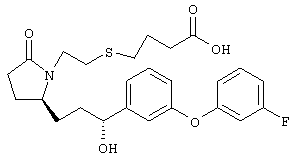
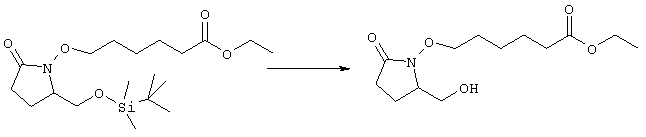Патент на изобретение №2296122
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ 2-ПИРРОЛИДОНА В КАЧЕСТВЕ ПРОСТАНОИДНЫХ АГОНИСТОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ
(57) Реферат:
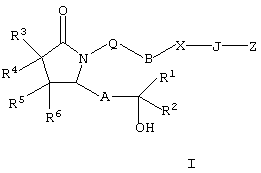
Изобретение относится к новым производным 2-пирролидона формулы I
где Q означает -CH2, -O-; В означает -CH2-, -(СН2)2, -(CH2)3-, -(CH2)4-, -(СН2)5-, -СН=СН-, -CH2-СН=СН-, -СН=СН-СН2-, -СН2-СН=СН-СН2-, при условии, что если В означает -СН=СН-, -СН=СН-СН2-, то Q означает -СН2-; Х означает -O-, -S-, -SO- или -SO2– или простую связь, при условии, что если Х означает простую связь, то Q означает кислород; J означает -(CRbRc)n-, где n означает целое число от 1 до 4; Rb и Rc оба означают водород или оба Rb и Rc означают (С1-С4)алкил, а другой из них означает Н, или Rb и Rc вместе с атомом углерода, к которому они присоединены, образуют С2-С5полиметиленовую группу, или -СН2-СН=СН-; А означает -СН2-СН2, -СН=СН- или -С Z означает -C(O)OR n означает 1, 2, 3 или 4; R p означает 0, 1, 2, 3, 4, 5; R2 означает Н, C1-С6алкил; R3, R4, R5 и R6 означают Н; или их фармацевтически приемлемые соли или сольваты, индивидуальные изомеры или рацемическая или нерацемическая смесь изомеров. Соединения I проявляют конкурентную аффинность связывания ЕР2, ЕР3 и EP4, что позволяет использовать их в фармацевтической композиции. 3 н. и 18 з.п. ф-лы, 1 табл.
Изобретение относится к некоторым производным 2-пирролидона, соответствующим фармацевтическим композициям, способам применения в качестве селективных агонистов простагландина ЕР4 и способам их получения. В литературе имеется множество публикаций, посвященных простагландинам или простаноидам (PGs). Термин простаноиды является международным непатентуемым названием природных и синтетических простагландинов и простагландинподобных соединений, относительно которых известно, что даже незначительные изменения в химической структуре или стереохимической конфигурации оказывают существенное влияние на их биологическую активность. Простагландины или простаноиды (PGs) представляют собой группу биологически активных соединений, выделенных из мембранных фосфолипидов, образованных из незаменимых жирных кислот, содержащих 20 атомов углерода и включающих циклопентановое кольцо. Эти соединения подразделяются на несколько основных классов, обозначаемых буквами, и различаются заместителями в циклопентановом кольце. Основные классы подразделяются на подвиды, обозначаемые индексами 1, 2 или 3, которые соответствуют их жирнокислотным предшественникам. Например, конкретным представителем простагландинов Е является PGE2 формулы
В настоящее время известно четыре различных подвида рецепторов PGE2, которые обозначаются EP1, EP2, ЕР3 и EP4. Применение соединений, характеризующихся высокой активностью связывания с рецептором PGE2, включает профилактику и/или лечение иммунологических заболеваний (аутоиммунные заболевания, трансплантация органов и т.п.), астмы, нарушения образования костной ткани, гибели нейронов, тромбоза и инсульта, гепатопатии, абортов, нарушений мужской и женской половой функции, преждевременных родов, воспаления, такого, как ревматоидный артрит, или невропатических нарушений сетчатки, таких, как глаукома. Более подробно простагландины и ассоциированные с ними рецепторы описаны, например, в статье М.Abramovitz и др., The Utilization of Recombinant Prostanoid Receptors to Determine the Affinities and Selectivities of Prostaglandins and Related Analogs, Biochimica et Biophysica Acta, 1483, 285-293 (2000). Участие агонистов рецептора простагландина Е в резорбции костной ткани описано, например, в статьях T.Suzawa и др., The Role of Prostaglandin Е Receptor Subtipes in Bone Resorption: An Analysis Using Specific Agonists for the Respective EPs, Endocrinology, 141, 1554-1559 (2000), K.Ono и др., Important Role of EP4, a Subtype of Prostaglandin (PG) E Receptor, in Osteoclast-like Cell Formation from Mouse Bone Marrow Cells Induced by PGE2, J. of Endocrinology, 158, R1-R5 (1998), M.Suda и др., Prostaglandin E Receptor Subtipes in Mouse Osteoblastic Cell Line, Endocrinology, 137, 1698-1705 (1996). Эти селективные агонисты рецептора простагландина Е используются также при лечении патологических изменений в желудочно-кишечном тракте, см., например, H.Araki и др., The Roles of Prostaglandin E Receptor Subtypes in the Cytoprotective Action of Prostaglandin E2 in Rat Stomach, Aliment. Pharmacol. Ther., 14 (Suppl.l), 116-124 (2000), T.Kunikata и др., E Type Prostaglandin Inhibits Indomethacin-Induced Small Intestinal Lesions Through ЕР3 and EP4 Receptors: A Study Using Rats and Knockout Mice, Gastroenterology, 118, absr. №3787. Другие примеры применения агонистов рецептора простагландина Е включают активацию функции почек, см., например, M.D.Breyer и др., Prostaglandin E Receptors and the Kidney, Am. J.PhysioL, 279, F12-F23 (2000), и K.E.Purdy и др., EP1 and EP4 Receptors Mediate Prostaglandin E2 Actions in the Microcirculation of Rat Kidney, Am. J. PhysioL, 279, F755-F764 (2000), лечение тромбоза и инсульта, а также других состояний, при которых благоприятное действие оказывает ингибирование агрегации тромбоцитов, см., например, B.Z.S.Paul и др., Distribution of Prostaglandin IP and EP Receptor Subtypes and Isoforms in Platelets and Human Umbilical Artery Smooth Muscle Cells, Br. J. HaematoL, 102, 1204-1211 (1998), противовоспалительное действие за счет ингибирования образования Лечение импотенции и/или эректильной дисфункции с использованием простагландинов, которые являются селективными лигандами рецептора ЕР2 и/или ЕР4, заявлено в опубликованной международной заявке WO 99/02164, поданной фирмой Pharmacia & Upjohn AB. Дополнительная информация по простагландинам и их рецепторам содержится в монографии Goodman and Gillman, The Pharmacological Basis of Therapeutics, 9th edition, McGraw-Hill, New York, гл.26, стр.601-616 (1996). Аналоги 8-аза-11-дезоксипростагландина, соответствующие PGE2, имеют следующее строение:
8-аза-11-дезоксипростагландин Замена атома углерода в положении 8 на атом азота приводит к изменению трехмерной конформации полученного простагландина, а поскольку соединение обладает биологической активностью такое изменение конформации оказывает существенное воздействие на биологическую активность. 8-Аза-11-дезоксипростагландины Е, содержащие природные боковые цепи, описаны в литературе, см., например, BE 841165 (Syntex USA Inc.). Соединения по настоящему изобретению представляют собой аналоги 8-азапростагландина, содержащие неприродную боковую цепь при атома азота (т.е. включающую гетероатом), который также изменяет конформацию полученных аналогов. Эти соединения обладают высокой селективностью действия в качестве агонистов рецептора ЕР4. Увеличение селективности снижает сильные побочные действия, часто наблюдаемые при введении неселективных агонистов простагландинов. Следовательно, существует необходимость в соединениях по настоящему изобретению. Настоящее изобретение относится к соединениям формулы I
где Q означает -СН2– или кислород, В означает -СН2-, -(СН2)2-, -(CH2)3-, -(СН2)4-, -(СН2)5-, -СН=СН-, -СН2-СН=СН-, -СН=СН-СН2– или -СН2-СН=СН-СН2-, при условии, что если В означает -СН=СН- или -СН=СН-СН2-, то Q означает -СН2-, Х означает -NRа– (где Ra означает водород, галоген, C1-С6алкил или C1-С6ацил), -O-, -S-, -SO- или -SO2– или простую связь, при условии, что если Х означает простую связь, то Q означает кислород, J означает -(CRbRc)n-, где n означает целое число от 1 до 4, a Rb и Rc оба означают водород или один или оба Rb и Rc означают (низш.)алкил, или один из них означает водород, или Rb и Rc вместе с атомом углерода, к которому они присоединены, образуют С2-С5полиметиленовую группу, или -СН2– СН=СН-, А означает -СН2-СН2-, -СН=СН- или -С Z означает СН2OH, -C(O)OR’, -C(O)NR’R”, -C(O)NSO2R’, -P(C1-С6)алкил(O)(OR’), -PO(OR’)2 или тетразол-5-ил, где R’ и R n означает 1,2,3 или 4, R1 означает -(CH2)pR7 или -(CH2)qOR8, где R7 и R8 каждый независимо друг от друга означает C1-С6алкил, галоген(С1-С6)алкил, С3-С8циклоалкил, гетероциклил, арил или гетероарил, р и q каждый независимо друг от друга означает 0, 1, 2, 3, 4 или 5, R2 означает водород, C1-С6алкил, C1-С6алкенил или C1-С6алкинил, а R3, R4, R5 и R6 независимо друг от друга означают водород или C1-С6алкил; или к их фармацевтически приемлемым солям. Указанные соединения включают сольват, пролекарство, индивидуальный изомер или рацемическую или нерацемическую смесь изомеров. Другим объектом изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество по меньшей мере одного соединения формулы I или его фармацевтически приемлемой соли или сольвата, пролекарства, индивидуального изомера или рацемической или нерацемической смеси изомеров в смеси по меньшей мере с одним пригодным носителем, разбавителем или эксципиентом. Еще одним объектом изобретения является способ лечения заболевания, прежде всего заболевания костной ткани, у млекопитающего, излечиваемого введением агониста рецептора простагландина ЕР4, причем способ включает введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Еще одним объектом изобретения является способ получения соединений формулы I. Если не указано иное, термины, используемые в описании заявки и формуле изобретения, имеют значения, указанные ниже. “Алкокси” означает радикал -OR, где R означает алкил, имеющий значения, указанные в тексте заявки, например метокси, этокси, пропокси, бутокси и т.п. “Алкил” означает линейный насыщенный одновалентный углеводородный радикал, содержащий от одного до шести атомов углерода, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести атомов углерода, например метил, этил, пропил, 2-пропил, н-бутил, изобутил, трет-бутил, пентил и т.п. “Алкилен” означает линейный насыщенный двухвалентный углеводородный радикал, содержащий от одного до шести атомов углерода, или разветвленный насыщенный двухвалентный углеводородный радикал, содержащий от трех до шести атомов углерода, например метилен, этилен, 2,2-диметилэтилен, пропилен, 2-метилпропилен, бутилен, пентилен и т.п. “Алкилтио” означает радикал -SR, где R означает алкил, имеющий значения, указанные выше, например метилтио, этилтио, пропилтио, бутилтио и и т.п. “Арил” означает одновалентный моноциклический или бициклический ароматический углеводородный радикал, который необязательно независимо друг от друга замещен одним или более заместителями, предпочтительно одним, двумя или тремя заместителями, выбранными из группы, включающей алкил, галогеналкил, галоген, нитро, циано, амино, метилендиокси, этилендиокси, фенил, необязательно замещенный группой Y, Y-гетероарил, Y-циклоалкил, -Y-гетероциклил, -Y-OR’, -Y-NR’R “Циклоалкил” означает насыщенный одновалентный циклический углеводородный радикал, содержащий в цикле от трех до семи атомов углерода, например циклопропил, циклобутил, циклогексил, 4-метилциклогексил и т.п. “Галоген” означает фтор, хлор, бром или иод, предпочтительно фтор и хлор. “Галогеналкил” означает алкил, замещенный одним или более идентичных или различных атомов галогена, например -СН2Cl, -CF3, -СН2CF3, -СН2CCl3 и т.