I), 1546 (амид II). Найдено %: С 80,24; H 7,61; N 5,54. Вычислено %: С 80,60; H 7,56; N 5,53. C17H19NO3. ВЭЖХ в условиях(3): индивидуальный пик, время удерживания 37,86 мин. Синтез проводили в соответствии с методикой Б, приведенной для соединения VII. Выход 0,20 г (77%). Rf 0,80 (1). Найдено %: С 80,39; H 7,53; N 5,30. Вычислено %: С 80,60; H 7,56; N 5,53. C17H19NO3. Пример 5 3-(п-гидроксифенил)пропионилфенилэтиламин (IX) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,20 г (90%). Rf (11) 0,4. Тпл= 102-104°С. Лит. [84] 102-104° [М]+ 269,6. 1H-ЯМР, CDCl3, ИК-Фурье, см-1: 3263 (вал. OH); 1618 (амид I); 1537 (амид II). Найдено %: С 75,57; H 6,93; N 5,09. C17H19NO2. Вычислено %: С 75,81; H 7,11; N 5,20. ВЭЖХ в условиях (1): индивидуальный пик, время удерживания 14,77 мин. Тесты на биологическую активность Пример 6 Исследование влияния соединений общей формулы I на метаболизм [14C]арахидоновой кислоты в бесклеточном гомогенате легочной ткани мыши in vitro Исследования метаболизма арахидоновой кислоты проводили на мышах-самках линии СВА, находившихся на стандартном рационе вивария. Животных (мышей) забивали, извлекали легкие, гомогенизировали в стеклянном гомогенизаторе фирмы “Wheaton” (США) при 40С в 10 объемах 0,05 М трис-HCl буфера. Аликвоты (0,5 мл) супернатанта инкубировали с 0,5 мкКю [1-С14]-арахидоновой кислоты ([C14]-АА), “Amersham”, Англия; удельная активность 50-60 мКю/ммоль) при 370С в течение 30 мин. Экстракцию неметаболизированной [C14]-АК и продуктов ее метаболизма осуществляли в 20 объемах смеси хлороформа и метанола (1:1), при эффективности экстракции не менее 90%, оцененной с помощью [C14]-ПГF2 Полученные данные представлены в Таблице 3.
Полученные данные по профилю эйкозаноидов демонстрируют способность соединений общей формулы I ингибировать циклооксигеназу на 22-44% и свидетельствуют об их перспективности в качестве потенциальных анальгетических и противовоспалительных средств. Пример 7 Анальгетическая и противовоспалительная активность соединений, соответствующих общей формуле (I) Исследование анальгетической активности на модели “уксусные корчи”
где Ск – количество корчей в контрольной группе, Со– количество корчей в опытной группе. Полученные данные представлены в Таблице 4.
* Р < 0,05 относительно контрольной группы ** Р < 0,01 относительно контрольной группы Соединения, соответствующие общей формуле I, в тесте “корчи” проявляют анальгетическую активность, близкую к диклофенаку – препарату сравнения (табл.4), при этом анальгетический эффект большинства соединений составляет от 38 до 68 %. Пример 8 Исследование анальгетической активности на модели “горячая пластина” Анальгетическое действие соединений, соответствующих общей формуле I, А-100% = Х, где А – фоновый показатель; Х – анальгетический эффект (%) А = (время через 0,5-4 часа ×100%): фоновое время В качестве препаратов сравнения использовали: анальгин (150 мг/кг), парацетамол (200 мг/кг), кеторол (10 мг/кг). Полученные данные представлены в Таблице 5.
