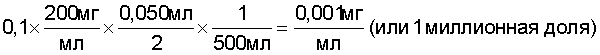Патент на изобретение №2300766
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБЫ ИЗМЕРЕНИЯ СКОРОСТИ РАСТВОРЕНИЯ АНАЛИЗИРУЕМОГО ВЕЩЕСТВА В НЕВОДНОЙ ЖИДКОЙ КОМПОЗИЦИИ
(57) Реферат:
Изобретение относится к области аналитических измерений. В способе используют вещество, которое предпочтительно представляет собой фармацевтически активный компонент в лекарственной форме с замедленным высвобождением. Способ предусматривает (i) контактирование разбавленной неводной композиции, содержащей анализируемое вещество и неводную основу, такую как воск или жир, с водной растворяющей средой в течение заранее определенного периода времени и (ii) измерение количества анализируемого вещества в водной растворяющей среде. Технический результат – повышение точности и надежности определения скорости растворения анализируемого вещества. 3 н. и 31 з.п. ф-лы, 8 ил., 3 табл.
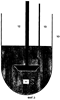
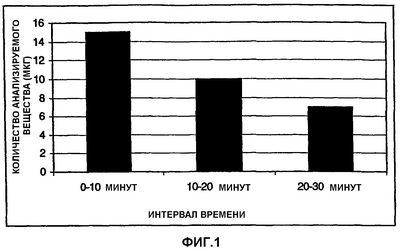
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ Изобретение относится к способу изучения свойств переноса анализируемого вещества из неводной жидкой композиции в водную среду и, в частности, к способу измерения in vitro растворения лекарственного средства из лекарственной формы с замедленным высвобождением. УРОВЕНЬ ТЕХНИКИ Одним важным аспектом изготовления фармацевтических композиций является фармакокинетическое поведение лекарственного средства. В зависимости от ряда факторов, таких как физическое состояние лекарственного средства (т.е. газ, жидкость, твердое вещество), его кристаллическая форма, размер его частиц, лекарственная форма и используемые наполнители, зависимое от времени высвобождение лекарственного средства в организме может резко меняться. Даже если в одной и той же лекарственной форме присутствует одно и то же лекарственное средство, могут наблюдаться различия между партиями. Для получения разрешения на применение лекарственного средства часто определяют его фармакокинетическое поведение путем введения лекарственного средства животным или людям и измерения количества лекарственного средства или его метаболитов в крови через определенные промежутки времени после введения. Указанный способ требует много времени и расходов и обычно не применяется для контроля качества лекарственных средств во время процесса производства. Был предложен ряд методик для оценки in vivo фармакокинетического поведения лекарственных средств в тестах in vitro. Некоторые из тестов стандартизированы и описаны в Фармакопее Соединенных Штатов (USP). Широко используются такие методики USP как способ корзины (способ USP I) и лопастной способ (способ USP II). Помимо указанных стандартизированных методик описан обширный ряд способов для конкретного индивидуального применения. Обзор ряда способов растворения можно найти, например, в G.K.Shiu, Drug Information Journal, 30, 1045-1054 (1996). Andonaegui et al. (Drug Development and Industrial Pharmacy, 25(11), 1199-1203 (1999)) описывают способ in vitro для прогнозирования поведения in vivo матриксных таблеток теофиллина с замедленным высвобождением, при введении натощак и при рационе с высоким содержанием жиров. Изучали профили растворения теофиллина в трех типах матриксных таблеток с замедленным высвобождением. Для улучшения корреляции in vitro/in vivo при рационе с высоким содержанием жиров таблетки предварительно обрабатывали смешиванием с арахисовым маслом перед тестом на растворение в ходе лопастного способа USP. В заявке на выдачу патента Японии JP 05-249097 описан тест на растворение для прогнозирования высвобождения in vivo таблетки с замедленным высвобождением. Таблетку изучали с использованием лопастного способа, извлекали, обрабатывали маслами, а затем возвращали в лопастной аппарат с гранулами в водной растворяющей среде или погружали в корзину. Указанный способ, как сообщается, прогнозирует концентрацию лекарственного средства в плазме крови в живом организме, и на него не влияет механизм контроля высвобождения таблетки с замедленным высвобождением. Различные способы растворения in vitro для систем доставки лекарственного средства с форме множества частиц сравнивают Conti et al. в Drug Development and Industrial Pharmacy, 21(10), 1233-1233 (1995). Изучалось влияние скорости перемешивания, ионной силы и присутствия поверхностно-активного агента. Также описаны способы растворения для испытания масляных лекарственных препаратов. Takahashi et al. (Chem. Pharm. Bull., 42(8), 1672-1675 (1994)) сравнивают лопастной способ и способ вращающейся диализной камеры. В разновидности способа вращающейся диализной камеры в качестве внешней фазы использовали октанол, в то время как в качестве внутренней фазы использовали кислый раствор. Machida et al. (Chem. Pharm. Bull., 34(6), 2637-2641 (1986)) описывают одну попытку преодолеть проблемы, с которыми сталкиваются при определении свойств растворения масляных лекарственных препаратов. Они предлагают использовать модификацию лопастного способа 2 Японской Фармакопеи с дополнительной вспомогательной лопастью для перемешивания поверхности водной растворяющей среды. Кроме того, для улучшения перемешивания добавляют бусы, а в качестве водной растворяющей среды используют раствор желчных солей. Фармакокинетическое поведение неводных фармацевтических композиций, в которых лекарственное средство растворено или диспергировано в неводной основе, трудно надежно прогнозировать при использовании известных способов. Точность и надежность in vitro измерений часто низкие, и результаты in vitro измерений не всегда коррелируют с поведением лекарственного средства in vivo. Таким образом, одной целью настоящего изобретения является создание надежного способа, с помощью которого можно определить скорость растворения анализируемого вещества в неводной жидкой композиции. Другой целью настоящего изобретения является создание соответствующего способа, в котором используется стандартизированный аппарат для растворения. КРАТКОЕ ОПИСАНИЕ ФИГУР Фиг.1 показывает одну возможность построения графика скорости растворения, если количество анализируемого вещества определяется более одного раза. Фиг.2 показывает другую возможность построения графика скорости растворения, если количество анализируемого вещества определяется более одного раза. Фиг.3 показывает схему типичного лопастного узла. Рисунок не масштабирован. Фиг.4 показывает вариации в поведении распределения, наблюдающегося в примере 1. Фиг.5 показывает взаимоотношения между продолжительностью замедленного высвобождения in vivo и лопастным способом in vitro, если неводную жидкую композицию не разбавляют неводным разбавителем при осуществлении лопастного способа. Фиг.6 показывает взаимоотношения между продолжительностью замедленного высвобождения in vivo и лопастным способом in vitro, если неводную жидкую композицию разбавляют неводным разбавителем при осуществлении лопастного способа. Фиг.7 иллюстрирует влияние размеры аликвоты на скорость растворения. Фиг.8 показывает линейность способа по настоящему изобретению. СУЩНОСТЬ ИЗОБРЕТЕНИЯ В одном варианте осуществления настоящее изобретение относится к способу определения скорости растворения анализируемого вещества из неводной жидкой композиции, предусматривающему стадии: (a) получения неводной жидкой композиции, содержащей анализируемое вещество и неводную основу; (b) добавления неводного разбавителя к неводной жидкой композиции для получения разбавленной неводной жидкой композиции; (c) помещения по меньшей мере части разбавленной неводной жидкой композиции и водной растворяющей среды в прибор для тестирования на растворение; (d) контактирования разбавленной неводной жидкой композиции и водной растворяющей среды в течение заранее определенного периода времени; и (e) определения количества анализируемого вещества в водной растворяющей среде. В другом варианте осуществления настоящее изобретение относится к способу определения скорости растворения анализируемого вещества из неводной жидкой композиции, предусматривающему стадии: (a) получения неводной жидкой композиции, содержащей анализируемое вещество и неводную основу; (b) помещения по меньшей мере части разбавленной неводной жидкой композиции и водной растворяющей среды в прибор для тестирования на растворение, причем указанная водная растворяющая среда включает буфер, имеющий молярность приблизительно от 0,1 мМ до 10 мМ; (c) контактирования неводной жидкой композиции и водной растворяющей среды в течение заранее определенного периода времени; и (d) определения количества анализируемого вещества в водной растворяющей среде. Описан также способ определения скорости растворения анализируемого вещества из неводной жидкой композиции, предусматривающий стадии: (a) получения неводной жидкой композиции, содержащей анализируемое вещество и неводную основу; (b) помещения по меньшей мере части разбавленной неводной жидкой композиции и водной растворяющей среды в прибор для тестирования на растворение, причем отношение объемов неводной жидкой композиции и водной растворяющей среды в аппарате для тестирования на растворение составляет приблизительно от 1:2000 до 1:100000; (c) контактирования неводной жидкой композиции и водной растворяющей среды в течение заранее определенного периода времени; и (d) определения количества анализируемого вещества в водной растворяющей среде. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Настоящее изобретение относится к надежным способам определения скорости растворения анализируемого вещества из неводной жидкой композиции. Хотя указанные способы предпочтительно используются для определения скорости растворения фармацевтически активного ингредиента из фармацевтической композиции, их также можно использовать в других областях аналитической химии, например, для определения скорости, с которой происходит утечка загрязняющих веществ из масел в окружающую среду, для определения скорости, с которой активные агенты, такие как ингибиторы коррозии и т.