п. “Гетероарил” означает одновалентный моноциклический или бициклический радикал, содержащий в цикле от 5 до 12 атомов и по меньшей мере один ароматический цикл, включающий в цикле один, два или три гетероатома, выбранных из ряда N, О или S, а остальные атомы в цикле являются атомами углерода, причем гетероарильный радикал присоединен к ароматическому циклу. Гетероарильный цикл необязательно замещен независимо друг от друга одним или более заместителями, предпочтительно одним или двумя заместителями, выбранными из ряда алкил, галогеналкил, галоген, нитро, циано, амино, метилендиокси, фенил, необязательно замещенный группой Y, Y-циклоалкил, -Y-гетероциклил, -Y-OR’, -YNR’R «Гетероциклил» означает насыщенный или ненасыщенный неароматический циклический радикал, содержащий от 3 до 8 атомов в цикле, из которых один или два атома в цикле являются гетероатомами, выбранными из ряда N, О или S(O)0-2, а остальные атомы в цикле являются атомами углерода, причем один или два атома углерода необязательно заменены на карбонильную группу. Гетероциклил необязательно замещен независимо друг от друга одним, двумя или тремя заместителями, выбранными из ряда алкил, галогеналкил, галоген, нитро, циано, фенил, необязательно замещенный группой Y, Y-гетероарил, Y-циклоалкил, -Y-OR’, -YNR’R “Уходящая группа” означает группу, название которой обычно ассоциируется с ее использованием в синтетической органической химии, т.е. означает атом или группу, которая замещается нуклеофилом и включает галоген (такой, как хлор, бром или иод), алкилсульфонилокси, арилсульфонилокси, алкилкарбонилокси (например, ацетокси), арилкарбонилокси, мезилокси, тозилокси, трифторметансульфонилокси, арилокси (например, 2,4-динитрофенокси), метокси, N,O-диметилгидроксиламино и т.п. “Необязательно замещенный фенил” означает фенильный цикл, который необязательно замещен независимо друг от друга одним или более заместителями, предпочтительно одним или двумя заместителями, выбранными из группы, включающей алкил, гидрокси, алкокси, галогеналкил, галогеналкокси, гетероалкил, галоген, нитро, циано, амино, метилендиокси, этилендиокси и ацил. “Изомерия” означает соединения с одинаковой химической формулой, но отличающиеся природой или последовательностью связей между атомами или пространственным расположением атомов. Изомеры, которые различаются пространственным расположением атомов, называются «стереоизомерами». Стереоизомеры, которые не являются зеркальными изображениями друг друга, называются «диастереоизомерами», а стереоизомеры, которые являются несовместимыми зеркальными изображениями называются «энантиомерами» или в некоторых случаях оптическими изомерами. Углеродный атом, связанный с четырьмя различными заместителями, называется “хиральным центром”. «Хиральный изомер» означает соединение с одним хиральным центром. Такое соединение имеет две энантиомерные формы с противоположной хиральностью и может существовать в виде индивидуального энантиомера или в виде смеси энантиомеров. Смесь, содержащая равные количества индивидуальных энантиомерных форм противоположной хиральности, называется «рацемической смесью». Соединение, содержащее более одного хирального центра, может существовать в виде 2n-1 энантиомерных пар, где n означает число хиральных центров. Соединение, содержащее более одного хирального центра, может существовать в виде индивидуального диастереомера или в виде смеси диастереомеров, которая назвается «диастереомерной смесью». При наличии одного хирального центра стереоизомер можно охарактеризовать абсолютной конфигурацией (R или S) указанного хирального центра. Абсолютная конфигурация означает пространственное расположение заместителей, присоединенных к хиральному центру. Заместители, присоединенные к указанному хиральному центру, классифицируют по правилу Кана, Ингольда и Прелога (Cahn и др., Angew. Chem. Inter. Edit., 5, 385, (1966) (список опечаток с.511), Cahn и др., Angew. Chem., 78, 413 (1966), Cahn и Ingold., J. Chem. Soc. (London), 612 (1951), Cahn и др., Experientia, 12, 81, (1956), Cahn, J. Chem. Educ., 41, 116 (1964)). “Геометрические изомеры” означают диастереомеры, образующиеся за счет заторможенного вращения вокруг двойных связей. Эти конфигурации обозначаются приставками цис- и транс-, или буквами Z и Е, которые означают, что группы располагаются по одну или по разные стороны плоскости двойных связей согласно правилам Кана-Ингольда-Прелога. «Атропические изомеры» означают изомеры, образующиеся за счет заторможенного вращения объемных групп вокруг центральной связи. Соединения по настоящему изобретению могут существовать в стереоизомерной форме, следовательно, их можно получать в виде фармацевтически приемлемого эксципиента, что означает эксципиент, который используют при получении фармацевтической композиции и который обычно является безопасным, нетоксичным, безвредным в биологическом или ином отношении, и включает носитель, приемлемый как в ветеринарии, так и в фармацевтике. “Фармацевтически приемлемый эксципиент”, используемый в описании и пунктах формулы изобретения, включает один или более таких эксципиентов. “Фармацевтически приемлемая соль” соединения означает соль, которая является фармацевтически приемлемой и которая обладает необходимой фармакологической активностью исходного соединения. Такие соли включают: (1) кислотно-аддитивные соли неорганических кислот, таких, как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или органических кислот, таких, как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфокислота, этансульфокислота, 1,2-этандисульфокислота, 2-гидроксиэтансульфокислота, бензолсульфокислота, 4-хлорбензолсульфокислота, 2-нафталинсульфокислота, 4-толуолсульфокислота, камфорсульфокислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтоевая кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п., или (2) соли, образующиеся при замене кислотного протона, присутствующего в исходном соединении, на ион металла, например, ион щелочного металла, ион щелочно-земельного металла или ион алюминия, или при образовании протоном координационного соединения с органическим основанием, таким, как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. Предполагается, что все ссылки на фармацевтически приемлемые соли включают их сольватированные (сольваты) или кристаллические (полиморфные модификации) формы указанной кислотно-аддитивной соли, как описано описано в тексте заявки. “Кристаллические формы” (или полиморфные модификации) означают кристаллические структуры, в которых соединение присутствует в разных кристаллических формах, имеющих одинаковый элементный состав. Такие кристаллические формы обычно характеризуются разными рентгеновскими диффракционными картинами, разными инфракрасными спектрами, имеют разные температуры плавления, иную плотность, прочность и форму кристаллов, различаются по оптическим и электрическим свойствам, по устойчивости и растворимости. На образование преобладающей кристаллической формы влияют различные факторы, такие, как растворитель, используемый для перекристаллизации, скорость кристаллизации и температура хранения. “Сольваты” означают сольватированные формы, которые содержат стехиометрическое или нестехиометрическое количество растворителя. Такие соединения в кристаллическом состоянии удерживают фиксированное молярное количество молекул растворителя, образуя сольват. Если в качестве растворителя используется вода, образующийся сольват называется гидратом, если растворителем является спирт, образующийся сольват называется алкоголятом. Гидраты образуются при комбинации одной или более молекул воды с молекулой вещества, причем вода сохраняет молекулярную форму Н2О, а такие комбинации способны образовывать один или более гидратов. Термин “пролекарство” означает любое соединение, которое при введении млекопитающему высвобождает активное исходное лекарственное средство формулы I in vivo, когда такое пролекарство вводится млекопитающему. Пролекарства соединения формулы I получают модификацией одной или нескольких функциональной групп, содержащихся в соединении формулы I, причем модифицированная группа (группы) может расщепляться in vivo с образованием исходного соединения. Пролекарства включают соединения формулы I, в которых гидрокси, амино, сульфгидрил, карбокси или карбонильная группа в составе соединения формулы I связана с любой группой, которая может отщепляться in vivo с образованием свободной гидроксильной, амино- или сульфгидрильной группы соответственно. Примеры пролекарств включают, без ограничения перечисленным, сложные эфиры (например, ацетат, диалкиламиноацетаты, формиаты, фосфаты, сульфаты и бензоаты) и карбаматы (например, N,N-диметиламинокарбонил) гидроксильных функциональных групп, сложноэфирные группы (например, этиловые эфиры, морфолиноэтанольные эфиры) карбоксильных функциональных групп, N-ацильные производные (например, N-ацетил) основания Манниха, Шиффовы основания и енаминоны функциональных аминогрупп, оксимы, ацетали, кетали и енольные эфиры кетонных и альдегидных функциональных групп в соединениях формулы I, и т.п.(см. Bundegaard H., Design of Prodrugs, Elsevier, New York-Oxford, 1-92 (1985)). “Защитная группа” означает группу атомов, которая при связывании с реакционноспособной группой блокирует ее, препятствуя ее участию в основной реакции. Примеры защитных групп можно найти в монографиях T.W.Green и P.G.M.Futs, Protective Groups in Organic Chemistry (Wiley, 3 ed. (1999), и Harrison и др., Compendium of Synthetic Organic Methods, т.1-8, J.Wiley и Sons (1971-1996). Типичные аминозащитные группы включают формил, ацетил, трифторацетил, бензил, бензилоксикарбонил (CBZ), трет-бутоксикарбонил (Boc), триметилсилил (TMS), 2-триметилсилилэтансульфонил (SES), тритил и замещенные тритильные группы, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (FMOC), нитровератрилоксикарбонил (NVOC) и т.п. Типичные гидроксизащитные группы включают такие группы, в которых гидроксил ацилирован или алкилирован группой, такой, как бензил, тритильные простые эфиры, а также алкильные простые эфиры, тетрагидропиранильные простые эфиры, триалкилсилиловые простые эфиры и аллильные эфиры. “Терапия” или “лечение” заболеания включает (1) профилактику заболевания, т.е. подавление развития клинических симптомов заболевания у субъекта, который подвержен или предрасположен к заболеванию, но у которого не обнаруживаются или не проявляются симптомы патологического состояния; (2) подавление заболевания, например подавление или снижение интенсивности развития заболевания или его клинических симптомов; (3) ослабление заболевания, например стимуляцию регрессии заболевания или его клинических симптомов. “Терапевтически эффективное количество” означает количество соединения, которое при введении млекопитающему для лечения заболевания, является достаточным, чтобы обеспечить указанное лечение. Терапевтически эффективное количество может изменяться в зависимости от типа соединения, типа заболевания, подлежащего лечению, тяжести заболевания, возраста и массы и т.п. млекопитающего, подлежащего лечению. “Аналог простагландина” означает неприродное соединение, которое в структурном отношении является близким простагландину. “Рецептор простагландина” или “простаноидный рецептор” означает природный белок, связывающий простагландины, и который после образования комплекса изменяет функционирование клетки. Простагландиновые рецепторы могут проявлять возбуждающие или расслабляющие свойства. Такие рецепторы включают, без ограничения перечисленным, EP1, ЕР2, ЕР3, EP4, DP, FP, IP, TP1 и ТР2. Кроме того, эти рецепторы обсуждаются в обзоре Coleman и др., Pharmacological Reviews, 6, №2, 205-229 (1994). Название и нумерация соединений по настоящему изобретению иллюстрируется ниже.