* Р<0,01, относительно фоновых показателей Полученные данные показывают, что соединения общей формулы I в тесте “горячая пластина” также демонстрируют значительную активность, достоверно увеличивая порог болевой чувствительности. Важно подчеркнуть, что сравнимый с эталонными препаратами анальгетический эффект достигается при использовании доз 0,1-10 мг/кг, преимущественно 1-10 мг/кг, которая, на один-два порядка ниже дозы препарата сравнения – парацетамола, обладающего болеутоляющим и жаропонижающим действием. Данные, представленные в таблице 5, также показывают, что анальгетический эффект соединений общей формулы I в среднем составляет от 50 и максимум до 140%, можно расценивать как пролонгированный, т.к. он сохраняется в течение длительного времени – более 4 часов. Таким образом, по степени выраженности обезболивающего эффекта соединения общей формулы I сравнимы с известными ненаркотическими анальгетиками (анальгин, парацетамол), а по длительности анальгетического действия превосходят препараты сравнения, причем их действующие дозы оказались на порядок ниже, чем у эталонных ненаркотических анальгетиков. Пример 9 Исследование воздействия вещества на каррагениновый отек лапы крыс В правую лапу крысы субплантарно вводили 1% раствор каррагенина (SERVA) 0,1 мл. Животные рассаживались в индивидуальные камеры. 1% Мазь наносили на лапу непосредственно после введения каррагенина, через 1 и 2 часа. Измерение объема лап проводили с помощью плетизмометра (Ugo Basile) через 4 часа после введения каррагенина. Эффект терапевтического воздействия мази оценивали по степени угнетения воспалительной реакции в сравнении с интактной левой лапой данного животного и реакцией лап крыс контрольной (нелеченой) группы. Торможение воспалительной реакции, выраженное в процентах, рассчитывали по формуле:
Полученные данные представлены в Таблице 6.
Результаты, представленные в таблице 6, демонстрируют выраженную локальную противовоспалительную активность соединений общей формулы I, сопоставимую с активностью эталонного препарата из группы НПВС – индометацина, при этом действующая доза соединения на порядок ниже, чем у препарата сравнения. Пример 10 Исследование ульцерогенного действия Тесты проводили на белых аутбредных крысах самках массой 300-320 г. Тестируемые соединения вводили в дозе 30 мк/кг, однократно, внутрижелудочно крысам, лишенным пищи на 24 часа до опыта. Животным в контрольной группе вводили дистиллированную воду в том же объеме. Через 24 часа животных забивали, извлекали желудки. В пустой желудок вводили 2% р-р формалина и помещали его в стакан с формалином. Через 30 минут вскрывали желудок по большой кривизне, расправляли на предметном столике, фиксировали и промывали дистиллированной водой. С помощью лупы МБС-9 (8-кр. ув.) измеряли длину и ширину дефектов слизистой желудка и подсчитывали общую площадь в мм2 (1 деление линейки лупы = 0,1 мм). Ульцерогенный эффект вещества оценивали по площади язвенного поражения слизистой желудка по методике, представленной в Руководстве по экспериментальному (доклиническому) изучению новых фармакологических веществ. / Москва, Ремедиум, 2000, 398 с. Полученные данные представлены в таблице 7.
Полученные данные показывают, что при внутрижелудочном введении соединений общей формулы I, в дозе 30 мг/кг, язвенное поражение слизистой оболочки желудка крыс отсутствует. ПРИМЕРЫ СТАНДАРТНЫХ ЛЕКАРСТВЕННЫХ ФОРМ ПРИМЕР 11 А. Таблетированная форма Таблетированную форму получают, используя приведенные ниже ингредиенты:
Компоненты смешивают и прессуют для образования таблеток весом 300 мг каждая. Б. Суппозитории Пример состава суппозитория:
При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями. В. Мази Пример состава мази:
Мази изготавливают по общеизвестной технологии. Г. Гели Пример состава геля:
