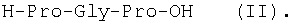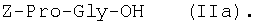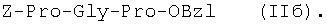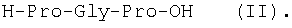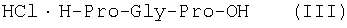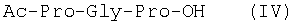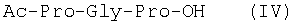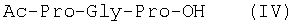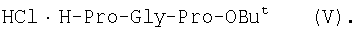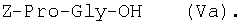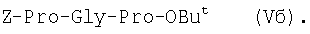Патент на изобретение №2303602
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ТРИПЕПТИДОВ
(57) Реферат:
Изобретение относится к области фармацевтической химии, конкретно к способу получения трипептидов общей формулы I: A-Pro-Gly-Pro-OX, где А – Н, Ас; Х – Н, Bzl, But и имеет своей целью упрощение процесса получения и повышение выхода целевых трипептидов. Сущность изобретения заключается в том, что синтез осуществляют жидкофазным методом путем конденсации С-концевого производного пролина общей формулы H-Pro-ОХ1, где X1 – Bzl, But с N-защищенным дипептидом общей формулы: Y-Pro-Gly-OH, где Y – Z, Boc и полученный защищенный трипептид общей формулы: Y-Pro-Gly-Pro-OX1, где Y и X1 имеют значения указанные выше, обрабатывают деблокирующими реагентами и полученный продукт в случае необходимости подвергают ацетилированию и необязательно получают гидрохлориды. 1 з.п. ф-лы.
Изобретение относится к фармацевтической химии, конкретно к способу получения трипептидов общей формулы: A-Pro-Gly-Pro-OX, где А – Н, Ас; Х – Н, Bzl, But, или их гидрохлоридов, и может найти применение в медицине для получения физиологически активных пептидов. Трипептиды, содержащие последовательность Pro-Gly-Pro, входят в состав известных регуляторных пепидов ( Известен способ получения бромгидрата H-Pro-Gly-Pro-OH путем ацилирования С-концевого дипептида хлорангидридом N-бензилоксикарбонил-L-пролина с последующим удалением N-концевой защитной группы обработкой раствором бромистого водорода в уксусной кислоте [3]. Недостатком этого способа является низкий выход целевого продукта и необходимость использования в синтезе хлорангидрида N-бензилоксикарбонил-L-пролина, который является нестабильным соединением; кроме того, при его получении используют высокотоксичные реагенты – тионилхлорид или пятихлористый фосфор [4]. Известен способ получения трипептида Ac-Pro-Gly-Pro-OH, твердофазным методом путем конденсации С-концевого пролина, связанного с полимерной подложкой, с Boc-Pro-Gly-OH с использованием N,N’-дициклогексилкарбодиимида в качестве конденсирующего агента с последующим отщеплением от трипептидилполимера N-концевой защитной группы, ацилированием уксусным ангидридом и отщеплением от полимерной положки обработкой безводным HF [5]. Недостатками этого способа являются необходимость использования специального аппаратурного оформления (твердофазного синтезатора и прибора для работы с жидким фтористым водородом) и применение в синтезе высокотоксичного фтористого водорода, что делает этот метод малопригодным для препаративной наработки целевых соединений. Наиболее близким по технической сущности к описываемому