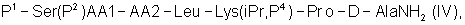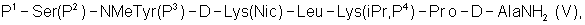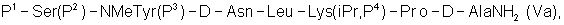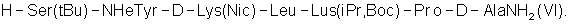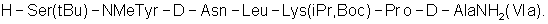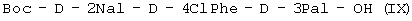Патент на изобретение №2307125
|
||||||||||||||||||||||||||
(54) ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА АНТАГОНИСТА РИЛИЗИНГ-ФАКТОРА ЛЮТЕИНИЗИРУЮЩЕГО ГОРМОНА (LHRH), СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТА LHRH
(57) Реферат:
Изобретение относится к способу получения трипептидов Ac-D-2Nal-D-4ClPhe-D-3Pal-OH и Boc-D-2Nal-D-4ClPhe-D-3Pal-OH, которые представляют собой промежуточные соединения в синтезе аналогов рилизинг-фактора лютеинизирующего гормона (LHRH), путем сочетания с подходящими гептапептидами, в частности с гептапептидами P1-Ser(P2)-NMeTyr(P3)-D-Lys(Nic)-Leu-Lys(iPr,P4)-Pro-D-AlaNH2 и P1-Ser(P2)-NMeTyr(P3)-D-Asn-Leu-Lys(iPr,P4)-Pro-D-AlaNH2. 3 н. и 4 з.п. ф-лы.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ Настоящее изобретение относится к промежуточным “соединениям для синтеза антагонистов LHRH, к способу получения этих промежуточных соединений и к способу получения антагонистов LHRH. ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ Рилизинг-фактор лютеинизирующего гормона (luteinizing hormone-releasing hormone, LHRH) контролирует секрецию фолликулостимулирующего гормона (FSH) и лютеинизирующего гормона (LH). Антагонисты LHRH представляют собой соединения, способные блокировать секрецию FSH и LH. Они, как правило, представляют собой нона- и декапептиды (но могут быть короче или длиннее), содержащие часть или полную структуру LHRH, где одна или несколько аминокислот замещены другими природными аминокислотами и/или аминокислотами, не обнаруживаемыми в природе. Синтетические антагонисты LHRH могут быть использованы для контрацепции и при лечении доброкачественной гиперплазии предстательной железы, гормонзависимых опухолей груди и яичников, дисменореи, эндометриоза и других состояний. Эти синтетические антагонисты LHRH имеют общую формулу
где Х представляет собой 5-6 природных и/или синтетических аминокислотных остатков. Более конкретно они имеют вышеупомянутую общую формулу, где Х представляет собой AA1-AA2-Leu-AA3-Pro-D-Ala, в частности, когда АА1 представляет собой природную или синтетическую аминокислоту и АА2 представляет собой природную или синтетическую аминокислоту или нуль, АА3 представляет собой природную или синтетическую аминокислоту. Хотя существует множество способов синтеза для получения аналогов LHRH, известных из уровня техники, существует необходимость в усовершенствовании, так как общий выход аналогов LHRH, получаемых с использованием известных способов, является невысоким и, в дополнение, продуктам может потребоваться трудоемкая очистка. Кроме того, способы синтеза аналогов LHRH, известные из уровня техники, являются весьма дорогостоящими. Стратегия синтеза, описанная в патенте США №5710246, для получения декапептидных или нонапептидных антагонистов LHRH включает в себя сочетание промежуточного соединения трипептида, представляющего аминокислотные остатки от 1 до 3 (отсчет начинается с N-конца пептида), с гептапептидом или