п., истощаются из масляных основ или для измерения скорости, с которой компоненты высвобождаются из пестицидов или удобрений. Термин “скорость растворения” представляет собой скорость, с которой анализируемое вещество растворяется в водной растворяющей среде. Если количество анализируемого вещества в водной растворяющей среде определяют только за один заранее определенный период времени, то скорость растворения представляет собой общее количество анализируемого вещества, которое растворилось до указанного заранее определенного момента времени (например, выраженное как масса), деленное на заранее определенный период времени. Например, если было определено, что 3 мкг анализируемого вещества растворились в течение 30 минут, то скорость растворения будет составлять 3 мкг/30 минут или 0,1 мкг в минуту. Если количество анализируемого вещества в водной растворяющей среде определяют более одного раза, то скорость растворения можно проиллюстрировать несколькими различными способами, которые известны специалистам. Одним распространенным способам является построение плоского графика данных, в котором ось х представляет собой линию времени, а ось y представляет количество анализируемого вещества, растворившегося между текущим и предыдущим анализом водной растворяющей среды. Другим распространенным способом является построение плоского графика данных, в котором ось х также представляет собой линию времени, а ось y представляет общее количество анализируемого вещества, растворившегося между началом измерения и текущим анализом водной растворяющей среды. Разумеется, ту же информацию можно представить в виде таблицы или в виде другой удобной формы, не являющейся плоским графиком, обсуждавшимся выше. Следующие серии экспериментов можно использовать в качестве примера: изучают неводную жидкую композицию и определяют количество анализируемого вещества, растворившегося в течение 10 минут (n=1), 20 минут (n=2) и 30 минут (n=3). За 10 минут растворилось 15 мкг анализируемого вещества, за 20 минут растворилось 25 мкг анализируемого вещества и за 30 минут растворилось в целом 32 мкг анализируемого вещества. В первом случае получают график, представленный на фиг.1, в то время как во втором случае получают график, представленный на фиг.2. Используемый в настоящем документе термин “неводная жидкая композиция” представляет собой любую композицию, которая является жидкой при температуре контакта и которая включает анализируемое вещество и неводную основу. Смесь анализируемого вещества и неводной основы может быть в любой форме, например они могут образовывать раствор, эмульсию или суспензию. Если анализируемое вещество суспендировано в неводной основе, размеры частиц анализируемого вещества обычно находятся в пределах приблизительно от 50 нм до 200 микрон, предпочтительно, приблизительно от 100 нм до 200 микрон. Концентрация анализируемого вещества в неводной жидкой композиции особо не ограничивается. Неводная жидкая композиция предпочтительно представляет собой фармацевтическую композицию. В способах по настоящему изобретению фармацевтическая композиция обычно будет представлять собой жидкость, подходящую для парентерального, перорального, сублингвального, интраназального, интрабронхиального, внутрилегочного, интрамаммарного, ректального, вагинального, внутриглазного или местного введения. Однако возможно также определение скорости растворения анализируемого вещества в фармацевтической композиции, которая заключена в капсулу. В данном случае оболочка капсулы будет распадаться при контакте с водной растворяющей средой и высвобождать ее содержимое. Термин “анализируемое вещество” относится к компоненту в неводной жидкой композиции, растворение которого необходимо охарактеризовать. Примерами анализируемых веществ являются, без ограничения, контаминант, активный компонент или неактивный компонент. В случае фармацевтических композиций анализируемое вещество обычно будет представлять собой активный ингредиент, но также он может представлять собой наполнитель или любой другой компонент фармацевтической композиции. Способ по настоящему изобретению не ограничивается определением единственного анализируемого вещества; если это желательно, можно определить растворение двух или более анализируемых веществ. Способ по настоящему изобретению не ограничивается определением анализируемых веществ, обладающих любыми конкретными физическими или химическими характеристиками. Фактически, с использованием способа по настоящему изобретению можно определять растворение любого анализируемого вещества, органического или неорганического, при условии, что анализируемое вещество по меньшей мере частично растворяется в водной растворяющей среде, выбранной для данного способа. Примеры анализируемых веществ, которые можно определить с использованием способа по настоящему изобретению, включают следующие иллюстративные, неограничивающие классы: ингибиторы АПФ, агонисты Следует понимать, что любая ссылка в настоящем документе на конкретное лекарственное соединение включает таутомеры, стереоизомеры, энантиомеры, соли и пролекарства указанного соединения, и не является специфичной для какого бы то ни было твердого состояния лекарственного средства. Способ по настоящему изобретению является особенно подходящим для определения скорости растворения цефалоспоринов, таких как третье поколение цефалоспоринов. Примеры включают, без ограничения, цефтиофур, цефепим, цефиксим, цефоперазон, цефотаксим, цефподоксим, цефтазидим, цефтизоксим, цефтриаксон, моксалактам, их фармацевтически приемлемые соли и производные. Особенно предпочтительным цефалоспорином является цефтиофур, его фармацевтически приемлемые соли и производные. Цефтиофур в настоящее время можно приобрести у компании Pharmacia под торговыми наименованиями Naxel® и Excenel®. Другой предпочтительной формой цефтиофура является кристаллическая свободная кислота цефтиофура (CCFA). Указанное соединение, а также его фармацевтические композиции описаны в патенте США №5721359, который целиком включен в настоящий документ в качестве ссылки. Неводная жидкая композиция также содержит неводную основу, которая обычно представляет собой жидкость при температуре контакта и может смешиваться, частично не смешиваться или не смешиваться с водой. Неводная основа может представлять собой липид или смесь липидов, таких как жиры, воски и стеролы. Липид может быть гидрированным или негидрированным, насыщенным, ненасыщенным или полиненасыщенным, и может быть дополнительно модифицирован с помощью способов, известных специалистам. Предпочтительно выбирать неводную основу из восков или жиров, как натуральных, так и синтетических. Используемый в настоящем документе термин “воски” относится к смесям эфиров длинноцепочечных карбоновых кислот и длинноцепочечных спиртов. Карбоновая кислота в воске обычно имеет четное количество атомов углерода от 16 до 36, в то время как спирт обычно имеет четное количество атомов углерода от 24 до 36. Используемый в настоящем документе термин “жиры” относится к эфирам длинноцепочечных карбоновых кислот и трехатомного спирта глицерина, которые могут быть натуральными или синтетическими; жиры могут быть жидкими, твердыми или полутвердыми при комнатной температуре (около 25°С). “Жиры” также называются глицеридами, триацилглицеролами и триглицеридами. Жир, который является жидким при комнатной температуре, также называют “маслом”. Таким образом, используемый в настоящем документе термин “жир” включает в себя “масло”. В настоящем изобретении более предпочтительно, чтобы неводная основа представляла собой натуральное или синтетическое масло. Иллюстративные примеры синтетических масел, подходящих в качестве неводной основы, включают триглицериды или диэфиры пропиленгликоля и насыщенных или ненасыщенных жирных кислот, имеющих от 6 до 24 атомов углерода. Указанные карбоновые кислоты включают такие карбоновые кислоты, которые имеют от 6 до 24 атомов углерода, такие как, например, гексановая кислота, октановая (каприловая), нонановая (пеларгоновая), декановая (каприновая), ундекановая, лауриновая, тридекановая, тетрадекановая (миристиновая), пентадекановая, гексадекановая (пальмитиновая), гептадекановая, октадекановая (стеариновая), нонадекановая, эйкозановая, генэйкозановая, докозановая и лигноцериновая кислота. Примеры ненасыщенных карбоновых кислот включают олеиновую, линолевую, линоленовую кислоту и т.п. Понятно, что триглицеридный носитель может включать моно-, ди- или триглицериловый эфир жирных кислот или смешанные глицериды и/или пропиленглиуолевые диэфиры, в которых по меньшей мере одна молекула глицерина эстерифицирована жирными кислотами с углеродными цепями различной длины. Примерами триглицериловых эфиров являются следующие соединения: триненасыщенные эфиры, включая триолеин, трилинолеин и трилиноленин; насыщенные тринасыщенные эфиры, включая трипальмитин, тристеарин и тридеканоин. Дополнительные примеры триглицериловых эфиров включают динасыщенные-мононенасыщенные типы: олеодинасыщенные эфиры, такие как 1,2-дипальмитоил-3-олеоил-рац-глицерин или 1,3-дипальмитоил-2-олеоил-рац-глицерин; линолеодинасыщенные эфиры, такие как 1,3-дипальмитоил-2-линолеоил-рац-глицерин. Дополнительными примерами триглицеридов являются мононасыщенные-диненасыщенные эфиры: такие как мононасыщенные олеолинолеиновые эфиры, включая 1-пальмитоил-2-олеоил-3-линолеоил-рац-глицерин и 1-линолеоил-2-олеоил-3-стеароил-рац-глицерин, и мононасыщенные линолеиновые эфиры, включая 1,2-дилинолеоил-3-пальмитоил-рац-глицерин. Примеры диглицериловых эфиров включают: диненасыщенные эфиры, такие как 1,2-диолеин или 1,3-диолеин, 1,2-дилинолеин или 1,3-диленолеин и 1,2-дилиноленин или 1,3-диленоленин; насыщенные динасыщенные эфиры, такие как 1,2-дипальмитин или 1,3-дипальмитин, 1,2-дистеарин или 1,3-дистеарин, и 1,2-дидеканоин или 1,3-дидеканоин; насыщенные-ненасыщенные диглицериловые эфиры, такие как 1-пальмитоил-2-олеоилглицерин или 1-олеоил-2-пальмитоилглицерин, 1-пальмитоил-2-линолеоилглицерин или 1-линолеоил-2-пальмитоилглицерин. Примеры моноглицериловых эфиров включают: ненасыщенные эфиры, такие как 1-олеин или 2-олеин, 1-линолеин или 2-линолеин и 1-линоленин или 2-линоленин; насыщенные эфиры, такие как 1-пальмитин или 2-пальмитин, 1-стеарин или 2-стеарин и 1-деканоин или 2-деканоин. Примеры диэфиров полиэтиленгликоля (PEG) включают: диненасыщенные эфиры, такие как 1,2-диолеин или 1,3-диолеин, 1,2-дилинолеин или 1,3-дилинолеин и 1,2-дилиноленин или 1,3-дилиноленин; насыщенные динасыщенные эфиры, такие как 1,2-дипальмитин или 1,3-дипальмитин, 1,2-дистеарин или 1,3-дистеарин и 1,2-дидеканоин или 1,3-дидеканоин. Другие примеры диэфиров PEG из насыщенных-ненасыщенных диглицериловых эфиров включают: 1-пальмитоил-2-олеоилглицерин или 1-олеоил-2-пальмитоилглицерин, 1-пальмитоил-2-линолеоилглицерин или 1-линолеоил-2-пальмитоилглицерин. Иллюстративными примерами натуральных масел являются масло канолы, кокосовое масло, кукурузное