способу является способ получения H-Pro-Gly-Pro-OH, основанный на конденсации метилового эфира L-пролина и N-карбобензокси-L-пролилглицина с последующей обработкой защищенного трипептида деблокирующими реагентами [6]. Недостатками известного способа является многостадийность и низкий выход целевого продукта. Задачей данного изобретения является упрощение процесса получения и повышения выхода целевых трипептидов. Решение поставленной задачи достигается заявленным способом, который заключается в том, что синтез трипептидов общей формулы (I) осуществляют в растворе путем конденсации производного пролина общей формулы: Н-Pro-ОХ1, где X1 – бензил (Bzl), трет.-бутил (But), с защищенным дипептидом общей формулы: Y-Pro-Gly-OH, где Y – бензилоксиакрбонил (Z), трет.-бутилоксикарбонил (Boc), и полученный защищенный трипептид общей формулы: Y-Pro-Gly-Pro-OX1, где Y и X1 имеют значения, указанные выше, обрабатывают деблокирующими реагентами и полученный продукт в случае необходимости подвергают ацетилированию. Список сокращений АсОН – уксусная кислота Ас – ацетил AcONp – пара-нитрофенилацетат Вос – трет.-бутилоксикарбонил But – трет.-бутил Bzl – бензил DCC – N,N’-дициклогексилкарбодиимид DCHU – N,N’-дициклогексилмочевина DMF – N,N-диметилформамид HOBt – 1-гидроксибензотриазол HONp – пара-нитрофенол HONSu – N-гидроксисукцинимид TFA – трифторуксусная кислота Z – бензилоксикарбонил ВЭЖХ – высокоэффективная жидкостная хроматография ТСХ – тонкослойная хроматография Экспериментальная часть В синтезе использованы производные L-аминокислот фирмы Bachem (Швейцария). DCC, TFA, HONSu, HONp фирмы Fluka (Швейцария). DMF очищали перегонкой над нингидрином и окисью бария. Аналитическую ВЭЖХ проводили на хроматографе фирмы Gilson (Франция). ТСХ проводили на стеклянных хроматографических пластинках Merck-Kiselgel 60 (Германия) в следующих системах растворителей:
1Н-ЯМР – спектры снимают на спектрометре WH-500 Bruker 500 МГц (ФРГ) в DMSO-d6 при 300 К. Химические сдвиги ( Масс-спектры регистрируют на масс-спектрометре Analytical Compact MALDI 4 фирмы Kratos (Великобритания). Описываемый способ иллюстрируется следующими примерами. Пример 1. 1. 4.1 г (55 ммоль) глицина растворяют в 55 мл 1н. раствора NaOH, добавляют при охлаждении раствор 15.6 г (50 ммоль) Boc-Pro-ONSu в 100 мл DMF и перемешивают в течение 24 ч, контролируя протекание реакции с помощью ТСХ в системе Б. Раствор упаривают, остаток растворяют в воде, водный раствор экстрагируют эфиром (2×20 мл), подкисляют до рН 2-3 12 н. раствором HCl и экстрагируют смесью этилацетата с бутанолом в соотношении 2:1 (2×150 мл). Объединенные органические вытяжки промывают насыщенным раствором NaCl и водой до нейтральной реакции и упаривают. Остаток кристаллизуют из смеси эфир-гексан 1:1, отфильтровывают, сушат в вакууме. В итоге получают 11.6 г (85%) Ia с Rf 0.57 (А), 0.71 (Б), 0.86 (В). 