гексапептидом, соответственно представляющим аминокислотные остатки 4-10 и 4-9 соответственно. Промежуточное соединение трипептид, описанный в US 5710246 А, представляет собой сложный эфир Boc-D-2Nal-D-4ClPhe-D-3Pal-O-Me или соответствующий бензиловый или аллиловый сложный эфир. ЗАДАЧИ ИЗОБРЕТЕНИЯ Таким образом, задача данного изобретения заключается в том, чтобы предложить промежуточное соединение трипептид для синтеза 3+7 и 3+6 аналогов LHRH, где улучшены выход и/или чистота продукта. Еще одна задача данного изобретения заключается в том, чтобы предложить способ получения такого промежуточного соединения трипептида. Еще одна задача данного изобретения заключается в том, чтобы предложить способ получения аналогов LHRH, при котором осуществляют сочетание трипептида с гепта- или гексапептидом. Другие задачи данного изобретения станут понятными из следующего краткого изложения сущности изобретения, описания предпочтительных воплощений и прилагаемой формулы изобретения. ОПРЕДЕЛЕНИЯ И СОКРАЩЕНИЯ Относительно определений и сокращений, используемых в данной заявке, и которые обычно приняты в области изобретения, ссылки даны, в частности, на US 5710246 А. КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ В соответствии с настоящим изобретением предложен трипептид, представляющий аминокислоты 1-3 антагониста LHRH, концевая аминогруппа которого защищена Boc или Ас и концевая карбоксильная группа которого (то есть концевая группа аминокислоты №3) не защищена. В соответствии с данным изобретением описан трипептид (I)
который представляет собой полезное промежуточное соединение в способе синтеза антагониста LHRH общей формулы (II)
где Х представляет собой от 5 до 7 природных и/или синтетических аминокислотных остатков, более предпочтительно AA1-AA2-Leu-AA3-Pro-D-Ala, в частности, где АА1 представляет собой природную или синтетическую аминокислоту и АА2 представляет собой природную или синтетическую аминокислоту или нуль, АА3 представляет собой природную или синтетическую аминокислоту. Предпочтительным является применение трипептида (I) в синтезе пептида общей формулы (IIa)
где АА1 и АА2 имеют значение, указанное выше, в частности антагонист LHRH формулы (III)
или еще более предпочтительно формулы (IIIa)
В соответствии сданным изобретением также описан трипептид
обладающий такой же полезностью. Кроме того, в соответствии с данным изобретением описан способ получения трипептида формулы (I)
или (IX)
при котором осуществляют следующие последовательные стадии получения (I): (а) осуществляют взаимодействие Boc-D-4ClPhe-OH с HONSu с образованием Boc-D-4ClPhe-OSu (VII); (б) осуществляют взаимодействие Boc-D-4ClPhe-OSu (VII) с H-D-3Pal-ОН с образованием Boc-D-4ClPhe-D-3Pal-OH (VIII); (в) осуществляют взаимодействие Boc-D-4ClPhe-D-3Pal-OH (VIII) с Boc-D-2Nal-OSu, полученным путем осуществления взаимодействия Boc-D-2Nal-OH с HONSu, с образованием Boc-D-2Nal-D-4ClPhe-D-3Pal-OH (IX); (г) осуществляют взаимодействие Boc-D-2Nal-D-4ClPhe-D-3Pal-OH (IX) с уксусной кислотой с образованием Ac-D-2Nal-4ClPhe-D-3Pal-OH (I); или последовательные стадии с (а) по (в) для получения (IX). При способе по данному изобретению для получения антагониста LHRH осуществляют стадию сочетания трипептида (I) с гептапептидом (IV) общей формулы