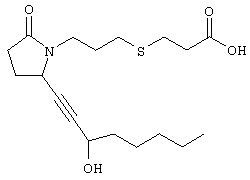
В общем случае, номенклатура, использованная в описании заявки, основана на программном обеспечении AUTONOM Например, соединение формулы I, где Q означает -СН2-, В означает -CH2-, Х означает -S-, J означает -(CH2)3-, Z означает -СООН, R2, R3, R4, R5 и R6 означают водород, А означает -СН=СН-, а R1 означает н-пентил, называется 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляная кислота. Предпочтительными являются соединения, в которых Z означает -C(O)OR’, CH2OH или тетразол-5-ил, более предпочтительны соединения, в которых Z означает СООН. Предпочтительными являются соединения, в которых Q и В вместе образуют -(CH2)n-, a n означает целое число от 2 до 5, более предпочтительно от 2 до 3. Предпочтительны соединения, в которых Х означает -О- или -S-. Предпочтительными являются соединения, в которых J означает -(СН2)3-, -(CHRa)3, где один из Ra означает (низш.)алкил, или J означает -СН2-СН=СН-. Предпочтительными являются соединения, в которых А означает -СН2-СН2-или СН=СН. Предпочтительными являются соединения, в которых R2, R3, R4, R5 и R6 означают водород. Среди соединений, в которых R7 означает арил или гетероарил, предпочтительны соединения, в которых R7 означает фенил, необязательно замещенный заместителем из ряда алкил, трифторметил, галоген, -Y-R9 или -Y-OR9, причем Y предпочтительно означает химическую связь, а R9 означает C1-С6алкил, галоген или необязательно замещенный фенил. В первом варианте осуществления изобретения Q, В, X, J, Z, А, R2, R3, R4, R5 и R6 имеют значения, указанные в кратком описании изобретения, R1 означает -(СН2)pR7, а R7 означает алкил или С3-С8циклоалкил. Предпочтительно R7 означает метил, а p равно 4. Во втором варианте изобретения Q, В, X, J, Z, A, R2, R3, R4, R5 и R6 имеют значения, указанные в кратком описании изобретения, а R1 означает -(CH2)pR7, где R7 означает арил, гетероарил или гетероциклил. Предпочтительно R7 означает необязательно замещенный фенил, в котором по меньшей мере один из заместителей выбирают из ряда C1-С6алкил, трифторметил, галоген, -Y-R9, -Y-OR9 и -Y-C(O)R9, где Y означает химическую связь или С1-С3алкиленовую группу, а R9 означает C1-С6алкил, арил, гетероарил или гетероциклил. Более предпочтительно R7 означает арил или гетероарил, а p равно 0. Наиболее предпочтительно R7 означает арил или гетероарил, а p равно 1. В третьем варианте изобретения R1, R2, R3, R4, R5 и R6, A, J и Z имеют значения, указанные в кратком описании изобретения, Q-B вместе означают -(СН2)2-6, а Х означает -NH-, -О- или -S-. Предпочтительно Q-B вместе означают -СН2-СН2-. Более предпочтительно в данном варианте R1 означает -(CH2)pR7, где R7 означает арил или гетероарил. В четвертом варианте изобретения R1, R2, R3, R4, R5, R6, A, B,J и Z имеют значения, указанные в кратком описании изобретения, а Q означает кислород. В пятом варианте изобретения R1, R2, R3, R4, R5, R6, A, Q, B, X и Z имеют значения, указанные в кратком описании изобретения, а J означает -(CH2)3-, -(CHRa)3, где один из Ra означает (низш.)алкил. Типичными соединениями формулы I являются следующие: 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляная кислота, {4-[2-[R-(3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутилсульфанил}уксусная кислота (Ro3308721), {4-[2-[R-(1Е)-(3S-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутилсульфанил}уксусная кислота, {4-[(R)-2-[(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусная кислота, (4-{(R)-2-[(Е)-3-гидрокси-3-(5-трифторметилфуран-2-ил)пропенил]-5-оксопирролидин-1-ил}бутилсульфанил)уксусная кислота, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этансульфинил}масляная кислота, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этансульфонил}масляная кислота, {(Z)-4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси} уксусная кислота, {4-[R-)-2-((Е)-(S)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусная кислота, (4-{(R)-2-[(S)-(Е)-3-гидрокси-4-(3-трифторметилфенил)бут-1-енил]-5-оксопирролидин-1-ил}бутокси)уксусная кислота, (4-{(R)-2-[(Е)-3-гидрокси-3-(2′-метилбифенил-3-ил)пропенил]-5-оксопирролидин-1-ил}бутокси)уксусная кислота, (4-{(R)-2-[(Е)-3-(4′-хлорбифенил-3-ил)-3-гидроксипропенил]-5-оксопирролидин-1-ил}бутокси)уксусная кислота, 4-{2-[(R)-2-((Е)-3-гидрокси-3-пентилокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил} масляная кислота, 4-{2-[(R)-2-((Е)-3-гидрокси-3-метилокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}масляная кислота, 4-[2-((S)-2-{(R)-3-[3-(3-фторфенокси)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляная кислота, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-3-метилмасляная кислота, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-2-метилмасляная кислота, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-4-метилмасляная кислота, 1-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанилметил}циклопропил)уксусная кислота, 5-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}уксусная кислота, 3-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}пропионовая кислота, 5-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}пентановая кислота, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-2-бутеновая кислота, 4-[2-((S)-2-{(R)-3-[3-(4′-хлор-2′-метилфенил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляная кислота, 4-[2-((S)-2-{(S)-3-[3-(4′-хлор-2-метилфенил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляная кислота, 4-[2-((S)-2-{(R)-3-[3-(3-(2′,4′-дифторфенил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляная кислота, 4-[2-((S)-2-{(R)-3-[3-(3-(4′-метокси-2′-метил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляная кислота, 6-[2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-илокси]гексановая кислота, 3-{3-[2-(3-гидроксиокт-1-инил)-5-оксопирролидин-1-ил]пропилсульфанил}пропионовая кислота. Соединения формулы I могут включать оптические изомеры, диастереомеры или энантиомеры указанной структуры или фармацевтически приемлемые соли, их биогидролизуемые амиды, сложные эфиры или имиды. Предпочтительная стереохимическая структура аналогична природному PGE2. Соединения по настоящему изобретению могут существовать в виде несольватированных форм, а также в виде сольватированных форм, включая гидратированные формы. В общем случае, сольватированные формы, включая, гидраты, эквивалентны несольватированным формам и также включены в объем настоящего изобретения. Кроме того, соединения формулы I образуют фармацевтически приемлемые основно-аддитивные соли. Такие формы также включены в объем настоящего изобретения. Соединения по изобретению получают методами, показанными ниже на схемах реакций. Для специалиста в данной области представляется очевидным, что в пределах объема изобретения возможны некоторые модификации указанных схем, например, некоторых стадий, включающих использование защитных групп для некоторых функциональных групп, которые несовместимы с конкретными условиями реакции. Исходные материалы и реагенты, используемые при получении указанных соединений, являются коммерческими препаратами или их получают методами, известными специалистам в данной области. Следующие схемы реакций лишь иллюстрируют некоторые методы синтеза соединений по настоящему изобретению, а различные модификации этих схем реакций будут использованы специалистом в данной области со ссылкой на материалы настоящей заявки. Если не указано иное, описанные реакции проводят при атмосферном давлении при температуре от приблизительно -78°С до приблизительно 150°С, более предпочтительно от приблизительно 0°С до приблизительно 125°С и наиболее предпочтительно приблизительно при комнатной температуре, приблизительно при 20°С. Схема А На схеме А показаны методы получения соединений общей формулы I, где Q означает -CH2-. Схема А
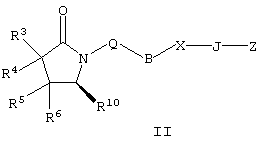
Соединения формулы а являются известными соединениями. Например, (R)-5-(гидроксиметил)-2-пирролидинон, в котором R3, R4, R5 и R6 означают водород, является коммерческим препаратом, а его получение описано в статье S.Saijo и др., Chem. Pharm. Bull., 28, 1449-1458 (1980), (R)-3,3-диметил-5-(гидроксиметил)-2-пирролидинон, в котором R3 и R4 означают метил, a R5 и R6 означают водород, можно получить по методике, описанной Y.Nakagawa и др., Tetrahedron, 54, 10295-10307 (1998), а 4,4-диметил-5-(гидроксиметил)-2-пирролидинон, в котором R3 и R4 означают водород, а R5 и R6 означают метил, можно получить по методике, описанной R.L.Mackman и др., J. Chem. Soc., PerkinTrans., 2111-2122 (1997). На первой стадии гидроксильную группу в соединении а защищают методами, известными в данной области и описанными выше. Методы защиты гидроксильной группы в соединении а в виде ацеталя описаны S. Saijo и др., Chem. Pharm. Bull., 28, 1449-1458 (1980). Другими защитными группами являются силиловые простые эфиры. Силиловые эфиры получают при взаимодействии с любым галогентриалкилсиланом, таким, например, как хлортриизопропилсилан, хлордиметилфенилсилан или трет-бутилхлордиметилсилан, в инертном органическом растворителе, таком, как, без ограничения перечисленным, дихлорметан, тетрагидрофуран или N,N-диметилформамид, в присутствии слабого основания, такого, как имидазол, триэтиламин или пиридин. 5-Гидроксиметил-2-пирролидинон с защищенной гидроксигруппой растворяют в полярном растворителе, таком, как тетрагидрофуран, N-метил-2-пирролидинон или N,N-диметилформамид, обрабатывают основанием, таким, как гидрид натрия, гексаметилдисилазид калия или трет-бутоксид калия, с образованием аниона, а затем вводят в реакцию с замещенным алкилом общей формулы b, где L означает уходящую группу, предпочтительно галоген, a Z означает сложноэфирную группу, указанную выше. Удаление защитной группы проводят в спирте, таком, как метанол, этанол или 2-пропанол, в присутствии каталитического количества кислоты, такой, как трифторуксусная кислота, пара-толуолсульфоновая кислота или хлористоводородная кислота, с образованием соединения с. Соединение с окисляют с использованием окислителя, который обеспечивает окисление гидроксильной группы до альдегидной без дальнейшего превращения в карбоксильную группу, с образованием альдегида общей формулы d. В качестве окислителей можно использовать, например, диметилсульфоксид-трифторуксусный ангидрид, а затем триэтиламин, гипохлорит натрия в присутствии в качестве катализатора радикала 2,2,6,6-тетраметил-1-пиперидинилокси, 1,1,1-триацетокси-1,1-дигидро-1,2-бензиодоксол-3-(1Н)-он, N-метилморфолин-N-оксид в присутствии в качестве катализатора перрутената тетрапропиламмония, или хлорхромат пиридиния в присутствии инертного носителя, такого, как целит При взаимодействии соединения d с На стадии 4 проводят восстановление кетона е гидридом, например борогидридом натрия, в растворителе, таком, как дихлорметан, толуол, этанол или тетрагидрофуран, с образованием диастереомерной смеси спиртов формулы f, где R2 означает водород. Если необходимо предпочтительное образование одного из диастереомеров, такого, как S-гидроксильный изомер, в котором R1 означает н-алкил, можно использовать стехиометрическую комбинацию алюмогидрид лития/этанол/(S)-(-)-бинафтол, как описано R.Noyori и др., J. Am. Chem. Soc., 106, 6717-6725 (1984), или, если необходим R-гидроксильный изомер, можно использовать комбинацию каталитического количества (R)-2-метил-CBS-оксазаборолидина со стехиометрическим количеством борна-диметилсульфида, как описано E.J.Corey и др., J. Am. Chem. Soc., 109, 7925-7926 (1987), или стехиометрических количеств (R)-3-пинанил-9-борабицикло[3.3.1]нонана, как описано М.М.Midland и др., J. Am. Chem. Soc., 102, 867-869 (1980). После каталитического гидрирования двойной связи в присутствии никеля Ренея или палладия на угле получают соединение общей формулы f с насыщенной боковой цепью. В другом варианте на стадии 5 соединение е вводят в реакцию с металлоорганическим соединением общей формулы R2М, где М означает металл или галогенид магния, более предпочтительно с реактивом Гриньяра общей формулы R2MgBr, где R2 имеет значения, указанные выше, с образованием соединения g. Если необходимо соединение с насыщенной боковой цепью, проводят восстановление двойной связи описанным выше методом. При необходимости сложноэфирную группу Z гидролизуют с образованием -С(O)ОН по методикам, хорошо известным специалисту в данной области, например, в присутствии основания, такого, как гидроксид лития, натрия или калия, или в присутствии кислоты, такой, как серная кислота или хлористоводородная кислота в протонном растворителе или в растворителе из группы простых эфиров, содержащем воду, или с использованием липазы типа VII в 0,05 М фосфатном буферном растворе с pH 6,8, как описано C.Luthy и др., J. Am. Chem. Soc., 100, 6211-6217 (1978). При получении соединений, в которых Z не означает C(O)OR’, алкилпроизводные b получают по известным методикам. Например, алкилпроизводные формулы b можно получить, а затем ввести группу Z в виде тетразол-5-ила, как описано W.S. Marshall и др., J. Med. Chem., 30, 682-689 (1987). Другим вариантом изобретения является промежуточное соединение формулы