масло, арахисовое масло, кунжутное масло, оливковое масло, пальмовое масло, масло сафлора красильного, соевое масло, хлопковое масло, рапсовое масло, подсолнечное масло и их смеси. Предпочтительным является хлопковое масло. Неводную основу можно модифицировать способами, известными специалистам. Например, в вариантах осуществления, в которых используется перокисленная ненасыщенная масляная основа, модифицированная основа может иметь пероксидное число приблизительно от 0,1 до 600, и, в некоторых вариантах осуществления, приблизительно 10, приблизительно 20, приблизительно 40 или приблизительно 80 или любую промежуточную величину. Используемые в настоящем документе пероксидные числа выражены как миллиэквиваленты (мэкв) пероксида на 1000 граммов образца масла. Помимо упомянутых выше компонентов, неводная жидкая композиция может также содержать дополнительные соединения. Например, если неводная жидкая композиция представляет собой фармацевтическую композицию, она может содержать любые фармацевтически приемлемые компоненты. Типичными дополнительными компонентами являются, например, фармацевтически активные ингредиенты, наполнители, добавки, суспендирующие агенты, консерванты, увлажняющие агенты, загустители, буферы и флоккулянты. Могут быть включены суспендирующие агенты, такие как камеди (например, аравийская камедь, караген, альгинат натрия и трагакант), производные целлюлозы (например, натрий-карбоксиметилцеллюлоза, микрокристаллическая целлюлоза и гидроксиэтилцеллюлоза) и глины (например, бентонит и коллоидный магний-алюминий). Могут быть добавлены консерванты, такие как метил- и пропилпарабен, бензиловый спирт, хлорбутанол и тимерозал. Могут быть использованы анионогенные поверхностно-активные агенты (например, докузат натрия и лаурилсульфат натрия), неионогенные поверхностно-активные агенты (например, полисорбаты, полиоксамеры, октоксинол-9) и катионогенные поверхностно-активные агенты (например, бромид триметилтетрадециламмония, хлорид бензалкония, хлорид бензетония, хлорид миристилгаммапиколиния). Могут быть добавлены загустители, такие как желатин, натуральные камеди и производные целлюлозы (такие как те, которые перечислены выше как суспендирующие агенты). Могут быть включены буферы, такие как цитратные и фосфатные буферные агенты, а также осмотические агенты, такие как хлорид натрия и маннит. Для фармацевтических композиций, которые предназначены для введения пероральным путем, можно использовать корригенты, подсластители (например, маннит, сахарозу, сорбит и декстрозу), окрашивающие агенты и отдушки. В фармацевтических композициях можно использовать наполнители, такие как моноолеат сорбитана (продается как Span 80® компанией Sigma-Aldrich) и фосфатидилхолин (продается как Phospholipon компанией American Lecithin Company). Перед тем как неводная жидкая композиция начнет контактировать с растворяющей средой для анализа на растворение, к неводной жидкой композиции добавляют неводный жидкий разбавитель, чтобы получить разбавленную неводную жидкую композицию. Неводный жидкий разбавитель обычно представляет собой жидкость при температуре контакта и может смешиваться, частично не смешиваться и не смешиваться с водой. Неводный разбавитель может быть выбран из той же группы соединений, которая упоминалась выше для неводной основы, и может быть таким же, как неводная основа, или отличаться от нее. Кроме того, неводный разбавитель может содержать органический растворитель. Разбавитель может также содержать поверхностно-активные агенты для влияния на поверхностное натяжение между образцом и средой высвобождения лекарственного средства. Неводный разбавитель может иметь плотность, которая больше или меньше плотности среды высвобождения лекарственного средства, но когда разбавитель объединен с образцом, объединенная композиция должная иметь плотность, которая меньше плотности среды высвобождения лекарственного средства. Неводный разбавитель не должен взаимодействовать неблагоприятным образом ни с какими компонентами неводной жидкой композиции или водной растворяющей среды. Неводный разбавитель предпочтительно выбирают из группы, состоящей из натуральных масел, синтетических масел и органических растворителей. Неводный разбавитель может также состоять из или содержать масла силиконового типа (например, полидиметилсилоксан и полиметилгидросилоксан). Органический растворитель может быть выбран из группы, состоящей из спиртов, алифатических углеводородов, ароматических углеводородов, хлорированных углеводородов, гликолей, гликолевых эфиров, сложных эфиров, эфиров, кетонов, нефтехимических продуктов, терпентина, диметилформамида и уайт-спиритов. Более предпочтительно, неводный разбавитель представляет собой натуральное или синтетическое масло. Иллюстративными примерами натуральных масел являются масло канолы, кокосовое масло, кукурузное масло, арахисовое масло, кунжутное масло, оливковое масло, пальмовое масло, масло сафлора красильного, соевое масло, хлопковое масло, рапсовое масло, подсолнечное масло и их смеси. Предпочтительным является кокосовое масло и хлопковое масло, и кокосовое масло является особенно предпочтительным. Неводный разбавитель можно модифицировать посредством перокисления или другими способами, известными специалистам, как описано выше для неводной основы. В неводный разбавитель также можно добавлять поверхностно-активный агент, чтобы манипулировать свободной энергией поверхности неводной фазы и поверхностным натяжением между неводным слоем и водной растворяющей средой. Типичными пригодными поверхностно-активными агентами являются неионогенные, катионогенные, анионогенные и амфотерные поверхностно-активные агенты. Иллюстративными примерами поверхностно-активных агентов, подходящих для применения в настоящем изобретении, являются додецилсульфат, моноолеат полиоксиэтиленсорбитана (Tween 80 Тип и количество поверхностно-активного агента будут зависеть от конкретной системы анализируемого вещества, неводной жидкой композиции и водной растворяющей среды и могут быть определены специалистом. Концентрации поверхностно-активного агента могут быть выше или ниже критической концентрации мицеллообразования. Типичные пределы концентраций поверхностно-активного агента составляют приблизительно от 0,001% до 1%. В предпочтительных вариантах осуществления настоящего изобретения неводный разбавитель представляет собой натуральное масло, необязательно, окисленное натуральное масло. Количество неводного разбавителя, которое добавляют к неводной жидкой композиции, особо не ограничивается, но оно должно быть таким, чтобы улучшать поведение распределения неводной жидкой композиции. Соотношение неводного разбавителя и неводной жидкой композиции обычно находится в пределах от 1:20 до 20:1 по объему, но может быть значительно ниже или выше. Точное количество может варьировать в зависимости от природы анализируемого вещества, неводной основы и растворяющей среды. Подходящее количество композиции и количество неводного разбавителя может определить специалист эмпирическим путем. Относительное количество композиции и разбавителя можно считать оптимальным, когда разбавленная композиция равномерно распределяется по поверхности среды высвобождения лекарственного средства, или когда получена адекватная точность повторных измерений. Не желая ограничиваться указанной теорией, заявители полагают, что добавление неводного разбавителя модифицирует и нормализует поведение распределения неводной жидкой композиции по поверхности водной растворяющей среды в аппарате для тестирования на растворение. Без добавления неводного разбавителя, даже если в повторных измерениях используется одна и та же неводная жидкая композиция, наблюдалось, что неводная жидкая композиция может распределяться в разной степени на водной растворяющей среде. Это, как полагают, приводит к вариациям в размере контактной площади между неводной жидкой композиций и водной растворяющей средой. Как следствие, на скорость растворения анализируемого вещества в водной растворяющей среде влияет изменяющаяся площадь контактной поверхности, и полученные результаты могут быть неточными и ненадежными. В случае, когда добавляют неводный разбавитель, разбавленная неводная жидкая композиция имеет тенденцию к распределению приблизительно в одной и той же степени, не только если один и тот же образец повторно наносят на поверхность водной растворяющей среды, но также если изучаются разные образцы аналогичных неводных жидких композиций. Таким образом, размер контактной площади между неводной жидкой композиций и водной растворяющей средой остается практически одним и тем же, а точность и надежность результатов улучшается. После добавления неводного разбавителя к неводной жидкой композиции и перемешивания по меньшей мере часть полученной разбавленной неводной жидкой композиции и водной растворяющей среды помещают в прибор для тестирования на растворение. Порядок добавления разбавленной неводной жидкой композиции и водной растворяющей среды не ограничивается. Их можно добавлять одновременно или последовательно. Обычно водную растворяющую среду будут помещать в прибор для тестирования на растворение первой, а затем будут добавлять разбавленную неводную жидкую композицию. Аппараты для тестирования на растворение хорошо известны специалистам-аналитикам, и некоторые из них стандартизированы, например, в различных фармакопеях, таких как Фармакопея Соединенных Штатов или Фармакопея Японии. Иллюстративными примерами аппаратов для тестирования на растворение являются способ вращающейся корзины (например, USP I), лопастной способ (например, USP II), различные проточные способы (например, USP IV), цилиндрический прибор с использованием возвратно-поступательного движения (например, USP III) и различные трансдермальные аппараты для тестирования на растворение (например, диффузионная камера Франца). Измерение высвобождения лекарственного средства из жидких образцов и, особенно, из неводных жидких лекарственных форм, часто является трудной задачей, и в настоящее время не приняты стандартизированные методики для изучения жидких образцов. В одном варианте осуществления настоящего изобретения в качестве аппарата для тестирования на растворение используется лопастной узел. Типичный лопастной узел показан на фиг.3. Он включает сосуд 10, который содержит водную растворяющую среду 11. В способах по настоящему изобретению разбавленную неводную жидкую композицию обычно наносят на поверхность водной растворяющей