где Р4 представляет собой Н или защитную группу для амино, такую как Boc, где АА1 и АА2 имеют вышеупомянутое значение, в частности, с гептапептидом (V) общей формулы P1-Ser(P2)-NMeTyr(P3)-D-Lys(Nic)-Leu-Lys(iPr,P4)-Pro-D-AlaNH2 (V), где Р1 выбран из Н или защитной группы для амино, Р2 и Р3 независимо выбраны из Н и защитной группы для -ОН, и Р4 имеет указанное выше значение, для получения антагониста LHRH Ac-D-2Nal-D-4ClPhe-D-3Pal-Ser-MeTyr-D-Lys(Nic)-Leu-Lys(iPr)-Pro-D-Ala-NH2 (III), более конкретно, с гептапептидом (Va) общей формулы P1-Ser(P2)-NMeTyr(P3)-D-Asn-Leu-Lys(iPr,P4)-Pro-D-AlaNH2 (Va), где Р1 выбран из Н или защитной группы для амино, Р2 и Р3 независимо выбраны из Н и защитной группы для -ОН, и Р4 имеет указанное выше значение, для получения антагониста LHRH Ac-D-2Nal-D-4ClPhe-D-3Pal-Ser-MeTyr-D-Asn-Leu-Lys(iPr)-Pro-D-Ala-NH2 (III). Гептапептид (V) описан в US 5710246 А. Гептапептид общей формулы (IV), включая гептапептид (Va), может быть синтезирован путем стандартных модификаций синтеза (V) или путем сочетания соответствующих Вос-аминокислот в аппарате для синтеза пептидов (Beckman Model 990), как описано в WO 94/40757, где также описан антагонист LHRH (III). В качестве альтернативы в способе по данному изобретению для получения антагониста LHRH осуществляют стадию сочетания трипептида (IX)
с гептапептидом (IV) общей формулы
где Р1, Р2, Р4, АА1 и АА2 имеют указанные выше значения, в частности, с гептапептидом (V) общей формулы
или еще более предпочтительно с гептапептидом общей формулы (Va)
где Р1 выбран из Н или защитной группы для амино, Р2 и Р3 независимо выбраны из Н и защитной группы для -ОН, и Р4 имеет указанное выше значение, с последующей заменой группы Boc на N-конце ацильной группой, в частности ацетильной группой. Более конкретно, гептапептид общей формулы (V) представляет собой гептапептид (VI)
или еще более предпочтительно гептапептид (VIa)
Особенное преимущество применения способа по данному изобретению заключается в том, что вместо сложного эфира H-Pal-OR·HCl, может применяться более дешевое исходное вещество H-D-Pal-OH·2HCl; отсутствует необходимость удалять защитную группу с исходного вещества. Таким образом, синтез по данному изобретению на одну стадию короче и позволяет избежать потери вещества на дополнительной стадии. Еще одно преимущество заключается в том, что избегают образования примесей на стадии омыления. Образование таких примесей хорошо известно. Например, основные условия на стадии гидролиза сложного эфира вызывают частичную рацемизацию D-Pal. Другая альтернатива удаления группы сложного эфира по предшествующему уровню техники путем каталитического гидрирования (в случае аллильных или бензильных сложноэфирных групп) вызывает риск потери Cl из 4ClPhe, приводя к образованию Phe. Хотя аллильные группы могут быть удалены путем применения других реагентов, полное удаление сложно контролировать. Данное изобретение более подробно объясняется далее со ссылкой на предпочтительное воплощение. ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВОПЛОЩЕНИЯ ДАННОГО ИЗОБРЕТЕНИЯ Синтез Ac-D-2Nal-D-4ClPhe-D-3Pal-OH (I) ПРИМЕР 1. Boc-D-4ClPhe-OSu. Boc-D-4ClPhe-OH (299,75 г; 1,0 экв.) и HONSu (184,1 г; 1,6 экв.) растворяют в 2-пропаноле (4,5 л). Смесь охлаждают до 0°С и добавляют DIC (164,1 г; 1,3 экв.). Смесь перемешивают в течение 16 часов, нагревая до комнатной температуры. Продукт отфильтровывают, промывают 2-пропанолом (1,5 л) и сушат. Выход: 85%. Чистота в соответствии с ВЭЖХ: 98,8%. ПРИМЕР 2. Boc-D-4ClPhe-D-3Pal-OH. H-D-3Pal-OH, 2HCl (251,1 г; 1,05 экв.) и Boc-D-4ClPhe-OSu (396,8 г; 1,0 экв.) растворяют в диметилсульфоксиде (ДМСО) (3,33 л) и добавляют NMM (318,8 г; 3,15 экв.). Смесь перемешивают в течение 16 часов при комнатной температуре. Добавляют 17 л воды и рН доводят до 4-4,5, что вызывает осаждение продукта. Смесь фильтруют и продукт промывают водой (3×5 л) для удаления следов ДМСО, H-D-3Pal-OH и Boc-D-4ClPhe-OH. Продукт сушат. Выход: 80%. Чистота в соответствии с ВЭЖХ: 97,8%. ПРИМЕР 3. Boc-D-2Nal-OSu. Boc-D-2Nal-OH (315,4 г; 1,0 экв.) растворяют в 2-пропаноле (6,8 л) при -10°С и добавляют IBC (157 г; 1,15 экв.) и NMM (116 г; 1,15 экв.). После перемешивания в течение 5-10 мин добавляют смесь HONSu (230,1 г; 2,0 экв.) в 2-пропаноле (1,4 л). Добавляют дополнительное количество NMM (10,1 г; 0,1 экв.). Через полчаса добавляют воду (0,82 л) для растворения осажденного NMM·HCl. Продукт выделяют путем фильтрования, промывают 2-пропанолом (1 л) и сушат. Выход: 90%. Чистота в соответствии с ВЭЖХ: 98,3%. ПРИМЕР 4. Boc-D-Nal-D-4ClPhe-D-3Pal-OH. (а) Удаление защиты. Boc-D-4ClPhe-D-3Pal-OH (447, 93 г; 1,0 экв.) растворяют в смеси этилацетата (3,4 л), уксусной кислоты (675 мл) и MSA (454 мл, 7,0 экв.) при 0°С и оставляют при этой температуре на два часа. Добавляют тетраэтиламмоний (ТЭА) (1669 мл; 12 экв.). (б) Конденсация. Boc-D-Nal-OSu (412,4 г; 1,0 экв.) добавляют к нейтрализованной смеси для удаления защиты при комнатной температуре. Реакционную смесь оставляют при этой температуре на 2-4 часа. Добавляют водный 25% NH3 (154 мл; 2,0 экв.) для гашения оставшегося количества гидроксисукцинимидного эфира. Добавляют 1-бутанол (4,5 л) для предотвращения осаждения при последующих экстракциях. (в) Очистка и выделение. Реакционную смесь дважды экстрагируют при рН 6 (2×4,5 л воды) для удаления ТЭА, при рН 9 (4,5 л воды), для удаления MSA и, наконец, при рН 7 (4,5 л воды). Экстракции осуществляют при 40-45°С для предотвращения осаждения. К органической фазе добавляют уксусную кислоту (4,5 л; 1 об.) и смесь концентрируют в вакууме и упаривают совместно с уксусной кислотой (4,5 л) с получением твердого вещества. ПРИМЕР 5. Ac-D-2Nal-D-4ClPhe-D-3Pal-ONa. (а) Удаление защиты. К твердому веществу Boc-D-2Nal-D-4ClPhe-D-3Pal-OH добавляют воду (90 мл), уксусную кислоту (1,8 л) и MSA (454 мл; 7,0 экв.) и смесь перемешивают в течение 1-2 часов при комнатной температуре. Смесь охлаждают до 0°С и нейтрализуют ТЭА (1071 мл; 7,7 экв.). Раствор концентрируют в вакууме и дважды упаривают совместно с толуолом (2×2,5 л) с получением масла. (б) Ацетилирование. Масло после стадии удаления защиты растворяют в толуоле (2,0 л) и добавляют ацетил и мидазол (132, 14 г). Смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют воду (100 мл) для гашения оставшегося ацетил и мидазола. (в) Очистка. Смесь после ацетилирования нагревают до 30-35°С