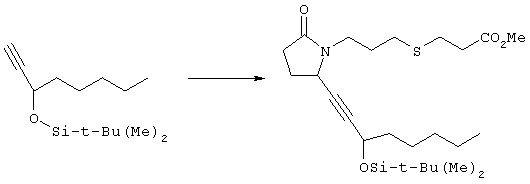
где Q, В, X, J, R3, R4, R5 и R6 имеют значения, указанные выше, Z означает C(O)OR’, где R’ означает водород или С1-С6алкил, а R10 означает -СН2ОН, -СНО, -CH=CH-C(O)R1. Указанное соединение формулы II представлено в виде общей формулы промежуточных соединений на схеме А. Схема Б На схеме Б показан общий метод получения соединений формулы I, в которых Q означает -СН2-, В означает -СН2-, R1 означает арил, a R2 означает Н. Схема Б
Соединения формулы ее получают по схеме А с использованием алкилгалогенида, такого, как триалкилсилиловый эфир Удаление гидроксилзащитной группы в соединении ff проводят в стандартных условиях, указанных выше, например в присутствии гидроксида натрия в водном метаноле или фторида тетрабутиламмония в тетрагидрофуране. Полученный диол можно селективно модифицировать по первичной гидроксильной группе при взаимодействии с бензолсульфонилхлоридом или метансульфонилхлоридом в присутствии триалкиламина, такого, как триэтиламин, при температуре от -25 до 0°С с образованием соответствующего моносульфоната. Затем указанный моносульфонат вводят в реакцию с нуклеофильным соединением литий- или калий-X-J-Z, таким, как тиолат калия, образующимся при взаимодействии метоксида калия с Схема В На схеме В показан общий метод получения фосфоната формулы k, который в свою очередь используется на схеме А для введения боковой цепи в виде групы (нижн.)алкил, в котором R1 означает арил. Схема В
В качестве исходного материала используют производные бензойной кислоты, в которых L означает уходящую группу, имеющую значения, указанные выше. Эти соединения являются коммерческими препаратами или их можно получить известными в данной области методами. Затем из них получают соединения формулы h или j соответственно. Условия получения соединений формулы h описаны D.A.Evans и др., Tetrahedron Lett., 39, 2937 (1998). Методы получения соединений формулы k описаны A.M.Echavarren и J.K.Stille., J. Am. Chem. Soc., 109, 5478-5486 (1987), N.Miyaura и A.Suzuki, Chem. Rev., 95, 2457-2483 (1995), и A.F.Littke и др., J. Am. Chem. Soc., 122, 4020-4028 (2000). Соединения h и J превращают в соединение k при взаимодействии с диалкилметилфосфонатом, который сначала обрабатывают основанием, таким, как н-бутиллитий или диизопропиламид лития, в инертном растворителе из группы простых эфиров, таком, как тетрагидрофуран или трет-бутилметиловый эфир. Схема Г На схеме Г показан общий метод получения соединений формулы I, в которых Q означает -O-, а Х означает химическую связь. Схема Г
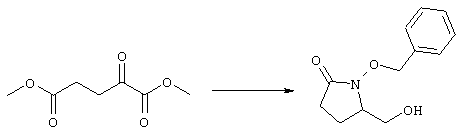
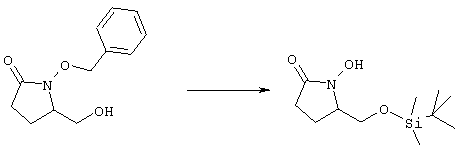
Исходные соединения формулы m являются известными соединениями. Например, диметил-2-оксоглутарат, в котором R3, R4, R5 и R6 означают водород, являются коммерческими препаратами. Последовательная обработка соединений m гидроксиламином, например, O-бензилгидроксиламином, в присутствии слабого основания, такого, как пиридин или ацетат натрия, а затем взаимодействие с восстанавливающим агентом, таким, как боран-пиридиновый комплекс или триацетоксиборогидрид натрия, приводит к получению соединения n, в котором защитная группа PG1 означает бензил. Обработка соединения n основанием, таким, как изопропилмагнийбромид или трет-бутоксид калия, и последующая обработка восстанавливающим агентом, таким, как борогидрид натрия, позволяет превратить сложноэфирную боковую цепь в лактам о. На стадии 3 гидроксил в соединении о сначала защищают, причем защитная группа PG1 может отщепляться в присутствии группы PG2. Лактам p, в котором, например, PG1 означает бензил, а PG2 означает трет-бутилдиметилсилил, получают в присутствии катализатора, такого, как палладий на угле или оксид платины, и газообразного водорода. N-Гидроксилактам p растворяют в полярном растворителе, таком, как тетрагидрофуран, N-метил-2-пирролидон или N,N-диметилформамид, и обрабатывают основанием, таким, как гидрид натрия, гексаметилдисилазид калия или трет-бутоксид калия, с образованием аниона, который затем взаимодействует с алкилпроизводным формулы
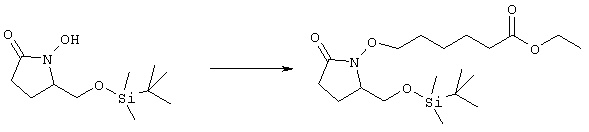
где L означает уходящую группу, предпочтительно галоген. Затем удаляют защитную группу, например, в присутствии фторида калия или тетрабутиламмония, с образованием соединений общей формулы cc. Из соединений формулы cc методом, показанным выше на схеме А, получают соединения общей формулы g. Схема Д На схеме Д показан метод получения соединения формулы I, в которой А означает угерод-углеродную тройную связь. Схема Д
В общем случае, алкилирование соединения г алкилгалогенидом общей формулы b, в котором L, В и Z имеют значения, указанные выше, a Q означает -СН2-, в полярном апротонном растворителе, таком, как N-метил-2-пирролидинон, N,N-диметилформамид, тетрагидрофуран или ацетонитрил, в присутствии слабого основания, такого, как метоксид натрия, карбонат калия или трет-бутоксид калия, приводит к образованию циклического имида формулы s. Алкоксилактамы общей формулы t получают восстановлением соединения s борогидридом лития или натрия в спиртовом растворителе в присутствии кислоты, такой, как лимонная кислота, трифторуксусная кислота, хлористоводородная кислота или бромистоводородная кислота, как описано J.C. Hubert и др., Tetrahedron, 31, 1437-1441 (1975). Взаимодействие соединения формулы t с алкинилоловоорганическим реагентом общей формулы и, где PG3 означает защитную группу, такую, например, как силиловый эфир, а М означает металл, обычно в присутствии кислоты Льюиса, такой, как галогенид магния, тригалогенид бора, соль титана (IV) или соль олова (IV), и последующее удаление защитной группы приводит к образованию соединения w. Соединение w, в котором Z означает -С(O)ОН, можно получить гидролизом соответствующих сложных эфиров, как показано на схеме А. Настоящее изобретение относится также к фармацевтическим композициям, включающим терапевтически эффективное количество соединения по настоящему изобретению в смеси по меньшей мере с одним пригодным носителем, разбавителем или эксципиентом. Соединения по настоящему изобретению могут применяться для получения лекарственного средства, прежде всего препарата для лечения заболевания, ассоциированного с дисфункциями костной ткани и более предпочтительно лекарственного средства для лечения остеопороза. Соединения по настоящему изобретению являются селективными агонистами простагландина ЕР4 и могут использоваться для лечения ряда патологических состояний, ассоциированных с заболеваниями, опосредованными рецептором простагландина ЕР4, прежде всего патологических состояний, ассоциированных с дисфункциями костной ткани, характеризующимися низкой костной массой и деградацией костной ткани с последующим увеличением ломкости кости и восприимчивостью к перелому, прежде всего таких состояний, при которых требуется существенное увеличение костной массы, объема костной ткани или прочности костной ткани. Состояния, ассоциированные с низкой костной массой, включают такие состояния, при которых наблюдается снижение костной массы ниже нормы, характерной для данного возраста. К таким состояниям относятся также детский идиопатический и первичный остеопороз. Лечение остеопороза включает профилактику или снижение интенсивности долгосрочных осложнений, таких, как искривление позвоночника, уменьшение роста, имплантируемое протезирование, заживление переломов и профилактика нарушения функции предстательной железы. Для специалиста в данной области очевидно, что термин масса костной ткани относится фактически к массе костной ткани на единицу площади, которую иногда называют плотностью костной ткани. Установлено, что аналоги 8-аза-11-дезоксипростагландина по настоящему изобретению могут применяться для лечения дисфункции костной ткани. Другие области применения указанных соединений включают профилактику и/или лечение аллергии, альвеолярного абсцесса, болезни Альцгеймера, бокового амиотрофического склероза (ALS), артрита, астмы, атопии, бронхита, ожогов, онкологических заболеваний, сердечно-сосудистого заболевания, болезни Крона, хронических обструктивных заболеваний легких, застойной сердечной недостаточности, гингивита, гломерулонефрита, гепатита, повреждений печени, острого гепатита, гипертонии (гипертензии), гиперцитокинемии, иммунных нарушений, воспалительного заболевания желудочно-кишечного тракта, болезни Кавасаки, недостаточности функции печени, заболевания печени, недостаточности функции легких, синдрома активации макрофагов, множественной недостаточности органов, рассеянного склероза, ишемии миокарда, нефрита, нейродегенерации, гибели нейронов, отторжения трансплантировнных органов, периодонтита, аггрегации тромбоцитов, повреждений легких, фиброза легких, эмфиземы легких, расстройства функции почек, почечной недостаточности, нарушений функции почек, легочной недостаточности, септицемии, сепсиса, шока, нарушения сна и нарушения агрегации трмбоцитов, болезни Стилла, системной гранулемы, тромбоза, язвенного колита и уремии или использование указанных соединений в качестве промотора остеогенеза. Соединения формулы I связываются с рецепторами ЕР4 и воздействуют на рецепторы ЕР4, которые являются подвидом рецептора PGE2. Действие соединений по настоящему изобретению определяют с использованием анализа связывания с клетками, экспрессирующими подвиды простаноидных рецепторов, как подробно описано в примере 10. Активность соединений определяют при конкурентном связывании указанных соединений с соответствующей мишенью, как описано в примере 11. Соединения по настоящему изобретению можно анализировать по их воздействию на плотность костной массы по методике, описанной Gunness-Hey и Hock, Metab. Bone Dis., 5, 177-181 (1984), как подробно описано в примере 12. В общем случае, соединения по настоящему изобретению можно вводить в терапевтически эффективном количестве любыми приемлемыми способами введения, принятыми для агентов аналогичного назначения. Фактическое количество соединения по настоящему изобретению, т.