среды, например, с помощью шприца или пипетки. Разбавленную неводную жидкую композицию и водную растворяющую среду перемешивают с использованием лопасти 12. Образцы водной растворяющей среды можно отбирать, например, с помощью шприца, или используя постоянную трубку для отбора образцов 13, которая, необязательно, присутствует в лопастном узле. Данные типы приборов для тестирования на растворение можно приобрести из ряда источников, например, VanKel (Varian Inc.), Distek Inc. и Hanson Research Corporation. Водной растворяющей средой может быть любая водная растворяющая среда, известная специалистам. Распространенными растворяющими средами являются вода, хлористоводородная кислота (например, имеющая концентрацию в пределах приблизительно от 0,001 моль до 0,1 моль HCl), имитированный желудочный сок с пепсином или без него, различные буферные растворы (глициновый, цитратный, ацетатный, фосфатный и боратный буферы), имитированные кишечные соки с ферментами или без них (например, 0,05 молярный фосфатный буфер с рН 7,5 с панкреатином или без него), вода, содержащая поверхностно-активный агент, буферные растворы, содержащие поверхностно-активный агент, и водные растворы спиртов (например, низкомолекулярных спиртов, растворимых в воде и обычно содержащих 5 или менее атомов углерода, которые действуют как сорастворители). Указанные различные параметры можно подобрать таким образом, чтобы изменить условия растворения для данного анализируемого вещества. Посредством повторных экспериментов возможно эмпирическим путем подобрать оптимальную композицию для среды высвобождения лекарственного средства, которая может позволить экспериментатору подобрать скорость высвобождения лекарственного средства in vitro в желательных пределах. Подбор условий растворения может также позволить экспериментатору распознавать in vitro объекты, которые ведут себя по-другому in vivo. В предпочтительном варианте осуществления настоящего изобретения буферный раствор, необязательно, содержащий поверхностно-активный агент, используется в качестве водной растворяющей среды. Тип буферного раствора особо не ограничивается, но должен быть выбран в зависимости от конкретной системы, подлежащей изучению. Буферные растворы можно выбирать таким образом, чтобы контролировать растворимость анализируемого вещества в среде высвобождения лекарственного средства, оптимизировать профиль высвобождения лекарственного средства и оптимизировать степень различий между важными образцами. Иллюстративными примерами буферных растворов являются 0,05 молярный глициновый буфер с рН от 2 до 3, 0,05 молярный цитратный буфер с рН 3, 0,05 молярный ацетатный буфер с рН от 4 до 5, 0,05 молярный ацетатный буфер в физиологическом растворе с рН от 5,5, 0,05 молярный фосфатный буфер с рН от 6 до 8, не содержащий калия 0,05 молярный фосфатный буфер с рН 6,8, 0,05 молярный фосфатный буфер в физиологическом растворе с рН 7,4, 0,05 молярный боратный буфер с рН от 8 до 10). Предпочтительными буферными растворами являются 0,05 молярные фосфатные буферы с рН от 6 до 7. Буфер может иметь любую подходящую молярность, например, приблизительно от 0,001 М до 0,5 М, предпочтительно, приблизительно от 0,01 до 0,1. Однако было установлено, что точность и надежность способов по настоящему изобретению можно дополнительно повысить, если использовать буфер, имеющий низкую молярность. Таким образом, в одном варианте осуществления настоящего изобретения молярность буфера находится в пределах приблизительно от 0,1 до 10 мМ, более предпочтительно, приблизительно от 0,5 до 2 мМ. Выбор буфера, имеющего низкую молярность, улучшает поведение распределения неводной жидкой композиции на поверхности среды высвобождения лекарственного средства и уменьшает нежелательные взаимодействия между неводной жидкой композицией и компонентами аппарата для высвобождения лекарственного средства (т.е. перемешивающего стержня). Путем улучшения однородности распределения и минимизации нежелательных физических взаимодействий, возможно улучшить точность и надежность аналитического метода. Информацию об изготовлении растворяющего буфера можно найти также в USP 24, стр.2231-2240, United States Pharmacopeial Convention Inc., 1 января 2000 г. В еще одном предпочтительном варианте осуществления настоящего изобретения водной растворяющей средой является вода, необязательно, содержащая поверхностно-активный агент. Необязательно, водная растворяющая среда может содержать поверхностно-активный агент, что представляет собой другой путь для манипулирования с растворимостью системы. Обычными подходящими поверхностно-активными агентами являются неионогенные, катионогенные, анионогенные и амфотерные поверхностно-активные агенты. Иллюстративными примерами поверхностно-активных агентов, подходящих для применения в настоящем изобретении, являются додецилсульфат, моноолеат полиоксиэтиленсорбитана (Tween 80 Тип и количество поверхностно-активного агента будут зависеть от конкретной системы анализируемого вещества, неводной жидкой композиции и водной растворяющей среды и могут быть определены специалистом. Концентрации поверхностно-активного агента могут быть выше или ниже критической концентрации мицеллообразования. Типичные пределы концентраций поверхностно-активного агента составляют приблизительно от 0,001% до 1%. рН водной растворяющей среды следует выбирать в зависимости от конкретной изучаемой системы. Обычно рН водной растворяющей среды будет находиться в пределах приблизительно от 1 до 10, предпочтительно, приблизительно от 2 до 8. Общеизвестно, что рН водной растворяющей среды может влиять на растворимость анализируемого вещества, и она представляет собой способ манипулирования с условиями погружения в ходе эксперимента. Путем оптимизации рН водной растворяющей среды возможно манипулировать с характеристиками растворения некоторых анализируемых веществ. В случае фармацевтических препаратов это может сделать возможным выявить корреляцию между характеристиками высвобождения лекарственного средства in vitro и фармакокинетическим поведением лекарственного средства in vivo. В качестве водной растворяющей среды особенно предпочтительной системой является водный буфер, имеющий оптимальную величину рН. Водные растворяющие среды, применяемые в способах по настоящему изобретению, можно изготовить с использованием любого типа воды, такого как деионизированная вода, вода двойной дистилляции или вода высокой чистоты (т.е. имеющая сопротивление по меньшей мере около 1 МОм, более предпочтительно, имеющая сопротивление по меньшей мере около 18 МОм). Хотя и не являясь предпочтительной, водопроводная вода также может быть использована, если ее составляющие не мешают измерениям. Предпочтительно, применяется вода двойной дистилляции или вода высокой чистоты, более предпочтительно вода высокой чистоты. Применение более чистой воды, особенно в комбинации с низкомолярным буфером, также, как было установлено, повышает точность и надежность результатов испытаний. Воду высокой чистоты можно получить, например, с использованием аппарата для очистки воды, такого как системы очистки воды Milli-Q от компании Millipore Corporation (Bedford, Massachusetts). Обычно полученная вода высокой чистоты имеет сопротивление около 18 МОм. Выбор воды высокой чистоты улучшает поведение распределения неводной жидкой композиции на поверхности среды, высвобождения лекарственного средства и уменьшает нежелательные взаимодействия между неводной жидкой композицией и компонентами аппарата для высвобождения лекарственного средства (т.е. перемешивающего стержня). Путем улучшения однородности распределения и минимизации нежелательных физических взаимодействий возможно улучшить точность и надежность аналитического метода. Количество неводной жидкой композиции, которое помещают в прибор для тестирования на растворение, может широко варьироваться в зависимости от различных факторов, таких как природа лекарственной формы (например, концентрация активного ингредиента, стандартная доза), объем растворяющей среды, размер поверхности контакта композиции с растворяющей средой. Обычно соотношение разбавленной неводной жидкой композиции и водной растворяющей среды составляет приблизительно от 1:20 до 1:500 (по объему). В одном варианте осуществления настоящего изобретения было сделано наблюдение, что корреляция высвобождения лекарственного средства in vitro с фармакокинетическим поведением его in vivo может быть превращена в обратную (т.е. отрицательная корреляция может становиться положительной корреляцией) только введением малых количеств неводной жидкой композиции в прибор для тестирования на растворение. В данном случае соотношение неводной жидкой композиции и водной растворяющей среды составляет приблизительно от 1:2000 до 1:100000 (по объему), предпочтительно, приблизительно от 1:20000 до 1:40000 (по объему). Когда разбавленную неводную жидкую композицию помещают в прибор для тестирования на растворение, разбавленная неводная жидкая композиция и водная растворяющая среда контактируют в течение заранее определенного периода времени. Для улучшения контакта между разбавленной неводной жидкой композицией и водной растворяющей средой их обычно смешивают, например, путем перемешивания. Продолжительность контакта может сильно варьироваться и будет зависеть, например, от интенсивности перемешивания, количества анализируемого вещества, неводной жидкой композиции, растворяющей среды, температуры, чувствительности метода обнаружения, используемого для определения количества анализируемого вещества, и от ряда других факторов. Кроме того, продолжительность контакта будет зависеть от желательности определения скоростей растворения за короткие, средние и продолжительные периоды времени. Обычно продолжительность контакта составляет от 5 минут до 24 часов, предпочтительно, до растворения приблизительно 90% всего количества анализируемого вещества. Обычно контакт будут осуществлять приблизительно от 15 минут до 120 минут, предпочтительно, приблизительно от 15 минут до 60 минут. Во время стадии контакта температуру водной растворяющей среды можно поддерживать на любом желательном уровне. Обычно температуру водной растворяющей среды поддерживают на постоянном уровне приблизительно 37°С. Однако можно использовать более высокие температуры для повышения и более низкие температуры для снижения скорости растворения. Поскольку температура растворяющей среды влияет на скорость растворения, если необходимо сравнить результаты более одного эксперимента, для каждого эксперимента следует выбирать одну и ту же температуру. В контексте настоящего изобретения “одна и та же температура” означает, что различия между температурами в различных экспериментах составляют, самое большое, 5°С, предпочтительно, самое большое, 2°С. Предпочтительно, температура контакта составляет 37°С. Интенсивность перемешивания во время контакта, такая как скорость перемешивания, также влияет на скорость растворения анализируемого вещества, и оптимальные условия следует определять на основе, например, размера и формы лопасти (если она имеется), геометрии аппарата для тестирования на растворение и количества и вязкости растворяющей среды. Оптимальные условия перемешивания специалист может определить экспериментально. Обычно оптимальные условия перемешивания дают возможность получить поверхность, которая является гладкой (без видимых всплесков или стоячих волн) от внешнего края сосуда до центра, включая площадь, на которой средство перемешивания контактирует с растворяющей средой (т.