2. К раствору 9.0 г (44 ммоль) H-Pro-OBzl в 100 мл DMF, охлажденному до -10°С, прибавляют при интенсивном перемешивании охлажденный до той же температуры раствор 10.9 г (40 ммоль) Ia, 6.7 г (44 ммоль) HOBt и 9.2 г (44 ммоль) DCC в DMF. Реакционную смесь перемешивают 10 мин при -10°С и оставляют при комнатной температуре на 24 ч. Полноту конденсации контролируют с помощью ТСХ в системах А и Г. По окончании реакции выпавший осадок DCHU отфильтровывают, фильтрат упаривают, остаток растворяют в этилацетате и промывают последовательно 2% H2SO4 (3×20 мл), водой, 2% раствором НаНСО3, водой до нейтральной реакции. Раствор упаривают, остаток растирают с гексаном, отфильтровывают, сушат в вакууме. В итоге получают 17.1 г (93%) Iб с Rf 0.80 (A), 0.88 (Б), 0.50 (Г). 3. 6 г (13.1 ммоль) трипептида Iб растворяют в 3 н. растворе HCl в АсОН в течение 1 ч. Полноту деблокирования контролируют с помощью ТСХ в системе Б. Через 1 ч реакционную смесь упаривают в вакууме. Остаток растирают с сухим холодным эфиром, отфильтровывают, сушат в вакууме. В итоге получают 5.0 г (97%) I. Гомогенность полученного продукта подтверждают с помощью аналитической ВЭЖХ в условиях градиентного элюирования буфером Б в буфере А (А – 0.1% TFA, Б – 80% ацетонитрила в буфере А) от 10 до 70% буфера Б за 30 мин на колонке Ultrasphere ODS 4.6×250 мм, размер зерна 5 мкм, детекция при 220 нм. Чистота, по данным ВЭЖХ 95%, Rt 17.5 мин (в указанных условиях). Rf 0.19 (А), 0.52 (Б), 0.76 (В). 1Н-ЯМР – спектр (DMSO-d6), Pro: 4.24 ( Gly: 8.72 (NH, 1H); 4.11, 3.98 ( Pro: 4.39 ( Пример 2. 1. 19,5 г (0,260 М) глицина растворяют в 260 мл 1 н. NaOH, полученный раствор добавляют к охлажденному до 0°С раствору 96,0 г (0,260 М) Z-Pro-ONp в 600 мл DMF. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, упаривают, остаток растворяют в 300 мл воды и трижды экстрагируют эфиром. Эфирный слой отделяют, к водному слою добавляют 150 мл 2 н. раствора HCl, выпавшее масло экстрагируют 600 мл этилацетатата. Органический слой отделяют и промывают водой до нейтральной реакции, сушат над Na2SO4, упаривают, остаток перекристаллизовывают из эфира. В итоге получают: 67,5 г (85%) IIa с Т.пл. 122-123°С; Rf 0,67 (A); Rf 0,81 (В), Rf 0,65 (С). 2. 0,920 г (3 мМ) соединения IIa растворяют в 40 мл DMF, к полученному раствору добавляют 0.33 мл (3 мМ) N-метилморфолина, охлаждают до -15°С и при перемешивании добавляют 0,39 мл (3 мМ) изобутилхлорформиата. Реакционную смесь перемешивают в течение 5 минут при -10°С, затем прибавляют к ней раствор 0,73 г (3 мМ) HCl·H-Pro-OBzl и 0.33 мл (3 мМ) N-метилморфолина в 20 мл DMF. Через час реакционную смесь упаривают, образовавшееся масло растворяют в 50 мл этилацетата и последовательно промывают 2% H2SO4, водой, 5% NaHCO3, водой. Органический слой упаривают. Получают 1.48 г (100%) соединения IIб в виде масла. Rf 0,87 (Б). 3. 1,48 г (3 мМ) соединения IIб растворяют в 60 мл этилового спирта и гидрируют над 5% Pd/C. Полноту процесса гидрирования контролируют с помощью ТСХ в системе Б. Катализатор отфильтровывают, трижды промывают этиловым спиртом, фильтрат упаривают, образовавшееся масло растирают в эфире, полученный осадок отфильтровывают. Получают 0,78 г (96%) соединения II с Т.пл 151-155°С, Rt 5,44 мин и чистотой 96,6% по данным аналитической ВЭЖХ. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М КН2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% Н2О; градиент: 10-70% В за 30 мин Rf 0,16 (В). MALDI-MS m/z, найдено: 270,23 [М+Н]+, вычислено: 269,3 1Н-ЯМР – спектр (DMSO-d6, для trans-конформера: Pro – 4,24 ( Gly – 8,70 (NH, 1H); 4,07/3,96 ( Pro – 4,24 ( Пример 3. 0,79 г (2 мМ) соединения I растворяют в 40 мл воды и гидрируют над 5% Pd/C. Полноту процесса гидрирования контролируют с помощью ТСХ в системе Б. Катализатор отфильтровывают, промывают на фильтре водой, фильтрат упаривают, оставшееся масло трижды упаривают с изопропиловым спиртом, растирают в эфире, полученный осадок отфильтровывают. Получают 0,57 г (93%) соединения III с Т.пл. 172-176°С, Rt 5,44 мин. и чистотой 96,6% по данным аналитической ВЭЖХ. Колонка Ultropac TSK ODS-120T 4.6×250 мм, 5 мкм. Буфер А: 0,05 М КН2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% H2O; градиент: 10-70% В за 30 мин Rf 0,16 (В). MALDI-MS m/r, найдено: 270,23 [М+Н]+, вычислено: 269,3 1Н-ЯМР – спектр (DMSO-d6, для trans-конформера: Pro – 4,24 ( Gly – 8,70 (NH, 1H); 4,07/3,96 ( Pro – 4,24 ( Пример 4. 0,61 г (2,25 мМ) соединения II растворяют в 5 мл DMF, к полученному раствору добавляют 0,25 мл (2,25 мМ) N-метилморфолина и 0,45 г (2,5 мМ) AcONp. Реакционную смесь выдерживают при комнатной температуре в течение 8 часов (полноту протекания реакции контролируют с помощью ТСХ в системе В). Затем реакционную смесь упаривают, образовавшееся масло растворяют в воде, дважды экстрагируют эфиром, эфирный слой отбрасывают, а водный подвергают очистке методом ВЭЖХ на колонке Диасорб 24×250 мм, 5 мкм. Буфер А: 0,1% TFA; буфер В: ацетонитрил; градиент: 0-40% В за 20 мин. Получают 0,67 г (95%) соединения IV с Т.пл 91-99°С, Rt 8,99 мин и чистотой 98,7% по данным аналитической ВЭЖХ. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М KH2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% Н2O; градиент: 10-70% В за 30 мин Rf 0,25 (В). MALDI-MS m/z, найдено: 312,4 [М+Н]+, вычислено: 311,3. 1Н-ЯМР – спектр (DMSO-d6, для cis-конформера: Pro – 4,37 ( Gly – 8,20 (NH, 1H); 4,06/3,79 ( Pro – 4,56 ( Ac – 1,89 (-СН3, 3Н) для trans-конформера: Pro – 4,329 ( Gly – 7,88 (NH, 1H); 3,93/3,81 ( Pro – 4,23 ( Ac – 1,96 (-СН3, 3Н) Пример 5. 0,61 г (2,25 мМ) соединения II растворяют в 5 мл DMF, к полученному раствору добавляют 0,25 мл (2,25 мМ) N-метилморфолина и 0,26 г (2,5 мМ) уксусного ангидрида. Реакционную смесь выдерживают при комнатной температуре в течение 1 часа (полноту протекания реакции контролируют с помощью ТСХ в системе В). Затем реакционную смесь упаривают, образовавшееся масло растворяют в воде и подвергают очистке методом ВЭЖХ на колонке Диасорб 24×250 мм, 5 мкм. Буфер А: 0,1% TFA; буфер В: ацетонитрил; градиент: 0-40% В за 20 мин. Получают 0,57 г (81%) соединения IV с Т. пл. 92-95°С, Rt 8,99 мин и чистотой 96,6% по данным аналитической ВЭЖХ. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М KH2РО4, рН 3,0; буфер В: 70% ацетонитрил + 30% Н2O; градиент: 10-70% В за 30 мин Rf 0,25 (В). MALDI-MS m/z, найдено: 312,4 [М+Н]+, вычислено: 311,3. 