Таким образом, настоящее изобретение относится к новым соединениям общей формулы I, простым и препаративным способам синтеза новых и известных соединений и их применению в качестве нестероидных противовоспалительных средств, ингибиторов циклооксигеназы, обладающих противовоспалительным и преимущественным анальгетическим действием, не проявляющих побочного ульцерогенного эффекта. Дополнительные данные Примеры синтеза соединений настоящего изобретения В качестве исходных соединений в синтезе использовали п-гидроксифенилуксусную кислоту (“Sigma”), метиловый и бензиловый эфиры L-тирозина, а также метиловый и бензиловый эфиры L-фенилаланина (“Bachem”, Германия), тирамин и 3-(п-гидроксифенил)пропионовую кислоту (“Fluka”). Все аминокислоты L-ряда. Индивидуальность полученных соединений проверяли методом ТСХ на пластинках “Kieselgel 60 F254” (“Merck”, Германия) в системе растворителей: хлороформ – метанол – этилацетат 6:1:3 (1), хлороформ – метанол – аммиак 6:3:0,5 (2). Хроматограммы проявляли хлор-толидиновым реактивом, нингидрином и по свечению в УФ-свете. Углы оптического вращения измеряли на поляриметре «Perkin Elmer 341» (Швеция). Аналитическую обращенно-фазовую ВЭЖХ проводили на хроматографе “Breeze”, детектор “Waters” (США) в условиях: детекция при 214 нм, скорость элюирования 1 мл/мин, колонка Symmetry 300 C18, 3,9×150 мм, 5 мкм, элюция 0,1%-ной водной TFA с градиентом ацетонитрила от 0% до 60% за 15 мин (1); в условиях, указанных выше с градиентом ацетонитрила от 0% до 60% за 18 мин (2). Пример 12 Бензиловый эфир п-гидроксифенилацетилтирозина Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0.59 г (55.7%). Rf 0.57 (1). [М+1]+ 406.0. [ п-Гидроксифенилацетилтирозин (II) К раствору 0.59 г (1.47 ммоль) бензилового эфира п-гидроксифенилацетилтирозина в 10 мл метанола прибавляли 0.20 г 10%-ного палладия на угле и при интенсивном перемешивании гидрировали в токе водорода в течение 1.5 ч. Катализатор отфильтровывали. Растворитель из фильтрата удаляли в вакууме. Маслообразный остаток растирали со смесью эфир-гексан (1:1). Образующийся белый осадок отфильтровывали и сушили в вакууме над CaCl2 и Р2O5. Получали 0.32 г (68%). Выход 37%. Rf 0.28 (2). [M+1]+ 316.07. [ Пример 13 Бензиловый эфир п-гидроксифенилацетилфенилаланина Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0.76 г (74%). Rf 0.87 (1). [M+1]+ 390.1. [ п-Гидроксифенилацетилфенилаланин (III) К раствору 0.65 г (1.67 ммоль) бензилового эфира п-гидроксифенилацетилфенилаланина в 10 мл метанола прибавляли 0.30 г 10%-ного палладия на угле и при интенсивном перемешивании гидрировали в токе водорода в течение 1.5 ч. Катализатор отфильтровывали. Растворитель из фильтрата удаляли в вакууме. Маслообразный остаток растирали с гексаном. Образующийся белый осадок отфильтровывали и сушили в вакууме над CaCl2 и Р2O5. Получали 0.27 г (53%). Выход 39.2%. Rf 0.42 (2). [M+1]+ 300.09. [ Пример 14 п-Гидроксифенилацетилтирозина метиловый эфир (IV) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0.17 г (39%). Rf 0.56 (1). [M]+ 329.85. [ Пример 15 п-Гидроксифенилацетилфенилаланина метиловый эфир (V) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0.40 г (39%), масло. Rf 0.70 (1). [M]+ 313.83. [ Пример 16 3-(п-Гидроксифенил)пропионилфенилаланина метиловый эфир (XIII) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0.37 г (38%), масло. Rf 0.73 (1). [М+1]+ 328.21. [ Пример 17 Фенилацетилтирамин (VI) Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0.35 г (37.6%). Rf 0.85 (1). Тпл 105-108°С. [M+1]+ 256.2. 1H-ЯМР, DMSO-d6, Тесты на биологическую активность Пример 18 Анальгетическая и противовоспалительная активность соединений, соответствующих общей формуле (I) Исследование анальгетической активности на модели “уксусные корчи” (см. табл.8).
Пример 19 Исследование анальгетической активности на модели “горячая пластина” (см. табл.9).