е. на поверхности отсутствует вихревой “завиток”, вызванный смещением по направлению книзу поверхности среды высвобождения лекарственного средства в результате перемешивания). Обычно скорость перемешивания будет находиться в пределах приблизительно от 25 до 100 об/мин, предпочтительно, приблизительно от 50 до 75 об/мин. Ранее предлагалось множество модификаций стандартизированного аппарата для тестирования на растворение, таких как лопастные узлы. В способах по настоящему изобретению стандартизированные аппараты для тестирования на растворение, известные специалистам как аппараты USP I и USP II, можно надежно использовать без каких бы то ни было модификаций. По истечении заранее определенного периода времени определяют количество анализируемого вещества в водной растворяющей среде. С помощью некоторых способов определения можно определить количество анализируемого вещества, в то время как водная растворяющая среда остается в аппарате для тестирования на растворение; обычно, однако, по меньшей мере часть водной растворяющей среды удаляют из аппарата для тестирования на растворение, например, с помощью шприца или трубки для отбора образцов 13. Несмотря на то, что возможно использование всей водной растворяющей среды для анализа, и это может быть необходимым для некоторых способов обнаружения, обычно будет использоваться только часть водной растворяющей среды. Размер образца, отбираемого для определения количества анализируемого вещества, будет зависеть от ряда факторов, особенно, от применяемого способа обнаружения, и может, например, составлять приблизительно от 0,1 до 100 мл, предпочтительно, приблизительно от 1 до 20 мл. Если желательно, образец водной растворяющей среды, который будет использоваться для определения количества анализируемого вещества, можно фильтровать после удаления из аппарата для тестирования на растворение. Это удаляет частицы инородных веществ и не растворившегося анализируемого вещества, которые могут помешать определению растворившегося анализируемого вещества и испортить измерение. Фильтрование можно осуществлять любыми подходящими средствами, такими как фильтрование через фильтр, имеющий средний размер пор приблизительно от 0,1 до 50 микрон, предпочтительно, приблизительно от 0,1 до 0,5 микрона. Указанные фильтры можно приобрести, например, под торговыми наименованиями Acrodisk® от компании Gelman Laboratory. После необязательной стадии фильтрования определяют количество анализируемого вещества в водной растворяющей среде. Можно использовать любой аналитический метод, подходящий для определения количества анализируемого вещества. Выбор аналитического метода будет зависеть от ряда параметров, включая природу анализируемого вещества, пределы его концентраций, растворяющую среду, а также от того, какие методы доступны в данной лаборатории. Иллюстративными примерами аналитических методов являются методики разделения (например, высокоэффективная жидкостная хроматография, жидкостная хроматография, тонкослойная хроматография, капиллярный электрофорез, газовая хроматография), фотометрические и спектрофотометрические методы (например, ультрафиолет-видимый (UV-Vis), трансформирующий инфракрасный Фурье (FTIR), атомное поглощение (АА), атомная эмиссия (АЕ), масс-спектрометрия (MS)). Хроматографические методы, в частности газовая хроматография (GC) и высокоэффективная жидкостная хроматография (HPLC), являются предпочтительными. Примерами подходящих хроматографических методов являются высокоэффективная жидкостная хроматография с обращенной фазой (RP-HPLC), и высокоэффективная жидкостная хроматография с нормальной фазой (NP-HPLC), включая любой из ряда методов обнаружения, известных специалистам. Примеры методов обнаружения, которые можно использовать в сочетании с подходящим хроматографическим методом, включают UV-Vis, индекс рефракции, масс-спектрометрию и обнаружение светорассеянием. Проточный инжекционный анализ (FIA) с UV-Vis обнаружением также можно использовать в качестве аналитического метода. FIA является особенно подходящим, когда необходима высокая пропускная способность для образцов, как в случае, когда необходимо характеризовать производственную систему во время процесса в реальном времени. Способы по настоящему изобретению были объяснены выше с точки зрения варианта осуществления настоящего изобретения, при котором определяют количество анализируемого вещества, растворившегося к одному, заранее определенному, моменту времени. Во многих случаях представляет интерес мониторинг скорости растворения в течение определенного периода времени, чтобы определить, постоянна ли скорость высвобождения анализируемого вещества или она меняется с течением времени (например, большое количество в начале тестирования на растворение, а затем меньшие количества с течением времени). В указанных случаях возможно использование достаточно большого аппарата для тестирования на растворение, чтобы отбирать два или более образцов в различные заранее определенные моменты времени и анализировать указанные образцы в индивидуальном порядке. Возможна также подготовка двух или более идентичных экспериментов и их контактирование при идентичных условиях, за исключением изменяющейся продолжительности перемешивания. Образцы водной растворяющей среды, отобранные в различные моменты времени, анализируют в индивидуальном порядке. Результаты затем можно использовать для определения зависимого от времени профиля скорости растворения. С использованием способов по настоящему изобретению в настоящее время возможно надежное и точное измерение скорости растворения анализируемого вещества в неводной жидкой композиции. Наблюдается достоверное уменьшение вариабельности результатов повторных измерений. При использовании в фармацевтических целях способы по настоящему изобретению делают возможным выявление полезной корреляции между способами in vitro и фармакокинетическими исследованиями in vivo. Таким образом, их можно использовать как прочный и надежный способ для контроля качества во время процесса производства фармацевтических препаратов, чтобы гарантировать адекватные биологические свойства и постоянные параметры партий лекарственного средства. Поскольку данные являются простыми, дешевыми и быстрыми и могут осуществляться с использованием стандартизированного аппарата для тестирования на растворение, их также можно выгодно использовать для разработки лекарственных средств и их лекарственных форм. ПРИМЕРЫ Следующие примеры представлены для иллюстрации изобретения. Однако они не должны истолковываться как ограничительные. Точность. Точность способов по настоящему изобретению можно определить расчетом относительного стандартного отклонения (RSD) повторных измерений. Обычно относительное стандартное отклонение определяют измерением скорости растворения анализируемого вещества при идентичных условиях в более чем двух повторностях. Затем рассчитывают относительное стандартное отклонение по следующей формуле:
где s.d. представляет собой стандартное отклонение, которое определяют как:
Х представляет собой индивидуальный результат; N представляет собой количество повторностей и Предпочтительно, относительное стандартное отклонение составляет 10% или менее, более предпочтительно, 2% или менее. Правильность. Правильность способов по настоящему изобретению можно определить измерение переноса анализируемого вещества из неводной жидкой композиции в водную среду, когда неводную жидкую композицию нагружают известным количеством анализируемого вещества. Нагруженную неводную жидкую композицию уравновешивают с водной средой высвобождения лекарственного средства встряхиванием или перемешиванием, после чего определяют количество анализируемого вещества в водной растворяющей среде. Концентрацию анализируемого вещества, который перешел в водную среду, затем сравнивают с концентрацией, которая должная получиться, теоретически, если состоялся перенос 100% анализируемого вещества (например, при допущении, что нет манипуляций с пипетками, ошибок при взвешивании или потерь, что 100% анализируемого вещества растворилось, и что 100% анализируемого вещества было обнаружено). Способы по настоящему изобретению являются правильными в пределах приблизительно от 70% до 100%, предпочтительно, приблизительно от 90% до 100%. Общая процедура растворения Если не указано иное, использовали следующую общую процедуру. Условия растворения. Прибор: USP II (вращающаяся лопасть) с закрытыми сосудами. Помещают зонды для отбора образцов на половине расстояния между поверхностью среды и лопастью. Устанавливают люэровские адаптеры на трубопровод зонда для отбора образцов, чтобы облегчить удаление образцов из аппарата. Все образцы должны удаляться через указанные адаптеры. Колбы и лопасти для растворения должны быть тщательно очищены. (см. процедуру очистки DRA). Остатки мыла или спирта могут повлиять на результаты. Размер колбы: 1000 мл Растворяющая жидкость: 500 мл 0,001 М рН 7,0 фосфата при 37°С±0,5°С Маточный буфер: растворяют 3,9 г одноосновного фосфата калия (KH2PO4) и 3,7 г двухосновного фосфата калия (K2HPO4) в воде Milli-Q или эквиваленте в волюметрической колбе объемом один литр. Разбавляют до объема водой Milli-Q или эквивалентом и перемешивают. Проверяют рН разбавление 10 мл маточного раствора до 500 мл водой Milli-Q. Величина рН должна составлять 7,0±0,1. Если необходимо, доводят рН маточного раствора 50% гидроксидом натрия или концентрированной