1Н-ЯМР – спектр (DMSO-d6, для cis-конформера: Pro – 4,37 ( Gly – 8,20 (NH, 1H); 4,06/3,79 ( Pro – 4,56 ( Ac – 1,89 (-СН3, 3Н) для trans-конформера: Pro – 4,329 ( Gly – 7,88 (NH, 1H); 3,93/3,81 ( Pro – 4,23 ( Ac – 1,96 (-СН3, 3Н) Пример 6. 0,89 г (2,25 мМ) соединения I растворяют в 10 мл DMF, к полученному раствору добавляют 0,25 мл (2,25 мМ) N-метилморфолина и 0,26 г (2,5 мМ) уксусного ангидрида. Реакционную смесь выдерживают при комнатной температуре в течение 1 часа (полноту протекания реакции контролируют с помощью ТСХ в системе В). Затем реакционную смесь упаривают, остаток растворяют в воде и гидрируют над 5% Pd/C. После завершения процесса гидрирования (контроль с помощью ТСХ в системе А) катализатор отфильтровывают, промывают на фильтре трижды водой, водный раствор целевого продукта упаривают до 1/3 объема и подвергают очистке методом ВЭЖХ на колонке Диасорб 24×250 мм, 5 мкм. Буфер А: 0,1% TFA; буфер В: ацетонитрил; градиент: 0-40% В за 20 мин. Получают 0,63 г (91%) соединения IV с Т.пл. 90-94°С, Rt 8,99 мин и чистотой 96,2% по данным аналитической ВЭЖХ. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М KH2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% Н2О; градиент: 10-70% В за 30 мин Rf 0,25 (В). MALDI-MS m/z, найдено: 312,4 [М+Н]+, вычислено: 311,3. 1Н-ЯМР – спектр (DMSO-d6, для cis-конформера: Pro – 4,37 ( Gly – 8,20 (NH, 1H); 4,06/3,79 ( Pro – 4,56 ( Ac – 1,89 (-СН3, 3Н) для trans-конформера: Pro – 4,329 ( Gly – 7,88 (NH, 1H); 3,93/3,81 ( Pro – 4,23 ( Ac – 1,96 (-СН3, 3Н) Пример 7. Гидрохлорид трет-бутилового эфира пролил-глицил-пролина 1. 19,5 г (260 ммоль) глицина растворяют в 260 мл 1 н. NaOH, полученный раствор добавляют к охлажденному до 0°С раствору 96,0 г (260 ммоль) Z-Pro-ONp в 600 мл DMF. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, упаривают, остаток растворяют в 300 мл воды и трижды экстрагируют эфиром. Эфирный слой отделяют, к водному слою добавляют 150 мл 2 н. раствора HCl, выпавшее масло экстрагируют 600 мл этилацетатата. Органический слой отделяют и промывают водой до нейтральной реакции, сушат над Na2SO4, упаривают, остаток перекристаллизовывают из эфира. В итоге получают: 67,5 г (85%) продукта с Т пл. 122-123°С; Rf 0,81 (Б), Rf 0,65 (Г). 2. К раствору 45.7 г (220 ммоль) H-Pro-Obut HCl в 500 мл DMF, охлажденному до -10°С, прибавляют при интенсивном перемешивании охлажденный до той же температуры раствор 67.5 г (220 ммоль) Va, 33.4 г (220 ммоль) HOBt и 45.3 г (220 ммоль) DCC в 100 мл DMF. Реакционную смесь перемешивают 10 мин при -10°С и оставляют при комнатной температуре на 24 ч. Полноту конденсации контролируют с помощью ТСХ в системах А и Г. По окончании реакции выпавший осадок DCHU отфильтровывают, фильтрат упаривают, остаток растворяют в этилацетате (800 мл) и промывают последовательно 2% H2SO4 (3×250 мл), водой (3×250 мл), 2% раствором NaHCO3 (3×250 мл), водой до нейтральной реакции. Раствор упаривают, остаток растирают с гексаном, отфильтровывают, сушат в вакууме. В итоге получают 90.9 г (90%) Vб, с Rf 0.82 (A), 0.91 (Б), 0.6 (Г). 