и добавляют 1-бутанол (4,5 л) для предотвращения осаждения. Смесь дважды экстрагируют при рН 5 (2×2,6 л воды) и дважды при рН 11 (2 х 2,6 л воды), используя NaOH для доведения рН до 11. К последним экстрактам добавляют метанол (2,25 л) для предотвращения осаждения. Добавляют NaCl (130 г) при первой и последней экстракциях для того, чтобы минимизировать потерю продукта в водных фазах. (г) Выделение. К тщательно перемешиваемой органической фазе после экстракций добавляют гептан (15 л) и получившуюся в результате суспензию оставляют перемешиваться при комнатной температуре в течение по меньшей мере 1 часа. Смесь фильтруют и продукт дважды промывают гептаном (2×3,5 л) и сушат. Выход: 75% (от Boc-D-4ClPhe-D-3Pal-OH). Чистота в соответствии с ВЭЖХ: 92%. Анализ аминокислот: 2Nal: 1,1; 4ClPhe: 1,0; 3Pal: 0,9. Масс-спектр (МС): масса 586. Na: 4,6%. ПРИМЕР 6. Ac-D-2Nal-D-4ClPhe-D-3Pal-OH·DCHA (а) Удаление защиты. К твердому веществу Boc-D-2Nal-D-4ClPhe-D-3Pal-OH добавляют воду (90 мл), уксусную кислоту (1,8 л) и MSA (454 мл; 7,0 экв.) и смесь перемешивают в течение 1-2 часов при комнатной температуре. Смесь охлаждают до 0°С и нейтрализуют ТЭА (1071 мл; 7,7 экв.). Раствор концентрируют в вакууме и дважды совместно упаривают с толуолом (2×2,5 л) с получением масла. (б) Ацетилирование. Масло после удаления защиты растворяют в толуоле (2,0 л) и добавляют ацетилимидазол (132,14 г). Смесь перемешивают при комнатной температуре в течение 1 часа с последующим добавлением воды (100 мл) для гашения оставшегося ацетилимидазола. (в) Очистка. Смесь нагревают до 30-35°С и добавляют 1-бутанол (4,5 л) для предотвращения осаждения. Смесь дважды экстрагируют при рН 7 (2×2,6 л воды), один раз при рН 9-9,5 (2,6 л воды) и один раз при рН 7 (2,6 л воды). Добавляют дициклогексиламин (DCHA) и смесь концентрируют в вакууме. Продукт суспендируют в 1-бутаноле (4,5 л) при 50°С и медленно добавляют к тщательно перемешиваемому гептану (27 л). Смесь перемешивают при 0°С в течение ночи, фильтруют и продукт дважды промывают смесью 1-бутанол/гептан (1:3; 2×4,8 л) и дважды гептаном (2×4,5 л). Выход: 65% (от Boc-D-4ClPhe-D-3Pal-OH). Чистота в соответствии с ВЭЖХ: 94,2%. Анализ аминокислот: 2Nal: 1,1; 4ClPhe: 1,0; 3Pal: 0,9. МС: масса 586 (свободный пептид). ПРИМЕР 7. Ac-D-2Nal-D-4ClPhe-D-3Pal-OH (а) Удаление защиты. К раствору твердого вещества Boc-D-2Nal-D-4ClPhe-D-3Pal-OH добавляют воду (90 мл), уксусную кислоту (1,8 л) и MSA (454 мл; 7,0 экв.) и смесь перемешивают в течение 1-2 часов при комнатной температуре. Смесь охлаждают до 0°С и нейтрализуют ТЭА (1071 мл; 7,7 экв.). Раствор концентрируют в вакууме и дважды упаривают совместно с толуолом (2×2,5 л) с получением масла. (б) Ацетилирование. Масло после удаления защиты растворяют в толуоле (2,0 л) и добавляют ацетилимидазол (132,14 г). Смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют воду (100 мл) для гашения оставшегося ацетил и мидазола. (в) Очистка. Смесь после ацетилирования нагревают до 30-35°С и добавляют 1-бутанол (4,5 л) для предотвращения осаждения. Смесь дважды экстрагируют при рН 7 (2×2,6 л воды), один раз при рН 9-9,5 (2,6 л воды) и один раз при рН 7 (2,6 л воды). Смесь концентрируют в вакууме с получением