Патент на изобретение №2158266
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ПУРИНОВЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМОЕ В НЕМ ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ
(57) Реферат: Изобретение относится к улучшенному способу получения пуриновых соединений общей формулы А, где Х означает водород, гидрокси, хлор, Ra и Rb означают водород, ацил. Указанные соединения находят применение в фармацевтической промышленности. Предлагаемый способ заключается в том, что соединение формулы II подвергают взаимодействию с соединением формулы V, где значения R1R2, R3 такие, как определено ниже для соединения формулы I, a L обозначает уходящую группу, с получением соединения формулы VI, которое затем декарбоксилируют. Полученное при этом соединение формулы 1, где R1 означает С1-С6-алкил или фенил-С1-С6-алкил, R2 означает хлор, R3 означает аминогруппу или защищенную аминогруппу, превращают в соединение формулы А путем превращения радикала R3 в незащищенную аминогруппу восстановлением сложно-эфирных групп CO2R1 в СН2ОН и, при необходимости, переводят их в соответствующие ацильные производные или, при желании, радикал R2 в соединении формулы I превращают в другое значение, соответствующее значению радикала Х в соединении формулы А. Соединение формулы I, являющееся промежуточным соединением для получения соединения формулы А, является новым и также является объектом притязаний. Предлагается также способ получения соединения формулы I. Изобретение позволяет упростить процесс и получить соединение формулы А с более высоким выходом, который возрастает с 10,6 до 40% по сравнению с известным способом. 3 с. и 4. з.п.ф-лы. 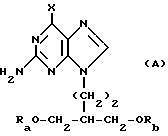 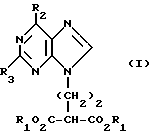 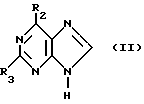 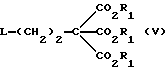 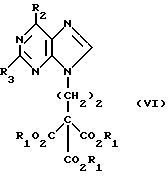
Данное изобретение относится к области получения химических соединений, в частности фармацевтических пуриновых соединений, обладающих антивирусной активностью. Известно, что 2-амино-6-хлорпурин (ACP) формулы 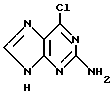 является полезным промежуточным соединением для получения антивирусных агентов, которые представляют собой аналоги нуклеозидов, таких как пенцикловир (пример 1) (ранее известный как BRL 39123) (EP-A-141927) и фамцикловир (пример 2) (ранее известный как BRL 42810) (EP-A-182024) соответственно. Известен (EP-A-0302644, C 07 D 473/32, C 07 D 473/18, C 07 D 473/40, 08.02.89) способ получения соединений формулы (A): 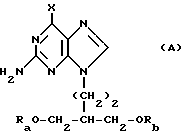 где X обозначает водород, гидрокси или хлор; Ra и Rb обозначают водород или ацил; при котором соединение формулы (II): 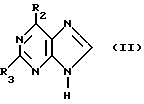 где R2 обозначает хлор, a R3 обозначает аминогруппу или защищенную аминогруппу, подвергают взаимодействию с соединением формулы (V): 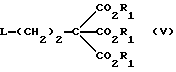 где L обозначает отщепляемую группу; R1 обозначает C1-6алкил или фенилC1-6алкил; с получением соединения формулы (VI): 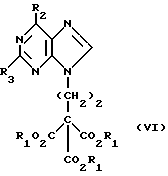 где R1, R2 и R3 имеют упомянутые выше значения, у которого затем удаляют хлор-группу в 6-ом положении молекулы, а полученное в результате соединение декарбоксилируют, а затем восстанавливают до соединения формулы (A). Недостатками указанного способа, особенностью которого является удаление хлоргруппы по 6-ому положению на ранней стадии процесса (смотри реакционные схемы 1 и 2 в конце описания), являются многостадийность, необходимость выделения промежуточных соединений с помощью технологически невыгодной колоночной хроматографии и, соответственно, низкий общий