3. 90.9 г (198 ммоль) Vб растворяют в 1000 мл EtOH и гидрируют над 5% Pd/C до исчезновения исходного соединения по ТСХ в системе Б. Затем катализатор отфильтровывают, к фильтрату добавляют 198 мл 1 н. водного раствора соляной кислоты и упаривают досуха, маслообразный остаток упаривают с изопропиловым спиртом, остаток обрабатывают эфиром. Выпавший осадок отфильтровывают, промывают на фильтре эфиром и сушат. В итоге получают 68.1 г (95%) V с Rf 0.15 (А), 0.45 (Б), 0.70 (В). Гомогенность полученного продукта также подтверждают с помощью аналитической ВЭЖХ в условиях градиентного элюирования буфером Б в буфере А (А – 0.1% TFA, Б – 80% ацетонитрила в буфере А) от 10 до 70% буфера Б за 30 мин на колонке Ultrasphere ODS 4.6×250 мм, размер зерна 5 мкм, детекция при 220 нм. Чистота, по данным ВЭЖХ, 96%, Rt 15.9 мин (в указанных условиях). 1Н-ЯМР – спектр (DMSO-d6), Pro: 4.23 ( Gly: 8.72 (NH, 1H); 4.11, 3.97 ( Pro: 4.37 ( Как видно из вышеприведенных примеров, описанный способ отличается простотой и позволяет получать целевые продукты в препаративном масштабе с высоким выходом. Литература 1. Успехи физиологических наук. Т.34, №1, с.14-19 (2003). 2. Патент РФ №2252779, кл. А61К 38/07 (2005). 3. J.Mol.Biol. V.17, р.255-272 (1966). 4. Гринштейн Дж., Виниц М. Химия аминокислот и пептидов. -М: Мир (1965). 5. Патент США 6310041, кл. 514/12 (2001). 6. Изв. АН СССР, Отд. хим. наук, 1959, №4, с.736-738.
Формула изобретения
1. Способ получения трипептидов общей формулы A-Pro-Gly-Pro-OX, где А – Н, Ас; Х – Н, бензил (Bzl), трет.-бутил (But), или их гидрохлоридов, отличающийся тем, что синтез осуществляют жидкофазным методом путем конденсации С-концевого производного пролина общей формулы Н-Pro-ОХ1, где X1 – Bzl, But с N-защищенным дипептидом общей формулы Y-Pro-Gly-OH, где Y – бензилоксикарбонил (Z), трет.-бутилоксикарбонил (Boc), и полученный защищенный трипептид общей формулы Y-Pro-Gly-Pro-OX1, где Y и Х1 имеют значения, указанные выше, обрабатывают деблокирующими реагентами и полученный продукт в случае необходимости подвергают ацетилированию и необязательно получают гидрохлориды. 2. Способ по п.1, отличающийся тем, что X1 представляет собой бензил (Bzl), Y – бензилоксикарбонил (Z), а деблокирование защищенного трипептида осуществляется каталитическим гидрогенолизом.
|
||||||||||||||||||||||||||
 -казоморфин, кальцитонин, гастрин) и целого ряда нейротропных пептидных препаратов, таких как семакс и селанк [1]. Кроме того, трипептид Pro-Gly-Pro обладает противоязвенной активностью [2].
-казоморфин, кальцитонин, гастрин) и целого ряда нейротропных пептидных препаратов, таких как семакс и селанк [1]. Кроме того, трипептид Pro-Gly-Pro обладает противоязвенной активностью [2]. , м.д.) измеряют относительно тетраметилсилана.
, м.д.) измеряют относительно тетраметилсилана.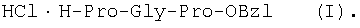
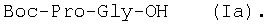
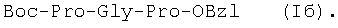
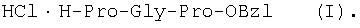
 -CH, 1H); 2.32, 1.88 (
-CH, 1H); 2.32, 1.88 ( -СН2, 2Н); 3.58, 3.54 (
-СН2, 2Н); 3.58, 3.54 (