хлористоводородной кислотой. Повторно проверяют, чтобы величина рН рабочего раствора составляла 7,0±0,1. Рабочий буфер. Разбавляют 10 мл маточного раствора до 500 мл водой Milli-Q. Перед применением дегазируют. Скорость перемешивания: 50 об/мин Объем образца: 10 мл Фильтр: Acrodisk (Gelman) 0,2 микрометра одноразовый (№4496) или эквивалент Изготовление рабочего стандарта. Точно взвешивают приблизительно 1 мг стандарта цефтиофура гидрохлорида и помещают в 100 мл волюметрическую колбу. Смачивают лекарственное средство приблизительно 1 мл метанола для растворения (обрабатывают ультразвуком, если необходимо). Разбавляют до объема рабочим буфером. Изготавливают по меньшей мере 2 рабочих стандартных раствора. Изготовление фармацевтического неводного образца: ресуспендируют каждый флакон суспензии кристаллической свободной кислоты цефтиофура (CCFA), изготовление которой описано в настоящем документе ниже, до тех пор, пока не исчезнут видимые признаки лекарственного средства на дне флакона. Разбавляют образец 1:1 (об./об.) гидрированным кокосовым маслом (доступен как Miglyol 812 от компании HulsAmerica) до проведения анализа на растворение следующим образом: используя калиброванную пипетку положительного перемещения, добавляют равные объемы суспензии CCFA и Miglyol в подходящий контейнер (например, 20 мл флакон с завинчивающейся крышкой). Истинный объем, который используется, не является принципиальным до тех пор, пока разведение составляет точно 1:1. Предложенные пределы объемов составляют от 1,0 мл до 5,0 мл для каждого компонента. Тщательно смешивают разбавленный образец вручную и вихревой мешалкой, затем извлекают 50 микролитров в калиброванный пипеттор с положительным перемещением. Промокают избыток суспензии с кончика и по каплям разливают содержимое на поверхность среды в каждую колбу для растворения при перемешивании. Наносят капли таким образом, чтобы кончик пипеттора находился на расстоянии приблизительно 1/2 дюйма от поверхности среды на половине расстояния между стенкой сосуда и зондом для отбора образцов. Погружают кончик пипеттора в среды для удаления оставшихся следов суспензии. Задерживают помещение образца в каждую последующую колбу с учетом времени на отбор образца. Все образцы должны быть разлиты по колбам для растворения как можно скорее после разбавления. По истечении определенных периодов времени (например, 15, 30, 60 и 120 минут) извлекают 10 мл растворяющей жидкости (удобно использовать 10 мл одноразовый шприц) и фильтруют через фильтр Acrodisk номер 4496. Сливают первые 5 мл фильтрата, а затем собирают подходящий объем фильтрата во флакон автоматического сэмплера для HPLC. Задерживают процесс отбора образцов таким же образом, как при нанесении образцов. Осуществляют количественный HPLC анализ. Условия хроматографии. Оборудование. Насос HPLC. Подходящий насос, способный к изократной работе при 3000 фунтах на кв. дюйм (например, Agilent 1100 от компании Agilent Technologies). Инжектор: Подходящий инжектор с малым объемом мертвого пространства Детектор: 254 нм Колонка: Waters Symmetry C8 3,9×50 мм, 5 микрон, или эквивалент Объем инжекции: около 20 мкл Рабочие параметры хроматографии. Затухание: подбирают нужное Скорость диаграммы: подбирают нужное Скорость потока: приблизительно 1,0 мл/мин (можно подбирать). Давление: приблизительно 2000 фунтов на кв. дюйм Мобильная фаза HPLC: для 1 литра мобильной фазы. Добавляют 3,85 г ацетата аммония и 13,5 мл 40% гидроксида тетрабутиламмония в подходящий контейнер. Разбавляют до 700 мл водой Milli-Q или водой для HPLC. Доводят рН до 6,7±0,1 ледяной уксусной кислотой. Фильтруют водный буфер через мембранный фильтр 0,45 микрометра. К 700 мл водного буфера добавляют 200 мл метанола и 110 мл ТГФ и смешивают. Для дегазации обрабатывают ультразвуком в условиях вакуума. Количественный анализ HPLC. Анализируют фильтрованные образцы с помощью HPLC. Соответствующие стандартные растворы необходимо помещать в начале и в конце каждого хроматографического цикла в количестве не менее шести стандартных инжекций на цикл. По сторонам каждого ряда из шести образцов помещают стандартные растворы. Подходящие контрольные растворы следует периодически анализировать, чтобы отслеживать инжекционную систему на предмет возможного перемещения. Тест на пригодность системы. Относительное стандартное отклонение стандартного фактора не должно превышать 2,0%. Стандартный фактор (SF) можно рассчитать по следующей формуле: SF=P×(Wstd/Rstd), где Р = чистота стандарта, выраженная в процентах; Wstd = вес стандарта; Rstd = площадь пика стандарта; Расчеты. Рассчитывают процент цефтиофура, высвобожденного в каждый момент времени, с использованием следующего уравнения, корригирующего удаленный объем:
где Dn = процент, растворенный в последовательный временной момент; Rsam = площадь пика образца; Rstd = площадь пика стандарта; Cs = концентрация рабочего стандарта в мг/мл; L = концентрация метки в суспензии CCFA (200 мг/мл); Р = чистота стандарта, выраженная в процентах; Vsus = объем использованной суспензии CCFA составляет 0,025 мл (поскольку использовали 50 мкл разведения 1:1); V = исходный объем растворяющей жидкости в мл; n = номер периода времени; SV = объем образца в мл; D1 = процент, растворившийся в первый период времени; D2 = процент, растворившийся во второй период времени; Dn-1 = процент, растворившийся во период времени (n-1). Процедура очистки DRA. Насыщают салфетки Kimwipes 3А спиртом и тщательно промокают лопасти для удаления остатков. Оставляют сушиться на воздухе. Расставляют образцы неводной композиции, содержащие водный буфер. Ополаскивают сосуд 3А спиртом и очищают большую часть остатков на колбе промоканием салфетками Kimwipes. Ополаскивают 3А спиртом и помещают сосуд назад в DRA. С помощью стеклянного шприца инъецируют приблизительно 10 мл диметилформамида (ДМФА) через линию отбора образцов из коллектора для образцов, собирая отходы в сосуд для высвобождения лекарственного средства. Затем следуют 10 мл 3А спирта. Удаляют сосуд и используют салфетки Kimwipes для впитывания смеси растворителей и очистки внутренней поверхности сосуда. Затем споласкивают 3А спиртом и сушат. Промывают систему трубок деионизированной водой, а затем продувают воздухом с помощью пустого шприца. Если растворители попали на лопасти во время очистки системы трубок, повторяют процедуру очистки лопастей. Тест-материалы Следующие процедуры использовали для изготовления экспериментальных фармацевтических неводных суспензий, использованных в примерах, описанных ниже. Суспензия кристаллической свободной кислоты цефтиофура (CCFA) 100 мг/мл в хлопковом масле. Лоты 40,620 и 40,700 изготавливали согласно одному и тому же процессу. Неводный носитель изготавливали накачиванием хлопкового масла в сосуд с кожухом и нагреванием до 115°С. Добавляли фосфолипон 90Н (0,05% мас.) (от компании American Lecithin Co.) и смешивали. Раствор охлаждали до 45°С. Добавляли моноолеат сорбитана (продается как Span 80® компанией Sigma-Aldrich) (0,15% мас.) и смешивали. Добавляли CCFA 100 мг/мл и смешивали с использованием триблендера до гомогенности суспензии. Суспензия рециркулировали через триблендер при работе и скрининге мешалки танка. Полученной суспензией наполняли стерильные флаконы, закупоривали и герметично запечатывали. Запечатанные флаконы стерилизовали гамма-облучением. Лоты помечали как 40,700 и 40,620. Суспензия кристаллической свободной кислоты цефтиофура (CCFA) 200 мг/мл в хлопковом масле с маслом Miglyol. Значительно перокисленное ненасыщенное масло получали из хлопкового масла. 105 объемных частей натурального хлопкового масла добавляли в сосуд, имеющий паровой кожух для нагревания. Пар подавали в кожух для нагревания масла до температуры приблизительно от 85 до 110°С. Масло барботировали воздухом при перемешивании. Скорость потока воздуха варьировала приблизительно от 1 стандартного кубического фута в час (SCFH)/литр до 20 SCFH/литр. Перемешивание было таким, что температура масла оставалась постоянной в течение периода нагревания. Масло нагревали в течение периода времени и при температуре, необходимых для достижения пероксидного числа, по измерению способом Фармакопеи США (USP 24 NF 19 на стр.1870) или способом AOCS, 8-53, а затем охлаждали, переносили в другой контейнер и хранили в атмосфере азота. Для достижения пероксидного числа около 10 при температуре приблизительно 89°С масло нагревали в течение приблизительно 9 часов, при температуре приблизительно 100°С масло нагревали в течение приблизительно 3 часов и при температуре приблизительно 105°С масло нагревали в течение приблизительно 2,3 часа. Для достижения пероксидного числа около 40 при температуре приблизительно 100°С масло нагревали в течение приблизительно 6,75 часа и при температуре приблизительно 105°С масло нагревали в течение приблизительно 5,5 часа. Для достижения пероксидного числа около 80 при температуре приблизительно 105°С масло нагревали в течение приблизительно 8 часов. Соотношение между временем и температурой масла по сравнению с его пероксидным числом, как представляется, является линейным, и специалист может получить желательное пероксидное число в зависимости от времени и температур, выбранных для обработки. Окисленное масло можно разбавлять свежим маслом для получения предпочтительного конечного пероксидного числа. После изготовления перокисленного ненасыщенного масла композицию 200 мг/мл CCFA составляли следующим образом: от 10 до 20 объемных частей перокисленного хлопкового масла, имеющего пероксидное число приблизительно 10-200, смешивали с 80-90 объемными частями Miglyol 812 (от компании HulsAmerica) для формирования носителя. Добавляли 0,2 массовых части CCFA и смешивали в течение 1-3 часов для формирования однородной суспензии таким образом, чтобы концентрация CCFA составляла 200 мг/мл. Суспензию нагревали приблизительно до 80-110°С в течение приблизительно от 0,1 до 10 дней и позволяли охлаждаться. Суспензию паковали и стерилизовали гамма-излучением, по желанию. Экспериментальные параметры для каждого из лотов в следующих примерах подробно представлены в таблице 1. Суспензия кристаллической свободной кислоты цефтиофура (CCFA) 100 мг/мл в хлопковом масле с маслом Miglyol. Процедуру, подробно описанную выше для композиции 200 мг/мл, повторяли за исключением того, что соотношение модифицированного хлопкового масла и Miglyol 812 составляло 10:90, а количество добавленной CCFA было таким, что концентрация CCFA составила 100 мг/мл.