выход, составляющий 10,6%. Соответственно в задачу настоящего изобретения входило создание нового, технологически более простого и эффективного способа получения соединений формулы (A), позволяющего существенно повысить выход целевого продукта. Поставленная задача решается тем, что предложен способ получения соединения формулы (A): 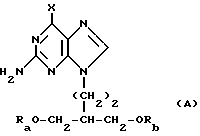 где X обозначает водород, гидрокси или хлор; Ra и Rb обозначают водород или ацил; при котором соединение формулы (II): 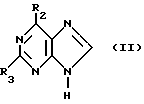 где R2 обозначает хлор, а R3 обозначает аминогруппу или защищенную аминогруппу, подвергают взаимодействию с соединением формулы (V): 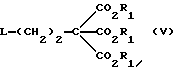 где L обозначает отщепляемую группу; R1 обозначает C1-6алкил или фенилC1-6алкил; затем полученное соединение формулы (VI): 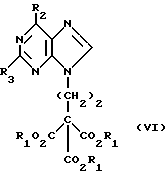 где R1, R2 и R3 имеют упомянутые выше значения, декарбоксилируют с получением соединения формулы (I): 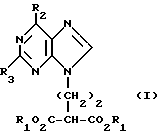 где R1, R2 и R3 имеют упомянутые выше значения, а R3, когда он не является аминогруппой, превращают в аминогруппу; и, если необходимо, соединение формулы (I), имеющее сложно-эфирные группы CO2R1, восстанавливают до соединения формулы (A), где Ra и Rb являются водородом и, если необходимо, превращают R2 в другие значения радикала X соединения формулы (A). Способом по изобретению получают, в частности, 9-(4-ацетокси-3-ацетоксиметилбут-1-ил)-2-аминопурин (фамцикловир) и 9-(-4-гидрокси-3-гидроксиметилбут-1-ил)гуанин (пенцикловир). В рамках решения поставленной задачи предложен также способ получения соединения формулы (I): 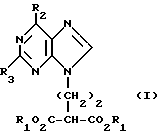 где R1, R2 и R3 имеют определенные выше значения, при котором соединение формулы (II): 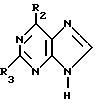 где R2 обозначает хлор, a R3 обозначает аминогруппу или защищенную аминогруппу, подвергают взаимодействию с соединением формулы (V): 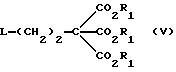 где L обозначает галоген; R1 обозначает C1-4алкил, затем полученное соединение формулы (VI): 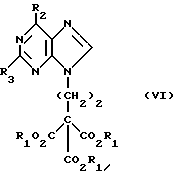 где R1, R2 и R3 имеют упомянутые выше значения, декарбоксилируют и, если необходимо, соединение формулы (I) превращают в другие соединения формулы (I). В рамках решения поставленной задачи предложено также соединение формулы (I) или его соль: 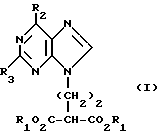 где R1, R2 и R3 такие, как определено выше. Предпочтительным является описанное выше соединение или его соль, где R1 обозначает метил или этил, a R3 обозначает аминогруппу. Наиболее предпочтительным является соединение, которое представляет собой 2-амино-6-хлор-9-(метил-2-карбометоксибутаноат-4-ил)пурин. Способ по изобретению, при котором хлоргруппу удаляют только на заключительной стадии, уже после стадии декарбоксилирования, обеспечивает значительные технологические преимущества, позволяет существенно упростить стадии процесса, в частности, за счет исключения стадий колоночной хроматографии, уменьшить их количество, а также значительно повысить выход целевого продукта – до 41%. Следующие примеры иллюстрируют изобретение. ПРИМЕР 1 (Продукт стадии 1) Получение 2-амино-6-хлор-9-(метил-2-карбометокси6утаноат-4-ил)пурина Смесь 2-амино-6-хлорпурина (9,18 г, 53,1 ммоль), триэтилового эфира 3-бромпропан-1,1,1-трикарбоновой кислоты (20,33 г, 57,3 ммоль), карбоната калия (11,1 г, 80,3 моль) и диметилформамида (190 мл) перемешивают при температуре от 60 до 63oC в течение 22 ч. Затем горячую реакционную смесь фильтруют через слой целита и осадок промывают диметилформамидом (30 мл). Фильтрат и промывную жидкость объединяют и