е. активного ингредиента, зависит от множества факторов, таких, как тяжесть заболевания, подлежащего излечению, возраст и относительное состояние здоровья субъекта, активность используемого соединения, способ и форма введения и др. факторы. Терапевтически эффективные количества соединений формулы I могут изменяться от приблизительно 0,0005-10 мг на 1 кг массы тела реципиента в сутки, предпочтительно приблизительно 0,001-1 мг/кг/сут. Таким образом, при введении пациенту массой 70 кг доза составит от приблизительно 0,1 мг до 70 мг в сутки. В общем случае, соединения по настоящему изобретению вводят в виде фармацевтических композиций одним из следующих способов: пероральным, системным (например, трансдермальным, интраназальным или с использованием суппозитория), или парентеральным (например, внутримышечным, внутривенным или подкожным) способом введения. Предпочтительным способом введения является пероральный при соответствующей суточной схеме приема лекарственного средства, которая назначается в соответствии с тяжестью заболевания. Композиции могут иметь форму таблеток, пилюль, капсул, полутвердых составов, порошков, составов для замедленного высвобождения, растворов, суспензий, эликсиров, аэрозолей или любых других пригодных композиций. Выбор состава зависит от множества факторов, таких, как способ введения лекарственного средства (например, для перорального введения предпочтительны составы в форме таблеток, пилюль или капсул) и биодоступности активного соединения. В последнее время разработаны фармацевтические составы прежде всего для лекарственных средств, характеризующихся низкой биодоступностью. Способ основан на том, что биодоступность можно повысить благодаря увеличению площади поверхности, т.е. за счет снижения размеров частиц. Например, в US 4107288 описан фармацевтический состав, содержащий частицы размером от 10 до 1000 нм, в которых активное соединение фиксировано на матрице из сшитого полимера. В US 5145684 описано получение фармацевтического состава, в котором лекарственное соединение измельчают до наночастиц (средний размер частиц составляет 400 нм) в присутствии модификатора поверхности, а затем диспергируют в жидкой среде с образованием фармацевтического состава, обладающего существенно более высокой биодоступностью. В общем случае, композиции включают соединение формулы I в комбинации по меньшей мере с одним фармацевтически примлемым эксципиентом. Пригодные эксципиенты являются нетоксичными, способствующими введению и не оказывающими отрицательного действия на лечебные свойства соединения формулы I. Такой эксципиент может представлять собой твердый, жидкий, полужидкий или, в случае аэрозольной композиции, газообразный эксципиент, обычно известный специалисту в данной области. Твердые фармацевтические эксципиенты включают крахмал, целлюлозу, тальк, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое обезжиренное молоко и т.п. Жидкие и полутвердые эксципиенты выбирают из ряда: глицерин, пропиленгликоль, вода, этанол и различные масла, включая минеральные, животные, растетльные или синтетические масла, например, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Предпочтительные жидкие носители, прежде всего для инъеционных растворов, включают воду, солевой раствор, водный раствор декстрозы и гликоли. Для диспергирования соединения по настоящему изобретению в аэрозольной форме используются сжатые газы. Инертными газами, пригодными для этих целей, являются азот, диоксид углерода и т.п. Прочие фармацевтически приемлемые носители и их составы описаны в книге Remington’s Pharmaceutical Sciences, ed. by E.W.Martin, Mack Publishing Company, 18th edition (1990). Содержание сосединения в составе может изменяться в диапазоне, известном специалисту в данной области. Обычно состав содержит от приблизительно 0,01 до приблизительно 99,99 мас.% соединения формулы I в расчете на общую массу состава с учетом массы одного или более пригодных фармацевтических эксципиентов. Предпочтительно содержание соединения составляет приблизительно 1-80 мас.%. Типичные фармацетические составы, содержащие соединение формулы I, описаны в примере 8. Примеры Приведенные ниже препараты и примеры приведены с целью предоставить возможность специалисту в данной области более подробно ознакомится с предлагаемым изобретением и использовать его на практике. Примеры приведены с целью иллюстрации и не ограничивают объема изобретения. Препарат 1 Метиловый эфир [(4-хлорбутил)тио]уксусной кислоты
К раствору метилтиогликолята (10,0 мл, 113 ммолей) в тетрагидрофуране (200 мл) при -78°С в атмосфере аргона по каплям добавляли н-бутиллитий (2,5 М раствор в гексане, 45 мл, 113 ммолей). После выдерживания при -78°С в течение 1 ч к опалесцирующему раствору быстро добавляли 1-бром-4-хлорбутан (13,0 мл, 113 ммолей) и нагревали при комнатной температуры в течение ночи. Реакционную смесь выливали в воду и гексан, промывали холодным раствором гидроксида натрия (0,1 М) и насыщенным раствором хлорида аммония, органический слой сушили над безводным сульфатом натрия. Летучие компоненты удаляли, остаток очищали хроматографией (элюент:гексан/этилацетат, 8:1), при этом получали метиловый эфир [(4-хлорбутил)тио]уксусной кислоты (10,7 г, 54,5 ммоля) в виде бесцветной жидкости. МС (EI): m/z 198 (М++37Cl), 196 (M++35Cl). Препарат 2 Метиловый эфир 4-[(2-хлорэтил)тио]масляной кислоты
Раствор 4-меркаптомасляной кислоты (3,85 г, 20 ммолей) в изопропаноле (70 мл) при 0°С в течение 20 мин обрабатывали четырьмя порциями гидрида натрия (95%, всего 1,56 г, 65 ммолей) и нагревали до комнатной температуры. Затем быстро добавляли 1-бром-2-хлорэтан (11 мл, 128 ммолей) и полученную суспензию интенсивно перемешивали в течение 2 сут. Летучие компоненты удаляли, остаток распределяли между разбавленной (5%) уксусной кислотой и этилацетатом. Объединенные органические экстракты промывали солевым раствором и сушили над сульфатом натрия. Экстракт фильтровали и летучие компоненты удаляли в вакууме. Остаток растворяли в метаноле (60 мл) и охлаждали до 0°C в атмосфере аргона. Затем добавляли по каплям тионилхлорид (5 мл, 69 ммоля) и раствор перемешивали при комнатной температуре. Через 2-3 ч летучие компоненты удаляли, добавляли толуол и удаляли летучие компоненты. Остаток очищали хроматографией, при этом получали метиловый эфир 4-[(2-хлорэтил)тио]масляной кислоты (2,93 г, 14 ммолей) в виде бесцветного масла. МС (NH3): m/z 199 (M+1++37Cl), 197 (M-1++35Cl). Препарат 3 Этиловый эфир Z-4-хлорбут-2-енилоксиуксусной кислоты
К раствору гидрида натрия (1,17 г, 48 ммолей) в 50 мл ДМФА при 0°С в атмосфере азота добавляли этилгликолят (4,5 мл, 48 ммолей) и реакционную смесь перемешивали в течение 1 ч. К смеси быстро добавляли Z-1,4-дихлор-2-бутен (7,6 мл, 72 ммоля), нагревали до комнатной температуры и перемешивали в течение ночи. Затем реакционную смесь выливали в воду, экстрагировали диэтиловым эфиром, сушили и концентрировали. Полученный продукт очищали хроматографией, при этом получали 2,7 г этилового эфира Z-(4-хлорбут-2-енилокси)уксусной кислоты в виде неочищенного масла. Препарат 4 Этиловый эфир (4-бромбутокси)уксусной кислоты
К раствору гидрида натрия в 40 мл ДМФА при 0°С и при перемешивании медленно добавляли этилгликолят (5 г, 48 ммолей). Через 1 ч добавляли 1,4-дибромбутан (8,6 мл, 72 ммоля) и реакционную смесь перемешивали в течение еще 2 ч. Затем смесь нагревали до комнатной температуры в течение 3 ч, добавляли раствор бикарбоната натрия, органический слой сушили над безводным сульфатом натрия и концентрировали. Остаток очищали хроматографией, при этом получали бесцветное масло. Пример 1 4-[(2-{(2R)-2-[(1Е,3S)-3-Гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляная кислота
Стадия 1 Метиловый эфир 4-({2-[(5R)-5-(гидроксиметил)-2-оксопирролидин-1-ил]этил}тио)масляной кислоты
К раствору (5R)-5-(гидроксиметил)пирролидин-2-она (фирма Aldrich, 8,86 г, 77 ммолей) в 70 мл хлороформа при комнатной температуре в атмосфере аргона добавляли этилвиниловый эфир (10,4 мл, 108 ммолей) и безводную трифторуксусную кислоту (0,2 мл) в качестве катализатора. Реакционную смесь перемешивали в течение 3 ч, распределяли между хлористым метиленом (150 мл) и водным раствором бикарбоната натрия (150 мл). Затем фазы разделяли, органическую фазу сушили (К2СО3) и фильтровали. Фильтрат упаривали при пониженном давлении, при этом получали 14,7 г (5R)-5-(1-этоксиэтоксиметил)пирролидин-2-она. К раствору (5R)-5-(1-этоксиэтоксиметил)пирролидин-2-она (3,31 г, 17,7 ммоля) в 50 мл безводного диметилформамида при 0°С в атмосфере аргона добавляли иодид калия (3,22 г, 19,4 ммоля) и гидрид натрия (95%, 0,44 г, 17,7 ммоля). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 30 мин. Затем к смеси добавляли метиловый эфир 4-[(2-хлорэтил)тио]масляной кислоты (3,46 г, 17,7 ммоля) в 5 мл безводного диметилформамида, нагревали до 50°С и перемешивали в течение 40 ч. Летучие компоненты удаляли перегонкой в вакууме, к неочищенному остатку добавляли этилацетат (150 мл) и водный раствор хлорида аммония (50 мл). Фазы разделяли, этилацетатный раствор сушили (над безводным Na2SO4) и упаривали при пониженном давлении. К раствору защищенного эфира (17,7 ммоля) в 70 мл безводного метанола при комнатной температуре в атмосфере аргона добавляли моногидрат пара-толуолсульфоновой кислоты (0,34 г, 1,8 ммоля) и реакционную смесь перемешивали в течение 5 ч. К смеси добавляли водный раствор бикарбоната натрия (50 мл) и упаривали при пониженном давлении. Затем добавляли этанол (50 мл) и раствор снова упаривали. Полученный материал очищали хроматографией на колонке, при этом получали метиловый эфир 4-({2-[(5R)-5-(гидроксиметил)-2-оксопирролидин-1-ил]этил}тио)масляной кислоты в виде масла желто-коричневого цвета (0,17 г, МС: 275 (М+). Стадия 2 Метиловый эфир 4-({2-[(2R)-2-формил-5-оксопирролидин-1-ил]этил}тио)масляной кислоты
Раствор безводного диметилсульфоксида (0,21 мл, 2,7 ммоля) в 5 мл хлористого метилена в атмосфере аргона охлаждали до -70°С и по каплям добавляли раствор трифторуксусного ангидрида (0,17 мл, 1,2 ммоля) в 1 мл хлористого метилена. Реакционную смесь перемешивали в течение 15 мин при указанной температуре, а затем добавляли раствор метилового эфира 4-({2-[(5R)-5-(гидроксиметил)-2-оксопирролидин-1-ил]этил}тио)масляной кислоты (0,15 г, 0,55 ммоля) в 2 мл хлористого метилена. Раствор перемешивали и выдерживали при температуре -70°С в течение 20 мин, а затем добавляли триэтиламин (0,27 мл, 1,9 ммоля). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 30 мин и реакцию останавливали добавлением разбавленной фосфорной кислоты (2%, 50 мл) и хлористого метилена (25 мл). Фазы разделяли, объединенные органические слои сушили (Na2SO4) и упаривали при пониженном давлении, при этом получали метиловый эфир 4-({2-[(2R)-2-формил-5-оксопирролидин-1-ил]этил}тио)масляной кислоты, который использовали на следующей стадии. Стадия 3 Метиловый эфир 4-[(2-{(5R)-2-оксо-5-[(1Е)-3-оксоокт-1-енил]пирролидин-1-ил}этил)тио]масляной кислоты