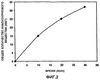
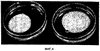
Пример 1 Данный пример иллюстрирует вариации в поведении распределения. Поведение распределения представляет собой способ описания феномена, который наблюдается, когда одна жидкая фаза помещается на поверхность другой несмешивающейся жидкой фазы. При контакте жидкость может образовывать тугонатянутый слой в форме линзы на поверхности другой жидкости или она может распределяться равномерно вдоль поверхности. Может также наблюдаться промежуточное и вариабельное распределение. Поведение распределения можно определить математически (например, поверхностной термодинамикой) или качественно. Для сравнения поведения распределения различных лотов суспензии CCFA один мл каждой суспензии осторожно наносили с помощью иглы калибра 18 в отдельные контейнеры (пластиковые чашки Петри), содержащие 25 мл среды высвобождения лекарственного средства. Образцы суспензии по каплям наносили на поверхность среды высвобождения лекарственного средства. Поведение распределения оценивали по размеру площади слоя суспензии поверх среды высвобождения лекарственного средства по истечении периода времени, достаточного для достижения квази-равновесия (около 21 часа). Фотографии образцов суспензии показаны на фигуре 4. Чашки Петри, содержащие лот 40,700, находятся слева; чашки Петри, содержащие лот 40,620, находятся справа. Через 21 час с помощью линейки измеряли диаметр слоя суспензии CCFA на среде высвобождения лекарственного средства. Диаметр линзы лота 40,700 составлял 4,8 см, в то время как диаметр линзы лота 40,620 составлял 6,0 см. Пример 2 Пример 2 показывает влияние разбавления неводной жидкой композиции неводным разбавителем. Непостоянное распределение суспензии на масляной основе на поверхности среды высвобождения лекарственного средства, показанное в примере 1, являлось значительным препятствием для разработки пригодного USP II анализа высвобождения лекарственного средства для суспензий CCFA на масляной основе. Вариабельное поведение распределения приводило в вариабельной площади поверхности суспензии при контакте со средой высвобождения лекарственного средства, что влияло на скорость растворения лекарственного средства. Это, в свою очередь, оказывало влияние на качество корреляции между высвобождением лекарственного средства in vitro и фармакокинетикой in vivo. Статистическая достоверность корреляции между высвобождением лекарственного средства in vitro и фармакокинетикой in vivo оценивали, как описано ниже. Корреляцию определяют как степень связи, или насколько хорошо одну переменную можно предсказать по другой переменной. Одним подходом для оценки степени корреляции между двумя переменными является статистический анализ тангенса соответствия наименьших квадратов. Достоверная корреляция между переменными наблюдается тогда, когда тангенс соответствия наименьших квадратов отличается от нуля при 95% доверительности (р Влияние вариабельного поведения распределения на корреляцию между высвобождением лекарственного средства in vitro и фармакокинетическим поведением in vivo можно видеть на фиг.5. Данные по высвобождению лекарственного средства in vitro для выбранных лотов CCFA нанесены на график в зависимости от из фармакокинетического поведения in vivo (т.е. продолжительности замедленного высвобождения в часах). Линия соответствия наименьших квадратов представлена на графике как сплошная линия на фиг.5. В данном случае анализ высвобождения лекарственного средства in vitro не включал разбавление неводной суспензии инертным маслом, и наблюдалось вариабельное поведение распределения лотов суспензии. Не наблюдалось достоверной корреляции между результатами высвобождения лекарственного средства in vitro и продолжительностью замедленного высвобождения. Тангенс соответствия наименьших квадратов достоверно не отличался от нуля (р=0,57). Разбавление композиции неводной суспензии 161 инертным маслом приводило к нормализации поведения распределения. Введение стадии предварительного разбавления в изобретение привело в разработке полезной корреляции in vitro/in vivo (IVIVC). Данные in vitro по выбранным лотам CCFA, полученные с использованием стадии предварительного разбавления, нанесены на график в зависимости от продолжительности замедленного высвобождения in vivo на фиг.6 вместе с линией соответствия наименьших квадратов. Достоверная корреляция наблюдалась между результатами высвобождения лекарственного средства in vitro и продолжительностью замедленного высвобождения. Тангенс соответствия наименьших квадратов достоверно отличался от нуля (р=0,04). Пример 3 Пример 3 иллюстрирует влияние ионной силы буфера на точность метода измерения. Во время тестирования высвобождения лекарственного средства in vitro из неводных суспензий неводная композиция, которая “плавает” на поверхности среды высвобождения лекарственного средства, может взаимодействовать с устройством для перемешивания или прилипать к нему. Адгезия или взаимодействие образца с устройством ингибирует однородное распределение суспензии на поверхности среды высвобождения лекарственного средства. Степень и продолжительность взаимодействия является переменной величиной, которая, в свою очередь, индуцирует нежелательную вариабельность результатов анализа. Минимизация ионной силы среды высвобождения лекарственного средства in vitro уменьшает и может устранить взаимодействие образца с устройством для перемешивания и усиливает распределение. Растворяющие буферы изготавливали в концентрациях 50 мМ, 5 мМ и 1 мМ. Единственный лот CCFA (SFH-95) анализировали множество раз с использованием каждого растворяющего буфера. Вариабельность анализа, с использованием трех растворяющих буферов, оценивали расчетом стандартного отклонения результатов, которые представлены в таблице 2.
Пример 4 В данном примере показан эффект размера образца. Исследования высвобождения лекарственного средства на лоте суспензии CCFA SFH-11 проводили, как описано в разделе “Общая процедура растворения”, выше, со следующей модификацией: объем неводной суспензии, которую наносили на поверхность среды высвобождения лекарственного средства, был изменен с 46 до 1000 микролитров. Результаты суммированы на фиг.7. Уменьшение размера образца повышало относительное количество лекарственного средства, растворившегося во время анализа. Пример 5 Пример 5 иллюстрирует линейность количественной аналитической процедуры HPLC, применявшейся в способе по настоящему изобретению. Изготавливали шесть растворов CCFA с концентрациями, которые варьировались в пределах от 1,27×10-4 до 2,68×10-2 мг/мл. Аликвоты из каждого раствора подвергали анализу и определяли площади пиков с использованием количественного метода HPLC и хроматографических параметров, описанных выше. Результаты суммированы на фиг.8. Пример 6 Пример 6 показывает извлечение анализируемого вещества из неводной жидкой композиции с использованием сред высвобождения лекарственного средства, описанных в разделе “Общая процедура растворения” выше. Извлечение массы лекарственного средства CCFA, растворившегося в средах высвобождения лекарственного средства, нагруженного 1:1 смесью плацебо лота SFH-10 и Miglyol 812, оценивали нагрузкой 15 микролитрами 1:1 смесью плацебо:Miglyol в 75 мл стандартных растворов CCFA. Данный уровень нагрузки (15 микролитров в 75 мл) соответствовал 100 микролитрам смеси плацебо:Miglyol на 500 мл водной среды. Это представляло двукратное увеличение относительной концентрации неводной фазы по сравнению с той, которая описана в разделе “Общая процедура растворения”, представляя, таким образом, “худший случай” или консервативный подход к оценке возможности негативного систематического отклонения (т.е. неполного извлечения) в анализе. Извлечение определяли при шести концентрациях CCFA, которые варьировались приблизительно от 1 до 15 миллионных долей CCFA в водной фазе. Для продукта CCFA 200 мг/мл данные концентрации соответствовали приблизительно 10-150% растворения. Общая процедура растворения описывает измерение высвобождения лекарственного средства из 50 микролитров (0,050 мл) 1:1 разведения суспензии CCFA в Miglyol 812 в 500 мл среды высвобождения лекарственного средства. Если растворилось 10% лекарственного средства, полученная концентрация CCFA в водной фазе должна составлять:
После нагрузки смеси уравновешивали встряхиванием на шейкере с платформой при комнатной температуре в течение двух часов. Нагруженные образцы фильтровали и определяли концентрацию фильтрата с использованием процедуры HPLC, описанной в разделе “Общая процедура растворения”. Результаты суммированы в таблице 3.