масла, которое растворяют в уксусной кислоте (750 мл), концентрируют, снова растворяют в уксусной кислоте (750 мл) и медленно добавляют к тщательно перемешиваемой смеси гептан/этилацетат (3:1; 3,6 л). Смесь оставляют перемешиваться при 0°С в течение ночи. Смесь фильтруют и продукт дважды промывают смесью этилацетат/гептан (1:3; 2×3,6 л) и дважды гептаном (2×3,6 л). Выход: 70% (от Boc-D-4ClPhe-D-3Pal-OH). Чистота в соответствии с ВЭЖХ: 93,9%. Анализ аминокислот: Nal; 1,1; 4ClPhe: 1,0; 3Pal: 0,9. МС: масса 586 (свободный пептид). ПРИМЕР 8. Ac-D-2Nal-D-3Pal-D-4ClPhe-Ser(tBu)-MeTyr-D-Asn-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 Z-Ser(tBu)-MeTyr(Bzl)-D-Asn-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 (122,8 г) растворяют в этаноле (1,2 л) и добавляют воду (400 мл). Гидрирование инициируют добавлением 5% Pd/C (18,4 г) и водород барботируют через реакционный сосуд. Непрерывным добавлением HCI (водн.) поддерживают постоянное значение рН 2,5-3,0 этой смеси. Через 1 час Pd/C удаляют фильтрованием, и рН фильтрата доводят до значения 3,6 добавлением NaOH (водн.). Фильтрат затем упаривают, к остатку добавляют этанол (360 мл) и снова проводят упаривание. Эту процедуру повторяют один раз с этанолом (360 мл) и один раз с этилацетатом (360 мл). Остаток суспендируют в этилацетате (1,25 л) и добавляют в раствор Ас-D-2Nal-D-3Pal-D-4ClPhe-OH, DCHA (дициклогексиламин) (60,9 г) в ДМСО (260 мл). HOBt (1-гидроксибензотриазол) (15,3 г) и CuCl2·Н2О (8,5 г) добавляют в раствор и его затем охлаждают до 0-5°С. DCC (дициклогексилкарбодиимид) (41,3 г) растворяют в этилацетате (33 мл) и добавляют в раствор пептида со скоростью, при которой поддерживается температура ниже 10°С. После окончания добавления реакционную смесь выдерживают при 0-5°С в течение 44 часов. Добавляют воду (77 мл), и эту реакционную смесь перемешивают в течение 30 минут и фильтруют. DCU-фильтровальный осадок промывают этилацетатом (2×60 мл). Объединенные органические фазы затем экстрагируют три раза. Сначала добавляют воду, и рН доводят до значения 9,1 добавлением NaOH (водн.). После перемешивания в течение 30 мин добавляют 25% NH3 (водн.) (46,2 мл), и фазам дают возможность разделиться. Водную фазу отбрасывают, а органическую фазу экстрагируют водой (1,54 л) и 25% NНз (водн.) (23,1 мл). После перемешивания в течение 30 минут фазы разделяют, и водную фазу отбрасывают. Органическую фазу окончательно экстрагируют водой (1,54 л). После разделения фаз водную фазу отбрасывают. Органическую фазу упаривают на роторном испарителе, и остаток растворяют в ацетоне (1,54 л) при 40°С и добавляют к гептану (460 мл), предварительно нагретому до 40°С. Смесь охлаждают до 4-8°С и оставляют стоять в течение 18 часов. Выпавший в осадок продукт выделяют фильтрованием, и остаток на фильтре промывают дважды смесью ацетона (208 мл) и гептана (92 мл), предварительно охлажденного до 2°С. Продукт затем сушат в вакуумном шкафу. Сухой продукт дважды подвергают перекристаллизации согласно следующей процедуре: продукт растворяют в этаноле и упаривают на роторном испарителе. Остаток затем растворяют в ацетоне, добавляют к гептану и охлаждают, фильтруют и промывают согласно процедуре, описанной выше. Выход 84 г (53,4%).