К раствору гидрида натрия (95%, 14 мг, 0,58 ммоля) в 3 мл безводного 1,2-диметоксиэтана при 0°С в атмосфере аргона добавляли диметиловый эфир 2-оксогептилфосфоната (ACROS, 0,13 мл, 0,61 ммоля). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 2 ч. Затем реакционную смесь охлаждали до 0°C и добавляли раствор метилового эфира 4-({2-[(2R)-2-формил-5-оксопирролидин-1-ил]этил}тио)масляной кислоты (0,55 ммоля) в безводном 1,2-диметоксиэтане. Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 2 ч, а затем реакцию останавливали добавлением насыщенного раствора хлорида аммония. К реакционной смеси добавляли этилацетат (50 мл) и смесь распределяли между двумя фазами. Органический раствор промывали солевым раствором, сушили (Na2SO4) и упаривали при пониженном давлении, остаток очищали хроматографией на колонке (SiO2, элюент: EtOAc/гексан, 1:1), при этом получали метиловый эфир 4-[(2-{(5R)-2-оксо-5-[(1Е)-3-оксоокт-1-енил]пирролидин-1-ил}этил)тио]масляной кислоты в виде масла, который непосредственно использовали на следующей стадии. Стадия 4 Метиловый эфир 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил1-5-оксопирролидин-1-ил}этил)тио]масляной кислоты
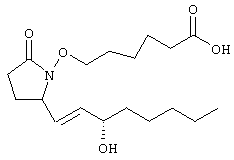
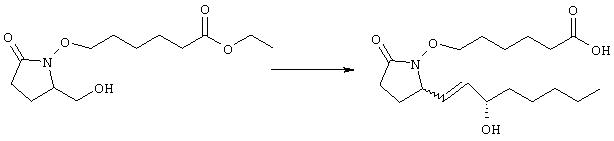
Раствор метилового эфира 4-[(2-{(5R)-2-оксо-5-[(1Е)-3-оксоокт-1-енил]пирролидин-1-ил}этил)тио]масляной кислоты (0,039 г, 0,1 ммоля) в 2 мл безводного толуола при температуре -30°С добавляли по каплям к раствору (R)-2-метил-CBS-оксазаборолидина (1 М в толуоле, 0,05 мл, 0,05 ммоля) и комплекса боран-метилсульфид (10 М, 0,01 мл, 0,1 ммоля). Реакционную смесь перемешивали при -30°С в течение 7 ч, добавляли раствор хлористоводородной кислоты в метаноле (2 М, 1-2 мл), нагревали до комнатной температуры и растворители удаляли при пониженном давлении. Остаток очищали хроматографией, при этом получали 11 мг метилового эфира 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляной кислоты в виде масла, который непосредственно использовали на следующей стадии. Стадия 5 4-[(2-{(2R)-2-[(1Е,3S)-3-Гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляная кислота
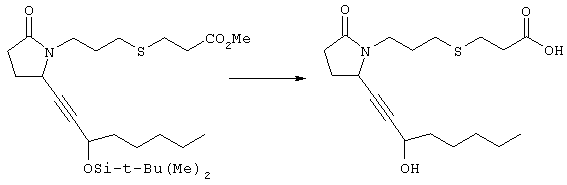
К раствору метилового эфира 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляной кислоты (0,011 г, 0,03 ммоля) в 2 мл метанола при комнатной температуре в атмосфере аргона добавляли водный раствор гидроксида натрия (1 М, 4-5 капель). Реакционную смесь перемешивали в течение 4 ч, упаривали при пониженном давлении и обрабатывали водным раствором хлористоводородной кислоты (1 М) до кислотной реакции. Остаток разбавляли водой (10 мл) и экстрагировали этилацетатом. Органический раствор промывали солевым раствором, сушили (Na2SO4) и упаривали, при этом получали 7,5 мг 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляной кислоты. МС: m/z 357 (М+). 1H ЯМР (CDCl3, 300 МГц,) частичный спектр: 5,74 (dd, J 5,7, 15,6 Гц, 1Н), 5,53 (dd, J 8,2, 15,6 Гц, 1Н), 4,11-4,20 (m, кажущийся q, J 6,0 Гц, 2Н), 3,62-3,81 (m dd, J 2,4, 10,2 Гц, 2Н), 3,08-3,20 (m, 1Н). Следующие соединения общей формулы I получали аналогично тому, как описано в примере 1, при замене соответствующих реагентов и стадий: {4-[2-[R-(3-гидроксиокт-1Е-енил)-5-оксопирролидин-1-ил]бутилсульфанил}уксусную кислоту (2), МС (ES): m/z 357 (M+), получали при замене метилового эфира 4-[(2-хлорэтил)тио]масляной кислоты (на стадии 1) на метиловый эфир [(4-хлорбутил)тио]уксусной кислоты, с последующим восстановлением борогидридом натрия на стадии 4, {4-[2-[R-(1Е-3S-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутилсульфанил} уксусную кислоту (3), МС (ES): m/z 357 (M+), получали при замене указанного выше реагента (на стадии 1) на метиловый эфир [(4-хлорбутил)тио]уксусной кислоты, {4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси} уксусную кислоту (4), МС: m/z 341 (M+), получали при замене указанного выше реагента (на стадии 1) на этиловый эфир (4-бромбутилокси)уксусной кислоты, а затем при замене диметилового эфира 2-оксогептилфосфоновой кислоты (на стадии 3) на диметиловый эфир [2-(5-трифторметилфуран-2-ил)-2-оксоэтил]фосфоновой кислоты (полученный, как описано в примере 5), получали (4-{(R)-2-[(Е)-3-гидрокси-3-(5-трифторметилфуран-2-ил)пропенил]-5-оксопирролидин-1-ил}бутилсульфанил)уксусную кислоту (5), МС: m/z 422 (M+), 4-{2-[(R)-2-((S)-(E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этансульфинил}масляную кислоту (6), МС: m/z 374 (M+), получали после обработки метилового эфира 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляной кислоты при -20°С раствором 3-хлорпербензойной кислоты в дихлорметане и последующего гидролиза, как описано на стадии 5, 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этансульфонил} масляную кислоту (7), МС: m/z 390 (M+), получали окислением метилового эфира 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляной кислоты при 0°С суспензией окислителя OXONE® в водном метаноле. Пример 2 {(Z)-4-[(R)-2-((E)-3-Гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси}уксусная кислота
Стадия 1 Этиловый эфир [(2)-4-((R)-2-гидроксиметил-5-оксопирролидин-1-ил)бут-2-енилокси] уксусной кислоты
К раствору (5R)-5-(1-этоксиэтоксиметил)пирролидин-2-она (2,5 г, 13,4 ммоля) в 10 мл безводного диметилформамида (ДМФА) при 0°С в атмосфере аргона добавляли раствор гидрида натрия (95%, 0,32 г, 13,4 ммоля) в 40 мл ДМФА. Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 60 мин. Затем добавляли раствор иодида калия (2,2 г, 13,4 ммоля) и этиловый эфир Z-4-хлорбут-2-еноксиуксусной кислоты (2,7 г, 21,3 ммоля) в 5 мл ДМФА, реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Затем добавляли насыщенный раствор NaHCO3, раствор экстрагировали, экстракт промывали солевым раствором, сушили (безводный Na2SO4) и упаривали при пониженном давлении. К раствору защищенного эфира (1,8 г, 5,24 ммоля) в 20 мл безводного метанола при комнатной температуре в атмосфере аргона добавляли моногидрат пара-толуолсульфоновой кислоты (0,1 г, 5,5 ммоля). Реакционную смесь перемешивали в течение ночи, затем добавляли водный раствор бикарбоната натрия (50 мл), экстрагировали, сушили и концентрировали, остаток очищали хроматографией, при этом получали этиловый эфир [(Z)-4-((R)-2-гидроксиметил-5-оксопирролидин-1-ил)бут-2-енилокси]уксусной кислоты. Стадия 2 Этиловый эфир [(Z)-4-((R)-2-формил-5-оксопирролидин-1-ил)бут-2-енилокси]уксусной кислоты
Раствор безводного диметилсульфоксида (0,45 мл, 5,7 ммоля) в 5 мл хлористого метилена в атмосфере азота охлаждали до -78°С и по каплям добавляли раствор трифторуксусного ангидрида (0,68 мл, 4,8 ммоля) в 2 мл хлористого метилена. Реакционную смесь перемешивали при указанной температуре в течение 15 мин, а затем добавляли этиловый эфир [(Z)-4-((R)-2-гидроксиметил-5-оксопирролидин-1-ил)бут-2-енилокси]уксусной кислоты (0,62 г, 2,3 ммоля) в 20 мл хлористого метилена. Раствор перемешивали, выдерживали при температуре -70°С в течение 20 мин и добавляли триэтиламин (0,96 мл, 6,9 ммоля). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 30 мин, а затем реакцию останавливали добавлением раствора хлорида аммония. Фазы разделяли, объединенные органические слои промывали солевым раствором, сушили (Na2SO4) и концентрировали, остаток очищали хроматографией, при этом получали этиловый эфир [(Z)-4-((R)-2-формил-5-оксопирролидин-1-ил)бут-2-енилокси]уксусной кислоты, который использовали на следующей стадии. Стадия 3 Этиловый эфир {(Z)-4-[(R)-2-оксо-5-((Е)-3-оксоокт-1-енил)пирролидин-1-ил]бут-2-енилокси}уксусной кислоты
К раствору гидрида натрия (95%, 0,02 г, 0,78 ммоля) в 10 мл безводного 1,2-диметоксиэтана при 0°С в атмосфере азота добавляли диметиловый эфир 2-оксогептилфосфоновой кислоты (0,17 мл, 0,78 ммоля) и реакционную смесь перемешивали в течение 1 ч. Затем добавляли раствор этилового эфира [(Z)-4-((R)-2-формил-5-оксопирролидин-1-ил)бут-2-енилокси]уксусной кислоты (0,21 г, 0,78 ммоля) в 2 мл безводного 1,2-диметоксиэтана. Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 3 ч, а затем реакцию останавливали добавлением насыщенного раствора хлорида аммония. Реакционную смесь экстрагировали этилацетатом, органический слой промывали солевым раствором, сушили (Na2SO4) и упаривали, остаток очищали хроматографией, при этом получали этиловый эфир {(Z)-4-[(R)-2-оксо-5-((E)-3-оксоокт-1-енил)пирролидин-1-ил] бут-2-енилокси} уксусной кислоты в виде масла, который непосредственно использовали на следующей стадии. Стадия 4 Этиловый эфир {(Z)-4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси}уксусной кислоты
Раствор этилового эфира {(Z)-4-[(R)-2-оксо-5-((E)-3-оксоокт-1-енил)пирролидин-1-ил]бут-2-енилокси}уксусной кислоты (0,7 г, 1,97 ммоля) в 15 мл этанола перемешивали при 0°С и добавляли 0,082 г NaBH4. Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 7 ч, а затем добавляли раствор хлористоводородной кислоты в метаноле (2 М, 1-2 мл) и экстрагировали этилацетатом. Органическую фазу сушили и концентрировали, остаток очищали хроматографией, при этом получали этиловый эфир {(Z)-4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси} уксусной кислоты, который непосредственно использовали на следующей стадии. Стадия 5 {(Z)-4-[(R)-2-((Е)-3-Гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси}уксусная кислота
К раствору этилового эфира {(Z)-4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси}уксусной кислоты (0,22 г, 0,6 ммоля) в смеси 5 мл метанола и 5 мл ТГФ при комнатной температуре в атмосфере азота добавляли раствор гидроксида лития (0,1 г, 2,4 ммоля) в 3 мл воды. Реакционную смесь перемешивали при 45°С в течение ночи, охлаждали до комнатной температуры и обрабатывали раствором хлористоводородной кислоты (1 М) до кислотной реакции. Остаток разбавляли водой (10 мл) и экстрагировали этилацетатом. Органический раствор промывали солевым раствором, сушили (Na2SO4) и упаривали, при этом получали {(Z)-4-[(R)-2-((E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бут-2-енилокси}уксусную кислоту. МС (ES): m/z 339 (М+). Пример 3 {4-[(R)-2-((Е)-(S)-3-Гидроксиокт-1 -енил)-5-оксопирролидин-1 -ил]бутокси}уксусная кислота
Стадия 1 Метиловый эфир {4-[(R)-2-((Е)-(S)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси} уксусной кислоты
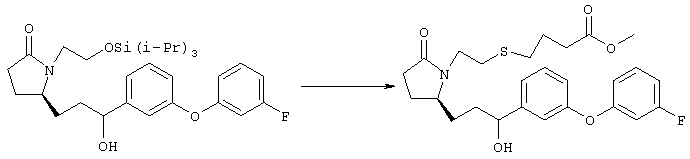
Раствор метилового эфира {4-[(R)-2-оксо-5-((Е)-3-оксоокт-1-енил)пирролидин-1-ил]бутокси}уксусной кислоты, полученного, как описано в примере 1 (250 мг, 0,71 ммоля), в 10 мл безводного толуола при -26°С добавляли по каплям к раствору (R)-метил-CBS-оксазаборолидина (фирма Aldrich, 1 M раствор в толуоле, 0,35 мл) и комплекса боран-метилсульфид (10 M, 0,07 мл). Реакционную смесь перемешивали в атмосфере азота при -26°С в течение 7 ч, затем добавляли раствор хлористоводородной кислоты в метаноле (2 M, 1-2 мл). Раствор нагревали до комнатной температуры и растворители удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле, при этом получали 11 мг метилового эфира {4-[(R)-2-((Е)-(S)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусной кислоты в виде масла, который непосредственно использовали на следующей стадии. Стадия 2 {4-[(R)-2-((Е)-(S)-3-Гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусная кислота
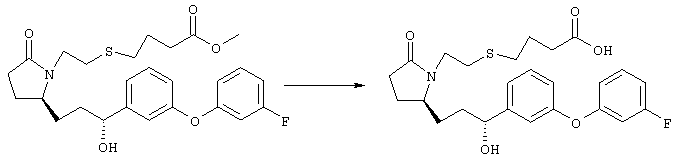