Среднее извлечение CCFA составляло 100,15%.
Формула изобретения
1. Способ определения скорости растворения анализируемого вещества из неводной жидкой композиции, предусматривающий стадии (a) получения неводной жидкой композиции, содержащей анализируемое вещество и неводную основу; (b) добавления неводного разбавителя к неводной жидкой композиции для получения разбавленной неводной жидкой композиции; (c) помещения по меньшей мере части разбавленной неводной жидкой композиции и водной растворяющей среды в прибор для тестирования на растворение; (d) контактирования разбавленной неводной жидкой композиции и водной растворяющей среды в течение заранее определенного периода времени; и (e) определения количества анализируемого вещества в водной растворяющей среде, где скорость растворения определяют измерением концентрации анализируемого вещества в заранее определенное время или несколькими измерениями концентрации анализируемого вещества. 2. Способ по п.1, в котором количество анализируемого вещества в водной растворяющей среде определяют в нескольких заранее определенных моментах времени. 3. Способ по п.1, дополнительно включающий на стадии (е) стадию фильтрования водной растворяющей среды, которая используется для определения количества анализируемого вещества в водной растворяющей среде, перед определением в ней количества анализируемого вещества. 4. Способ по п.3, в котором размер пор фильтра находится в пределах приблизительно от 0,1 до 50 мкм. 5. Способ по п.1, в котором неводная жидкая композиция представляет собой фармацевтическую композицию. 6. Способ по п.5, в котором анализируемое вещество представляет собой фармацевтически активный компонент. 7. Способ по п.5, в котором фармацевтическая композиция представляет собой лекарственную форму с замедленным высвобождением. 8. Способ по п.5, в котором фармацевтическая композиция дополнительно содержит фармацевтически приемлемые компоненты, выбранные из группы, состоящей из наполнителей, добавок, суспендирующих агентов, консервантов, увлажняющих агентов, загустителей, буферов, флоккулянтов, корригентов, подсластителей, окрашивающих агентов и отдушек. 9. Способ по п.1, в котором анализируемое вещество выбрано из группы, состоящей из ингибитора АПФ, агониста 10. Способ по п.1, в котором анализируемое вещество представляет собой цефалоспорин, выбранный из группы, состоящей из цефтиофура, цефепима, цефиксима, цефоперазона, цефотаксима, цефподоксима, цефтазидима, цефтизоксима, цефтриаксона, моксалактама, их фармацевтически приемлемых солей и производных. 11. Способ по п.10, в котором анализируемое вещество представляет собой цефтиофур, его фармацевтически приемлемую соль или производное. 12. Способ по п.1, в котором неводная основа выбрана из жира или воска. 13. Способ по п.12, в котором неводная основа представляет собой жир, который является маслом. 14. Способ по п.13, в котором масло выбрано из группы, состоящей из масла канолы, кокосового масла, кукурузного масла, арахисового масла, кунжутного масла, оливкового масла, пальмового масла, масла сафлора красильного, соевого масла, хлопкового масла, рапсового масла, подсолнечного масла и их смесей. 15. Способ по п.13, в котором масло представляет собой хлопковое масло. 16. Способ по п.1, в котором неводная жидкая композиция представляет собой суспензию, раствор или эмульсию. 17. Способ по п.1, в котором неводная жидкая композиция представляет собой суспензию. 18. Способ по п.1, в котором неводный разбавитель выбран из группы, состоящей из масел и органических растворителей. 19. Способ по п.18, в котором неводный разбавитель представляет собой масло. 20. Способ по п.19, в котором масло представляет собой кокосовое масло или хлопковое масло. 21. Способ по п.1, в котором количество неводного разбавителя составляет приблизительно от 0,25 до 10 мас.ч. относительно количества неводной жидкой композиции. 22. Способ по п.1, в котором контактирование осуществляют в течение заранее определенного периода времени для растворения приблизительно от 10 до 100% общего количества анализируемого вещества, которое исходно присутствовало в неводной жидкой композиции, в водной растворяющей среде. 23. Способ по п.22, в котором перемешивание осуществляют в течение заранее определенного периода времени для растворения приблизительно от 10 до 100% общего количества анализируемого вещества, которое исходно присутствовало в неводной жидкой композиции, в водной растворяющей среде. 24. Способ по п.1, в котором водную растворяющую среду изготавливают с использованием воды высокой чистоты. 25. Способ по п.1, в котором водная растворяющая среда выбрана из группы, состоящей из воды, раствора хлористоводородной кислоты, имитированного желудочного сока, буферного раствора, имитированного кишечного сока, воды, содержащей поверхностно-активный агент, буферного раствора, содержащего поверхностно-активный агент, и водного раствора спирта. 26. Способ по п.25, в котором водная растворяющая среда представляет собой буферный раствор. 27. Способ по п.26, в котором буферный раствор выбран из группы, состоящей из глицинового буфера с рН от 2 до 3, цитратного буфера с рН 3, ацетатного буфера с рН от 4 до 5, ацетатного буфера в физиологическом растворе с рН от 5,5, фосфатного буфера с рН от 6 до 8, не содержащего калия фосфатного буфера с рН 6,8, фосфатного буфера в физиологическом растворе с рН 7,4 и боратного буфера с рН от 8 до 10. 28. Способ по п.27, в котором буферный раствор имеет молярность приблизительно от 1 до 10 мМ. 29. Способ по п.27, в котором буфер имеет молярность приблизительно от 1 до 5 мМ. 30. Способ по п.1, в котором на стадии (d) соотношение неводной жидкой композиции и водной растворяющей среды составляет приблизительно от 1:2000 до 1:100000 по объему. 31. Способ по п.30, в котором на стадии (d) соотношение неводной жидкой композиции и водной растворяющей среды составляет приблизительно от 1:5000 до 1:40000 по объему. 32. Способ по п.1, в котором прибор для тестирования на растворение представляет собой лопастной узел. 33. Способ определения скорости растворения анализируемого вещества в неводной жидкой композиции, предусматривающий стадии (a) получения неводной жидкой композиции, содержащей анализируемое вещество и неводную основу; (b) помещения по меньшей мере части неводной жидкой композиции и водной растворяющей среды в прибор для тестирования на растворение, причем указанная водная растворяющая среда включает буфер, имеющий молярность приблизительно от 0,1 до 10 мМ; (c) контактирования неводной жидкой композиции и водной растворяющей среды в течение заранее определенного периода времени; и (d) определения количества анализируемого вещества в водной растворяющей среде, где скорость растворения определяют измерением концентрации анализируемого вещества в заранее определенное время или несколькими измерениями концентрации анализируемого вещества. 34. Способ определения скорости растворения анализируемого вещества из неводной жидкой композиции, предусматривающий стадии (a) получения неводной жидкой композиции, содержащей анализируемое вещество и неводную основу; (b) помещения по меньшей мере части неводной жидкой композиции и водной растворяющей среды в прибор для тестирования на растворение, причем соотношение объемов неводной жидкой композиции и водной растворяющей среды в аппарате для тестирования на растворение составляет приблизительно от 1:2000 до 1:100000; (c) контактирования неводной жидкой композиции и водной растворяющей среды в течение заранее определенного периода времени; и (d) определения количества анализируемого вещества в водной растворяющей среде, где скорость растворения определяют измерением концентрации анализируемого вещества в заранее определенное время или несколькими измерениями концентрации анализируемого вещества.
РИСУНКИ
TK4A – Поправки к публикациям сведений об изобретениях в бюллетенях “Изобретения (заявки и патенты)” и “Изобретения. Полезные модели”
Напечатано: (57) 9. …, иммунодепрессанта, слабительного агента, мистического агента, …
Следует читать: (57) 9. …, иммунодепрессанта, слабительного агента, миотического агента, …
Номер и год публикации бюллетеня: 16-2007
Код раздела: FG4A
Извещение опубликовано: 27.08.2007 БИ: 24/2007
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 11.09.2008
Извещение опубликовано: 27.07.2010 БИ: 21/2010
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 -адренергических рецепторов, агонисты
-адренергических рецепторов, агонисты  -адренергических рецепторов, блокаторы
-адренергических рецепторов, блокаторы  ), хенодезоксихолевая кислота, натриевая соль гликохолевой кислоты, монолаурат поли(оксиэтилен)n-сорбитана (Tween 20
), хенодезоксихолевая кислота, натриевая соль гликохолевой кислоты, монолаурат поли(оксиэтилен)n-сорбитана (Tween 20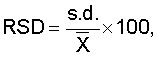
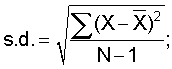
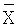 представляет собой среднюю величину.
представляет собой среднюю величину.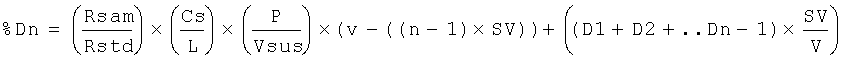
 0,05). Если тангенс не отличается от нуля при 95% доверительности (р>0,05), корреляция не является достоверной.
0,05). Если тангенс не отличается от нуля при 95% доверительности (р>0,05), корреляция не является достоверной.