Формула изобретения
1. Способ получения трипептида, включая его соль, формулы (I)
или (IX)
при котором последовательно осуществляют следующие стадии получения соединения (I): (а) осуществляют взаимодействие Boc-D-4ClPhe-OH с HONSu с образованием (б) осуществляют взаимодействие (в) удаляют защиту (г) осуществляют взаимодействие или последовательные стадии с (а) по (в) для получения (IX). 2. Способ получения антагониста рилизинг-фактора лютеинизирующего гормона (LHRH) или его фармацевтически приемлемой соли, при котором осуществляют стадии способа по п.1 с получением трипептида
где Р1 выбран из Н или защитной группы для амино, Р2 представляет собой Н или защитную группу для ОН, Р4 представляет собой Н или защитную группу для амино, такую как Boc, АА1 представляет собой природную или синтетическую аминокислоту, и АА2 представляет собой природную или синтетическую аминокислоту или отсутствует. 3. Способ по п.2, где гептапептид общей формулы (IV) представляет собой гептапептид общей формулы
где Р3 представляет собой Н или защитную группу для ОН. 4. Способ по п.2, где гептапептид общей формулы (IV) представляет собой гептапептид общей формулы
где Р3 представляет собой Н или защитную группу для ОН. 5. Способ по п.3, где гептапептид общей формулы (V) представляет собой гептапептид формулы
6. Способ по п.4, где гептапептид формулы (VI) представляет собой гептапептид формулы
7. Трипептид
TK4A – Поправки к публикациям сведений об изобретениях в бюллетенях “Изобретения (заявки и патенты)” и “Изобретения. Полезные модели”
Напечатано: (57) 5. … H-Ser(tBu)-NHeTyr-D- …
Следует читать: (57) 5. … H-Ser(tBu)-NMeTyr-D- …
Номер и год публикации бюллетеня: 27-2007
Код раздела: FG4A
Извещение опубликовано: 20.01.2008 БИ: 02/2008
|
||||||||||||||||||||||||||
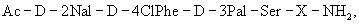
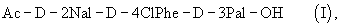
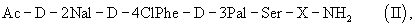
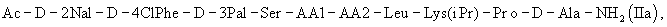
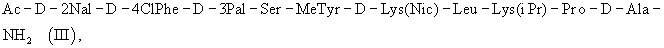
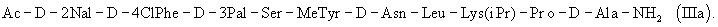

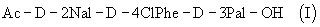
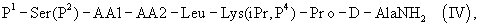
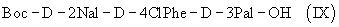
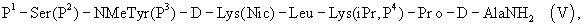
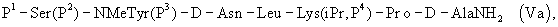
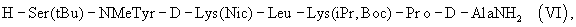
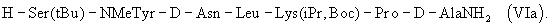
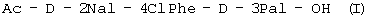
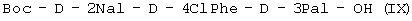
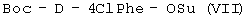 ;
;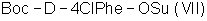 с H-D-3Pal-OH с образованием
с H-D-3Pal-OH с образованием 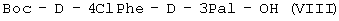 ;
;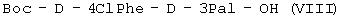 и осуществляют взаимодействие с Boc-D-2Nal-OSu, полученным путем взаимодействия Boc-D-2Nal-OH с HONSu, с образованием
и осуществляют взаимодействие с Boc-D-2Nal-OSu, полученным путем взаимодействия Boc-D-2Nal-OH с HONSu, с образованием 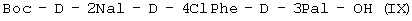
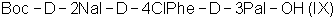 с уксусной кислотой и затем ацетилируют с образованием
с уксусной кислотой и затем ацетилируют с образованием 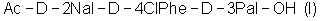 и далее осуществляют взаимодействие полученного трипептида с гептапептидом (IV) общей формулы
и далее осуществляют взаимодействие полученного трипептида с гептапептидом (IV) общей формулы