растворитель удаляют при помощи дистилляции в высоком вакууме с получением неочищенного красновато-коричневого масла. Его растворяют в метанолу (140 мл), охлаждают до 20oC и затем при перемешивании добавляют раствор метоксида натрия (1,2 г) в метаноле (40 мл). Через примерно 20 мин образуется осадок, а перемешивание продолжают всего в течение 1 ч. Реакционную смесь затем охлаждают до 15oC и выдерживают при этой температуре в течение 30 мин. Продукт отфильтровывают, промывают метанолом (10 мл) и высушивают при 40oC в течение 16 ч под вакуумом. Выход: 12,0 г продукта 95%-ной чистоты. ПРИМЕР 2 (продукт стадии 2) Получение 9-(4-ацетокси-3-ацетоксиметилбут-1-ил) -2-амино-6-хлор-пурина Смесь 2-амино-6-хлор-9-(метил-2-карбометоксибутаноат-4-ил)пурина (32,7 г, 0,1 моль), борогидрида натрия (11,5 г, 0,3 моль) и метилендихлорида (125 мл) перемешивают при 20oC. В течение 2,0 ч по каплями добавляют метанол (75 мл), в то время как температуру реакционной смеси поддерживают равной 20-22oC путем охлаждения. Оставляют реакционную смесь перемешиваться в течение еще 1,5 ч. Добавляют воду (100 мл), а затем по каплям – концентрированную соляную кислоту (20-22 мл) до pH в пределах 6,7-7,0, поддерживая температуру реакционной смеси около 20-22oC. Метилендихлорид и метанол удаляют под вакуумом, пока объем реакционной смеси не достигнет 150 мл. Реакционную смесь охлаждают до 5oC и перемешивают при этой температуре 30 мин. Полученный осадок отфильтровывают и оставшийся на фильтре продукт промывают холодной водой (20 мл). Полученное влажное твердое вещество (40-50 г) перемешивают с триэтиламином (15 мл), 4-диметиламинопиридином (1,0 г) в метилендихлориде (250 мл). По каплям в течение 20-30 мин добавляют уксусный ангидрид (75 мл, 0,79 моль) с такой скоростью, чтобы контролировать дефлегмацию. Реакционную смесь кипятят с обратным холодильником еще 1,5 ч. Реакционную смесь охлаждают до 20oC и нейтрализуют 20%-ным (мас./мас.) раствором гидроксида натрия до pH 6,4-6,5. Слой метилендихлорида отделяют и водную фазу экстрагируют метилендихлоридом (100 мл). Объединенные метилендихлоридные фазы выпаривают до сухого состояния. Неочищенное влажное твердое вещество перекристаллизовывают из смеси 3:1 метанол: вода (75 мл), охлаждают осадок до -5oC в течение 1 ч перед фильтрацией. Продукт промывают холодной смесью 3:1 метанол:вода (0oC) и высушивают при 40oC в течение 16 ч в вакуумной печи. Выход: 23 г продукта 97-98%-ной чистоты. ПРИМЕР 3 (продукт стадии 3) а) Получение 9-(4-ацетокси-3-ацетоксиметилбут-1-ил)- 2-аминопурина-фамцикловира Смесь 9-(4-ацетокси-3-ацетоксиметилбут-1-ил)-2-амино-6-хлорпурина (15,4 г, 43 ммоль), 5%-ного палладия на угле (6,16 г), триэтиламина (6,6 мл, 47 ммоль) и этилацетата (77 мл) перемешивают при 50oC в атмосфере водорода при давлении 1 бар (100 кПа) в автоклаве в течение 3-5 ч. После завершения реакции смесь удаляют из автоклава, который промывают этилацетатом (30 мл), сохраняя промывные жидкости при 50oC. Через слой целита фильтруют основную реакционную смесь, затем промывные жидкости и наконец этилацетат (30 мл). Воду (46 мл) добавляют к объединенной смеси этилацетатного фильтрата и промывных жидкостей. Этилацетат выпаривают до сухого состояния с получением неочищенного белого твердого вещества. Его перекристаллизовывают из н-бутанола (62 мл), перемешивают охлажденный раствор при 0-5oC в течение 3 ч перед фильтрацией. Отфильтровывают продукт и промывают маточными растворами. Твердое вещество ресуспендируют в н-гептане (50 мл), перемешивают 30 мин и фильтруют. Продукт высушивают под вакуумом при 40oC в течение 16 ч. Выход: 11-11,3 г. б) получение 9-(4-гидрокси-3-гидроксиметилбут-1-ил)гуанина – пенцикловира Смесь 9-(4-ацетокси-3-ацетоксиметилбут-1-ил)-2-амино-6-хлорпурина (10 г, 28,1 ммоль), муравьиной кислоты (96%, 6,3 мл) и воды (55 мл) перемешивают и кипятят с обратным холодильником в течение примерно 4 ч. После охлаждения раствор подщелачивают, смешивая с раствором гидроксида натрия (12,5 М, 27 мл), и образовавшийся раствор перемешивают 1,5 ч. Раствор нейтрализуют добавлением муравьиной кислоты. Образовавшуюся суспензию нагревают до температуры дефлегмации (около 105oC), затем охлаждают до 40-45oC и перемешивают около 3 ч. Неочищенный продукт выделяют и промывают водой (20 мл). Выделенный продукт растворяют в растворе гидроксида натрия (3М, 80 мл). Добавляют уголь (около 1,5 г) и перемешивают суспензию около 1 ч, затем уголь удаляют путем фильтрации и промывают водой (20 мл). Раствор нейтрализуют путем добавления уксусной кислоты, образовавшийся осадок повторно растворяют путем нагревания до примерно 100oC, а затем охлаждают. Выпавший в осадок продукт перемешивают около 3 ч, затем выделяют, промывают водой (2х20 мл) и высушивают. Выход 5,3-5,5 г. Формула изобретения
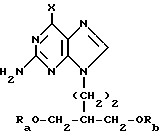 где Х обозначает водород, гидрокси или хлор; Ra и Rb обозначают водород или ацил, при котором соединение формулы II 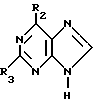 где R2 обозначает хлор; R3 обозначает аминогруппу или защищенную аминогруппу, подвергают взаимодействию с соединением формулы V 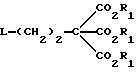 где L обозначает отщепляемую группу; R1 обозначает С1-6алкил или фенилС1-6алкил; отличающийся тем, что затем полученное соединение формулы VI 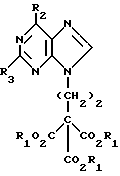 где R1, R2 и R3 имеют упомянутые выше значения, декарбоксилируют с получением соединения формулы I 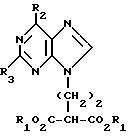 где R1, R2 и R3 имеют упомянутые выше значения; R3 когда он не является аминогруппой, превращают в аминогруппу, и, если необходимо, соединение формулы I, имеющее сложно-эфирные группы СО2R1, восстанавливают до соединения формулы А, где Ra и Rb являются водородом, и, если необходимо, превращают R2 в другие значения радикала Х соединения формулы А. 2. Способ по п.1, отличающийся тем, что получают 9-(4-ацетокси-3-ацетоксиметилбут-1-ил)-2-аминопурин (фамцикловир). 3. Способ по п.1, отличающийся тем, что получают 9-(4-гидрокси-3-гидроксиметилбут-1-ил)гуанин(пенцикловир). 4. Способ получения соединения формулы I 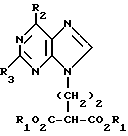 где R1, R2 и R3 имеют значения, определенные в п.1, при котором соединение формулы II 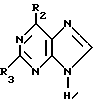 где R2 обозначает хлор; R3 обозначает аминогруппу или защищенную аминогруппу, подвергают взаимодействию с соединением формулы V 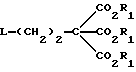 где L обозначает галоген; R1 обозначает С1-4алкил, отличающийся тем, что затем полученное соединение формулы VI 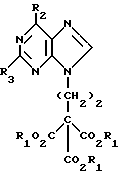 где R1, R2 и R3 имеют упомянутые выше значения, декарбоксилируют и, если необходимо, соединение формулы I превращают в другие соединения формулы I. 5. Соединение формулы I или его соль 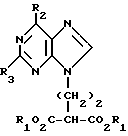 где R1, R2 и R3 такие, как определено в п.1. 6. Соединение по п.5 или его соль, где R1 обозначает метил или этил, а R3 обозначает аминогруппу. 7. Соединение по п.6, отличающееся тем, что оно представляет собой 2-амино-6-хлор-9-(метил-2-карбометоксибутаноат-4-ил)пурин. РИСУНКИ
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
Номер и год публикации бюллетеня: 5-2002
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 04.10.2001 № 13235
Извещение опубликовано: 20.02.2002
|
||||||||||||||||||||||||||