Формула изобретения
1. Фенилсодержащие N-ацильные производные аминов общей формулы I
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1; при условии, что 1) когда п равно 1, R1 представляет водород и R2 представляет -СООН, -COOR4, где R4 представляет метил или этил, то R3 не является гидроксигруппой; 2) когда п равно 1, R1 и R3 одновременно представляют водород, то R2 не является -СООН, -COOR4, где R4 представляет метил или этил; 3) когда п равно 1, R2 представляет водород, R1 и R3 одновременно не являются водородом; 4) когда п равно 1, R1 и R2 одновременно представляют водород, то R3 не является гидроксигруппой; и их фармацевтически приемлемые соли. 2. Соединение по п.1, в котором R2 представляет -СООН, -СООСН3. 3. Соединение по п.1, выбранное из п-гидроксифенилацетилтирозина, п-гидроксифенилацетилфенилаланина, п-гидроксифенилацетилтирозина метилового эфира, п-гидроксифенилацетилфенилаланина метилового эфира, п-гидроксифенилацетилтирамина, п-гидроксифенилацетилфенилэтиламина, и их фармацевтически приемлемых солей. 4. Соединение по любому из пп.1-3, обладающее ингибирующей циклооксигеназу активностью. 5. Соединение по п.4, обладающее анальгетическим и противовоспалительным действием. 6. Способ получения соединений общей формулы I
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; или их фармацевтически приемлемых солей, включающий активацию карбоксильной группы п-гидроксифенилуксусной кислоты или фенилуксусной кислоты общей формулы
взаимодействием с дифенилфосфорилазидом и триэтиламином в органическом растворителе при охлаждении с последующим взаимодействием с аминосоединением общей формулы
где R1-R3 принимают значения, определенные для соединений общей формулы I. 7. Способ по п.6, в котором используют 1-1,2 эквивалента дифенилфосфорилазида и триэтиламина. 8. Способ по п.6 или 7, в котором в качестве аминопроизводных используют эфиры тирозина или фенилаланина. 9. Способ получения соединений по п.1, или их фармацевтически приемлемых солей, включающий превращение п-гидроксифенилуксусной кислоты или фенилуксусной кислоты общей формулы
в активированный N-оксисукцинимидный эфир общей формулы
N’-дициклогексилкарбодиимидным методом, с последующим взаимодействием активированного N-сукцинимидного эфира с аминопроизводным общей формулы
где R1-R3 принимают значения, определенные для соединений общей формулы I в п.1. 10. Способ по п.9, в котором в качестве аминопроизводных используют эфиры тирозина или фенилаланина. 11. Фармацевтическая композиция, обладающая ингибирующей циклооксигеназу активностью, включающая в качестве активного агента эффективное количество соединения общей формулы (I)
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель. 12. Фармацевтическая композиция по п.11, обладающая анальгетическими и противовоспалительными свойствами. 13. Средство, обладающее ингибирующим циклооксигеназу действием, включающее соединение общей формулы (I)
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; или его фармацевтически приемлемую соль. 14. Средство по п.13, обладающее анальгетическими и противовоспалительным действием. 15. Применение соединений общей формулы (I)
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; или их фармацевтически приемлемых солей, в качестве ингибиторов циклооксигеназы. 16. Применение соединений по п.15, в качестве анальгетических и противовоспалительных средств. 17. Способ ингибирования циклооксигеназы, включающий введение млекопитающему эффективного количества соединения общей формулы (I)
где R1 представляет водород или гидроксигруппу; R2 представляет водород, -СООН, -COOR4, где R4 представляет C1-С6алкил; R3 представляет водород, гидроксигруппу; n равно 1 или 2; или его фармацевтически приемлемой соли. 18. Способ по п.17, для лечения болевых синдромов, воспалительных и воспалительно-дегеративных заболеваний суставов и соединительной ткани, а также костно-мышечной системы, других заболеваний, сопровождающихся воспалением. 19. Способ по п.18, для лечения послеоперационной боли, посттравматической боли, а также болевых синдромов гинекологической, неврологической, онкологической, стоматологической природы, ревматоидного артрита, артропатии, болезни Бехтерева, неспецифических спондилоартритов, подагрического артрита, остеоартроза, внесуставного ревматизма и тромбофлебита.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
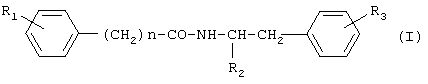
 Sceletium alkaloids in Sceletium subvelutinumsimsia cronquistii
Sceletium alkaloids in Sceletium subvelutinumsimsia cronquistii 1H-ЯМР спектроскопии и элементного анализа.