К раствору метилового эфира {4-[(R)-2-((Е)-(S)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусной кислоты (0,14 г, 0,4 ммоля) в смеси 2 мл метанола и 2 мл ТГФ при комнатной температуре в атмосфере азота добавляли раствор гидроксида лития (0,066 г, 1,6 ммоля) в 1 мл воды. Реакционную смесь перемешивали при 45°С в течение ночи, охлаждали до комнатной температуры и добавляли 1 М соляную кислоту до кислотного значения pH. Остаток разбавляли водой (10 мл) и экстрагировали этилацетатом. Органический раствор промывали солевым раствором, сушили (Na2SO4) и упаривали, остаток очищали хроматографией, при этом получали 28 мг {4-[(R)-2-((Е)-(S)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусной кислоты. МС (ES): m/z 341 (М+1) 341. Соединения общей формулы I получали по методикам, описанным в примере 3, при замене соответствующих реагентов: (4-{(R)-2-[(S)-(Е)-3-гидрокси-4-(3-трифторметилфенил)бут-1-енил]-5-оксопирролидин-1-ил}бутокси)уксусную кислоту, МС: m/z 430 (М), получали при использовании на стадии 1 этилового эфира {4-[(R)-2-оксо-5-[(Е)-3-оксо-4-(3-трифторметилфенил)бут-1-енил]пирролидин-1-ил]бутокси}уксусной кислоты, (4-{(R)-2-[(Е)-3-гидрокси-3-(2′-метилбифенил-3-ил)пропенил]-5-оксопирролидин-1-ил}бутокси)уксусную кислоту, МС: m/z 438 (М+1), получали при использовании на стадии 1 этилового эфира {4-[(R)-2-оксо-5-[(Е)-3-(2′-метилбифенил-3-ил)-3-оксопроп-1-енил]пирролидин-1-ил]бутокси}уксусной кислоты и замене (R)-2-метил-CBS-оксазаборолидина/борана-диметилсульфида на боргидрид натрия-хлорид церия (III), (4-{(R)-2-[(Е)-3-(4′-хлорбифенил-3-ил)-3-гидроксипропенил]-5-оксопирролидин-1-ил}бутокси)уксусную кислоту (12), МС: m/z 459 (М+1), получали при использовании на стадии 1 этилового эфира {4-[(R)-2-оксо-5-[(Е)-3-(4′-хлорбифенил-3-ил)-3-оксопроп-1-енил]пирролидин-1-ил]бутокси}уксусной кислоты. Пример 4 4-{2-[(R)-2-((Е’)-3-Гидрокси-3-пентилокт-1-енил)-5-оксопирролидин-1-ил]этансульфанил}масляная кислота
Стадия 1
Раствор енона (полученного, как описано в примере 1, стадия 3, 1,52 г) в тетрагидрофуране (38 мл) при -24°С обрабатывали бромидом н-пентилмагния (фирма Aldrich, 2 М раствор в эфире, 1,8 мл). Через 1 ч смесь выливали в водный раствор хлорида аммония и экстрагировали этилацетатом. Экстракт сушили, фильтровали и упаривали, остаток очищали хроматографией на силикагеле (элюент: метанол/этилацетат, 5:95), при этом получали требуемый спирт (780 мг). Стадия 2
Эфир (273 мг) растворяли в тетрагидрофуране (6 мл) и воде (1,2 мл), обрабатывали LiOH/H2O (126 мг) и перемешивали при комнатной температуре в течение 16 ч. Затем смесь распределяли между водой и этиловым эфиром, водный слой подкисляли ледяной уксусной кислотой и экстрагировали этилацетатом. Экстракты объединяли, сушили над сульфатом натрия и фильтровали. Указанную в заголовке кислоту (215 мг) получали в виде масла. МС: m/z 428 (М+1). Соединения общей формулы I получали по методике, описанной в примере 4, при замене соответствующих реагентов: 4-{2-[(R)-2-((Е)-3-гидрокси-3-метилокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}масляную кислоту, МС: m/z 372 (М+1), получали при замене на стадии 1 бромида н-пентилмагния на бромид метилмагния. Препарат 5 Диметиловый эфир {2-[3-(3-фторфенокси)фенил]-2-оксоэтил}фосфоновой кислоты
Стадия 1
Суспензию метилового эфира 3-гидроксибензойной кислоты (5,4 г, 35,5 ммоля), 3-фторфенилбороновой кислоты (5,5 г, 35, 5 ммоля), ацетата меди (7,1 г, 35,5 ммоля), молекулярных сит 3 Å (9 г), пиридина (12 мл, 145 ммолей) в дихлорметане (220 мл) перемешивали при комнатной температуре и атмосферном давлении в течение 11 сут. Затем смесь фильтровали через целит и из фильтрата удаляли летучие компоненты. Остаток очищали хроматографией на колонке с силикагелем (элюент: гексан/этилацетат, 5:1), требуемый эфир (3,68 г) использовали на следующей стадии. Стадия 2
Раствор диметилового эфира метилфосфоновой кислоты (4,0 мл, 37,5 ммоля) в тетрагидрофуране (100 мл) охлаждали до -78°С в атмосфере аргона, обрабатывали н-бутиллитием (15,0 мл, 2,5 М раствор в гексане, 37,5 ммоля) и перемешивали в течение 45 мин. К полученному раствору при -78°С добавляли раствор эфира, полученного на стадии 1, (4,62 г, 18,7 ммоля), в тетрагидрофуране (15 мл), и полученную смесь перемешивали при 0°С в течение 1 ч. Образовавшийся раствор желтого цвета распределяли между водным раствором хлорида аммония (100 мл) и этиловым эфиром (200 мл). Органическую фракцию промывали водой (3×30 мл) и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и удаляли летучие компоненты в вакууме, при этом получали требуемый 1H ЯМР (CDCl3, 300 МГц): Пример 5 4-[2-((S)-2-{(R)-3-[3-(3-Фторфенокси)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляная кислота
Стадия 1
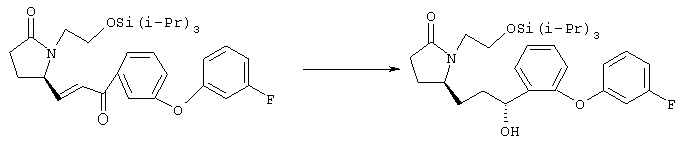
Раствор енона (950 мл, 1,8 ммоля) в этилацетате (70 мл) при комнатной температуре обрабатывали 10% палладием на угле (150 мг) и интенсивно перемешивали в атмосфере газообразного водорода при атмосферном давлении в течение 90 мин. Суспензию фильтровали через целит, летучие компоненты удаляли в вакууме. Остаток (945 мг) растворяли в безводном толуоле (10 мл) и добавляли к предварительно перемешанному при 0°С раствору (S)-2-метил-CBS-оксазаборолидина (фирма Aldrich, 0,20 мл, 1 М раствор в толуоле) и комплекса боран-диметилсульфид (0,25 мл, 5 М раствор в этиловом эфире) в безводном толуоле (20 мл). Раствор желтого цвета перемешивали при 0°С в течение 1,3 ч и обрабатывали соляной кислотой (1 мл, 2 М раствор в метаноле). Летучие компоненты удаляли на роторном испарителе, добавляли 10 мл метанола, летучие компоненты удаляли, добавляли 20 мл толуола и снова удаляли летучие компоненты. Полученную пасту белого цвета очищали хроматографией на силикагеле. Требуемый спирт (620 мг, 1,2 ммоля) элюировали в градиенте 2-пропанола (2-4%) в гексане/этилацетате, 2:1. Стадия 2
Раствор силилового эфира (620 мг, 1,2 ммоля) в тетрагидрофуране (5 мл) при комнатной температуре обрабатывали гидратом фторида тетрабутиламмония (443 мг, 1,4 ммоля) и перемешивали в течение 2,5 ч. Затем смесь разбавляли 10 мл гексана и наносили на слой силикагеля. Требуемый диол (380 мг) элюировали 5-10% этанола в этилацетате и получали в виде стекловидного вещества. Диол (375 мг) растворяли в тетрагидрофуране (10 мл), охлаждали до -20°С в атмосфере аргона и последовательно обрабатывали триэтиламином (0,17 мл) и метансульфонилхлоридом (0,08 мл), при этом получали суспензию. В отдельном сосуде смесь безводного метанола (1 мл) и безводного тетрагидрофурана (5 мл) в атмосфере аргона обрабатывали трет-бутоксидом калия (фирма Aldrich, 3,0 мл, 1 М раствор в тетрагидрофуране) и слегка теплый раствор перемешивали в течение 10 мин. Затем к полученному раствору одной порцией добавляли Стадия 3
Раствор сложного эфира (350 мг, 0,72 ммоля) в метаноле (10 мл) обрабатывали гидроксидом натрия (0,5 мл, 5 М раствор) и перемешивали при комнатной температуре в течение 4-5 ч. Летучие компоненты удаляли в атмосфере азота и смесь распределяли между водой и этиловым эфиром. Водный слой подкисляли 12 М соляной кислотой и экстрагировали этилацетатом (4×15 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и летучие компоненты удаляли в вакууме, при этом получали требуемую кислоту (299 мг) в виде масла. МС: m/z 476 (М+1). 1H ЯМР (CDCl3, 300 МГц) (частичный спектр): Соединения общей формулы I получали аналогично тому, как описано в примере 5, при замене соответствующих реагентов и стадий: при использовании на стадии 1 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-он, при исключении стадии каталитического гидрирования и замене (S)-2-метил-CBS-оксазаборолидина на (R)-2-метил-CBS-оксазаборолидин и последующем использовании метилового эфира 4-меркапто-3-метилмасляной кислоты на стадии 2, получали 4-{2-[(R)-2-((S)-(E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-3-метилмасляную кислоту (16), МС: m/z 372 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира 4-меркапто-2-метилмасляной кислоты на стадии 2 получали 4-{2-[(R)-2-((S)-(E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-2-метилмасляную кислоту (17), МС: m/z 372 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира 4-меркапто-4-метил-масляной кислоты на стадии 2 получали 4-{2-[(R)-2-((S)-(E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-4-метилмасляную кислоту (18), МС: m/z 372 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира 4-меркапто-2-метилбутеновой кислоты на стадии 2 получали 4-{2-[(R)-2-((S)-(E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-2-метилбутеновую кислоту (19), МС: m/z 356 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира 2-[1-(меркаптометил)циклопропил]уксусной кислоты на стадии 2 получали (1-{2-[(R)-2-((S)-(E)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанилметил}циклопропил)уксусную кислоту (20), МС: m/z 384 (M+1), при использовании на стадии 2 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира тиогликолевой кислоты получали 5-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}уксусную кислоту (21), МС: m/z 330 (М+1), при использовании на стадии 2 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира 3-меркаптопропионовой кислоты, и липазы на стадии 3 получали 3-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}пропионовую кислоту (22), МС: m/z 344 (М+1), при использовании на стадии 2 (R)-5-{(Е)-3-оксоокт-1-енил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и метилового эфира 5-меркаптопентановой кислоты, получали 5-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}пентановую кислоту кислоту (23), МС: m/z 372 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-[3-(4′-хлор-2′-метилфенил)фенил]-3-оксопропил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она получали 4-[2-((S)-2-{(R)-3-[3-(4′-хлор-2′-метилфенил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляную кислоту (24), МС: m/z 491 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-[3-(4′-хлор-2′-метилфенил)фенил]-3-оксопропил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и (R)-2-метил-CBS-оксазаборолидина получали 4-[2-((S)-2-{(S)-3-[3-(4′-хлор-2′-метилфенил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил] масляную кислоту (25), МС: m/z 491 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-[3-(2′,4′-дифторфенил)фенил]-3-оксопропил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и (S)-2-метил-CBS-оксазаборолидина получали 4-[2-((S)-2-{(R)-3-[3-(3-(2′,4′-дифторфенил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляную кислоту (26), МС: m/z 478 (М+1), при использовании на стадии 1 (R)-5-{(Е)-3-[3-(4′-метокси-2′-метилфенил)фенил]-3-оксопропил}-1-(2-триизопропилсилилоксиэтил)пирролидин-2-она и (S)-2-метил-CBS-оксазаборолидина получали 4-[2-((S)-2-{(R)-3-[3-(3-(4′-метокси-2′-метил)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил] масляную кислоту (27), МС: m/z 486 (М+1). Пример 6 6-[2-((S)-(Е)-3-Гидроксиокт-1-енил)-5-оксопирролидин-1-илокси]гексановая кислота
Стадия 1
Диметиловый эфир 2-(O-бензилоксиамино)глутаровой кислоты (4,7 г, 16,7 ммоля), полученной из диметилового эфира 2-оксоглутаровой кислоты при использовании методов, описанных в статье C.Fuganti и др., J.Org. Chem., 49, 543-546, растворяли в сухом тетрагидрофуране (170 мл) и охлаждали на ледяной бане. Затем медленно добавляли хлорид изопропилмагния (8,35 мл 2М раствора в тетрагидрофуране) и реакционную смесь перемешивали при нагревании до комнатной температуры в течение 16 ч. Реакцию останавливали добавлением насыщенного раствора хлорида аммония и воды и смесь экстрагировали этилацетатом. Слои разделяли и водной слой экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (230-400 меш) (элюент: 30% этилацетат/гексан). Фракции, содержащие продукт, собирали, при этом получали 2,85 г этилового эфира 1-бензилокси-5-пирролидинон-2-карбоновой кислоты в виде твердого вещества белого цвета. Этот способ был ранее описан для межмолекулярного амидирования (см. J.M.Williams и др., Tet.Lett, 36(31), 5461-5464). При использовании селективного восстановления этилового эфира 1-бензилокси-5-пирролидинон-2-карбоновой кислоты, как описано в статье М.Miller и др., J.Org. Chem., 47, 4928-4933, получали 1-бензилокси-5-гидроксиметил-2-пирролидинон. tпл. 121-123°С. МС: m/z 222 (М+1). Стадия 2