1H-ЯМР спектроскопии и элементного анализа.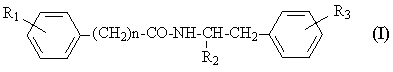
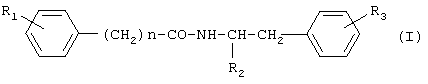
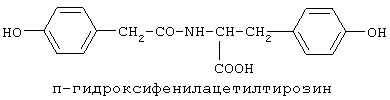
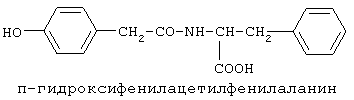
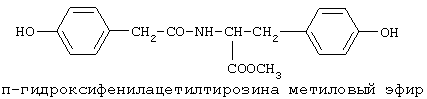
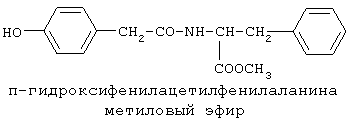
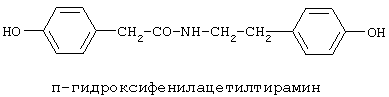
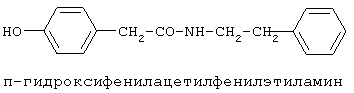
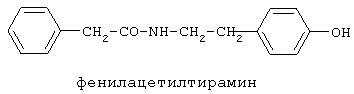
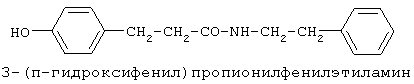
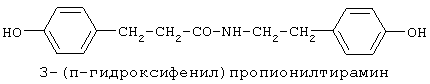
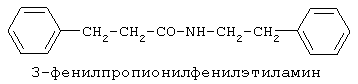
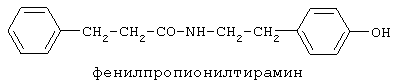
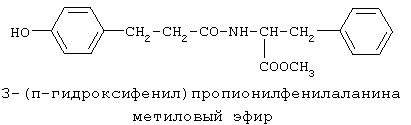
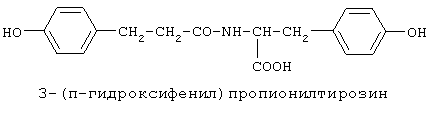
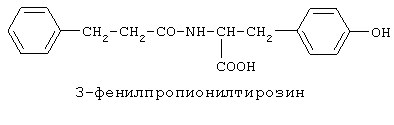
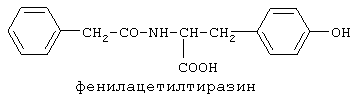
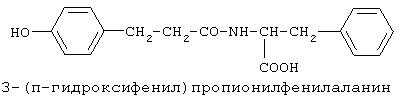
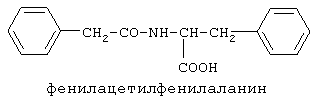
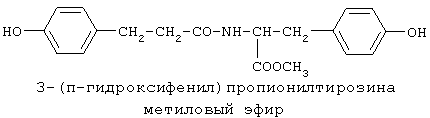
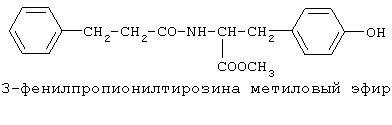
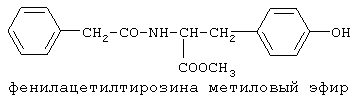
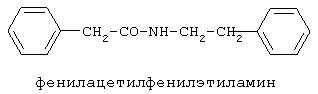
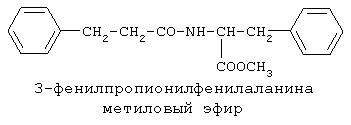
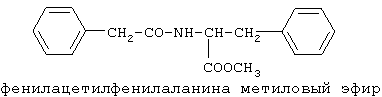
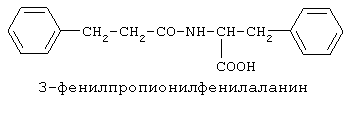
 90%).
90%).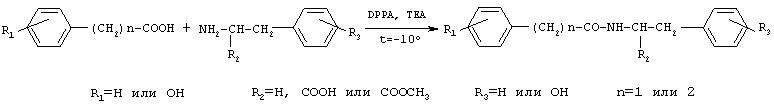
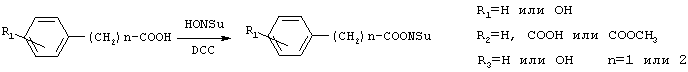
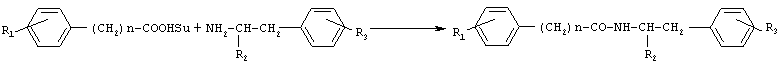
 III (Bruker, Германия).
III (Bruker, Германия). , м.д.: 2,65 (т, J=7 Гц, 2H,
, м.д.: 2,65 (т, J=7 Гц, 2H,  -CH2-TA), 3,29-3,32 (м, 4H,
-CH2-TA), 3,29-3,32 (м, 4H,