1-Бензилоксилактам (1,1 г, 4,97 ммоля) растворяли в сухом ДМФА (12 мл) и охлаждали на ледяной бане. К раствору добавляли имидазол (0,37 г, 5,5 ммоля), а затем трет-бутилдиметилсилилхлорид (0,97 г, 6,46 ммоля). Ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч. Затем добавляли этилацетат и воду, слои разделяли, органический слой дважды промывали 0,5 М HCl, водой и насыщенным раствором NaHCO3. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (230-400 меш) (элюент: 25-35% этилацетат/гексан). Фракции, содержащие продукт, концентрировали, при этом получали 1,54 г масла. Масло растворяли в 20% тетрагидрофуране/этаноле (20 мл), добавляли 1,9 г 10% Pd на угле и реакционную смесь перемешивали в атмосфере водорода (1 атм) в течение 2 ч. Реакционную смесь продували азотом, фильтровали через целит и концентрировали, при этом получали 1,1 г требуемого лактама гидроксамовой кислоты, МС: m/z 246 (М+1), который использовали без дополнительной очистки. Стадия 3
К сухому ДМФА (11 мл) добавляли иодид калия (измельченный, 0,89 г, 1,2 ммоля) и гидрид натрия (60% дисперсия в минеральном масле, 0,2 г, 1,1 ммоля). Смесь охлаждали на ледяной бане и по каплям добавляли раствор гидроксаматлактама (1,1 г, 4,47 ммоля) в 6 мл ДМФА. Реакционную смесь перемешивали в течение 10 мин и медленно добавляли раствор этилового эфира 6-бромгексановой кислоты (фирма Aldrich, 0,95 мл, 1,2 ммоля) в 5 мл ДМФА. Ледяную баню удаляли и реакционную смесь нагревали на бане при 50°С в течение 16 ч. Затем добавляли раствор хлорида аммония, воду и этилацетат, органический слой отделяли. Водный слой экстрагировали этилацетатом, объединенные органические слои промывали водой и солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток очищали хроматографией на силикагеле (элюент: 40% этилацетат/гексан), при этом получали 1,45 г этилового эфира в виде прозрачного масла. МС: m/z 388 (М+1). Стадия 4
К раствору силилового эфира (1,4 г, 1,44 ммоля), охлажденного на ледяной бане, в 18 мл сухого тетрагидрофурана добавляли фторид тетрабутиламмония (9,3 мл, 1 М раствор в ТГФ). Реакционную смесь перемешивали в течение 10 мин, ледяную баню удаляли и реакционную смесь перемешивали при КТ в течение 2 ч. Реакционную смесь охлаждали на ледяной бане и добавляли этилацетат, раствор хлорида аммония и воду. Слои разделяли и водный слой снова экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, упаривали и остаток очищали хроматографией на силикагеле (элюент: 3% метанол/дихлорметан), при этом получали гидроксиметилпроизводное (970 мг) в виде прозрачного масла. МС: m/z 274 Стадия 5
Индивидуальные диастереомеры разделяли на стадии восстановления енона (как описано в примере 3, стадия 1, с использованием реагента R-2-метил-CBS и с последующей хроматографией на силикагеле). Более полярный 12-диастереомер, 6-[2-((3S)-(Е)-3-гидроксиокст-1-енил)-5-оксопирролидин-1-илокси]гексановую кислоту, получали в виде твердого вещества белого цвета. tпл.87,9-89,4°С. МС: m/z 340 (М-1). Элементный анализ Рассчитано для C18H31NO5, %: С 63,32, Н 9,15, N 4,10. Найдено, %: С 63,08, Н 9,11, N 4,25. Менее полярный 12-эпимер, 6-[2-((3S)-(Е)-3-гидроксиокст-1-енил)-5-оксопирролидин-1-илокси]гексановую кислоту, получали в виде воскообразного твердого вещества белого цвета. tпл.54,7-60°С. МС: m/z 340 (М-1). Пример 7 3-{3-[2-(3-Гидроксиокст-1-инил)-5-оксопирролидин-1-ил]пропилсульфанил}пропионовая кислота
Стадия 1 и 2
Суспензию карбоната калия (5,9 г), сукцинимида (3,7 г), тетрабутиламмонийиодида (500 мг) и метилового эфира 7-хлор-4-тиагептановой кислоты (6,7 г) в диметилформамиде (100 мл) перемешивали при комнатной температуре в течение 44 ч. Смесь распределяли между водой (400 мл) и эфиром/гексаном (1:1, 4×100 мл) и сушили над сульфатом натрия. Летучие компоненты удаляли на роторном испарителе, неочищенную смесь очищали хроматографией на силикагеле (элюент:гексан/этилацетат, 3:2). Требуемый продукт (4,7 г) получали в виде масла. Сукцинимид (4,4 г, 17 ммолей) растворяли в метаноле (100 мл) и охлаждали до -5°С в атмосфере аргона. Затем одной порцией добавляли борогидрид натрия (911 мг, 24 ммоля) и по каплям в течение 3 ч добавляли соляную кислоту (2 М раствор в метаноле) с такой скоростью, чтобы температура смеси не превышала +5°С. Затем добавляли дополнительную порцию борогидрида натрия (275 мг, 7,2 ммоля), перемешивали в течение 4 ч и добавляли HCl/MeOH до pH 3-5. Летучие компоненты удаляли, остаток обрабатывали триметилортоформиатом (15 мл) и гидратом пара-толуолсульфоновой кислоты (700 мг) и перемешивали при КТ в течение приблизительно 18 ч. Летучие компоненты удаляли на роторном испарителе, остаток очищали хроматографией на силикагеле. Требуемый 5-метоксилактам (1,14 г) элюировали гексаном/ацетоном, 3:1. МС (ES): m/z 245 (М+ минус ОМе). Стадии 3, 4 и 5
Раствор 3-трет-бутилдиметилсилилокси-1-октина (4,0 г, 16,7 ммоля) в тетрагидрофуране (50 мл) охлаждали до -78°С и обрабатывали н-бутиллитием (фирма Aldrich, 7,4 мл, 2,5 М раствор в гексане) и нагревали до 0°С. Через 30 мин раствор снова охлаждали до -78°С и обрабатывали хлоридом три-н-бутилолова (фирма Aldrich, 4,8 мл). Охлаждающую баню удаляли и раствор желтого цвета перемешивали при комнатной температуре в течение 2 ч. Смесь распределяли между водным раствором хлорида аммония и гексаном (3×100 мл) и сушили над сульфатом натрия. Летучие компоненты удаляли на роторном испарителе в вакууме (2 мм рт. ст., КТ, 2 ч), при этом получали требуемый станнан (8,06 г, 15,2 ммоля). 5-Метилоксилактам (670 мг) и станнан (3,2 г) растворяли в безводном дихлорметане (10 мл) и охлаждали до 0°С в атмосфере аргона. Затем двумя порциями добавляли эфират трифторида бора (1,2 мл, 9,6 ммоля) и полученную суспензию перемешивали при комнатной температуре. Через 18 ч смесь распределяли между буферным раствором, pH 4, и дихлорметаном (2×50 мл) и сушили над сульфатом натрия. Требуемый алкин (77 мг) выделяли хроматографией на силикагеле (элюент: гексан/этилацетат, 3:2). Стадии 6 и 7
Силиловый эфир (77 мг) растворяли в тетрагидрофуране (5 мл) и уксусной кислоте (0,03 мл) при комнатной температуре, обрабатывали фторидом тетрабутиламмония (фирма Aldrich, 0,24 мл, 1 М раствор в ТГФ) и смесь перемешивали в течение 16 ч. Раствор разбавляли гексаном (10 мл) и наносили на слой силикагеля. Спирт (40 мг) элюировали этилацетатом/гексаном, 3:1, и суспендировали в 10 мл 10 мМ фосфатного буферного раствора (pH 6,5) при комнатной температуре. Суспензию обрабатывали липазой, тип VII (500 мг из С. rugosa, фирма Sigma), и интенсивно перемешивали в течение 4-5 ч. Смесь разбавляли этилацетатом (15 мл) и уксусной кислотой (0,5 мл) и наносили на слой целита. Осадок на фильтре промывали этилацетатом (40 мл), фильтрат упаривали, при этом получали указанную в заголовке кислоту в виде масла (5,9 мг), МС: m/z 356 (М+1). 1H ЯМР (CDCl3, 300 МГц) (частичный спектр): Пример 8 Ниже представлены фармацевтические составы, содержащие соединение формулы I. Состав таблетки Следующие ингредиенты смешивают до гомогенного состояния и прессуют таблетки с одной насечкой.
Состав капсулы Следующие ингредиенты смешивают до гомогенного состояния и смесью заполняют желатиновые капсулы с твердой оболочкой.
Состав суспензии Следующие ингредиенты смешивают до образования суспензии для перорального введения.
Состав раствора для инъекций Следующие ингредиенты смешивали до образования состава для инъекций
Пример 9 Определение функциональной активности рецептора ЕР4 (или ЕР2) с использованием метода анализа люциферазы Получение устойчивых трансфектированных клонов люциферазы ЕР4 Препарат кДНК простаноидного рецептора ЕР4, кодирующей полную цепь белка, субклонировали в соответствующих участках вектора экспрессии pcDNA.1(+)/Zeo млекопитающих (фирма Invitrogen). Последовательность, содержащую цАМФ-респонсивный элемент (CRE) и ген люциферазы, клонировали в вектор рХР1. Затем проводили одновременное трансфектирование клеток СНО препаратом pcDNA, содержащим EP4R, и препаратом рХР1, содержащим CRE-люциферазу, при соотношении ДНК 5:1 с использованием фугена (фирма Roche Molecular) в среде F-12 (фирма Gibco), содержащей 10% фетальную телячью сыворотку (фирмы Gibco), инактивированную нагреванием. Через 3 сут после трасфектирования среду культивирования заменяют на свежую среду, содержащую зеоцин. Культуру выдерживают в течение одного месяца до тех пор, пока не образуются устойчивые клоны. Метод определения цАМФ-зависимого гена люциферазы Функциональную активность лиганда-агониста ЕР4 определяли по связыванию с рецептором и образованию внеклеточного цАМФ. В данном случае уровень цАМФ определяют косвенным способом по трансляции репортерного гена люциферазы в ЕР4-люциферазных клонах. Клетки ЕР4-люциферазных клонов субклонировали в 200 мкл среды F-12 (фирма Gibco, BRL), содержащей 10% ФБС (фирма Gibco, BRL) и 25 мМ HEPES, в 96-луночных планшетах (фирма Packard) при концентрации 40000 клеток в лунке. Клетки культивировали при при 37°С в течение ночи в атмосфере СО2/воздух, 5%:95%, и на следующее утро заменяли среду. Клетки промывали дважды порциями по 100 мкл буферным раствором Хенкса и снова добавляли по 90 мкл среды F-12, содержащей 0,1% БСА. После предварительного инкубирования культуры при 37°С в течение от 1,5 до 3 ч в атмосфере СО2/воздух, 5%:95%, в лунки добавляли по 10 мкл раствора исследуемого соединения (в концентрации в 10 раз выше требуемой) и инкубирование продолжали при 37°С в течение еще 3 ч. Обычно для определения максимальной стимуляции люциферазы, опосредованной рецептором ЕР4, в каждый анализ включали 0,1 мкМ PGE-2 в качестве контроля полного агонистического действия. После завершения инкубирования среду выливали и лунки промокали досуха. Такие планшеты готовы для определения люциферазы. Количественное определение активности люциферазы Для количественного определения активности люциферазы использовали набор для анализа LucLite фирмы Packard. За 30 мин до завершения инкубирования субстрат LucLite и субстратный буферный раствор фирмы Packard уравновешивали при комнатной температуре. Субстрат растворяли в субстратном буферном растворе и смешивали переворачиванием пробирки. Для использования на следующей стадии смешивали равные объемы фосфатно-солевого буферного раствора Дальбекко (ДФСБ, фирма Gibco BRL), содержащего 1 мМ MgCl2 и 1 мМ CaCl2, и раствора субстрата. В каждую лунку 96-луночного планшета добавляли по 100 мкл смешанного раствора и планшет встряхивали при 300 об/мин на качалке для планшетов в течение 3 мин. Крышку планшета снимали и заменяли на герметизирующую пленку для планшетов (фирма Packard) для измерения радиоактивности в сцинтилляционном счетчике. Величину ЕС50 для соединения определяли при использовании четырехфакторной программы построения кривых KaleidaGraph. Пример 10 Анализ конкурентного связывания [3H]-PGE2 с рецептором rEP1, rEP2, rEP3 и rEP4 Культивирование клеток и трансфектирование Устойчивые трансфектированные клетки, экспрессирующие ЕР3, культивировали в среде F-12 (фирма Gibco), содержащей 10% фетальную телячью сыворотку, инактивированную нагреванием (фирма Gibco), и осаждали. Препарат кДНК простаноидного рецептора ЕР2 или ЕР4, кодирующей полную цепь белка, субклонировали в соответствующих участках вектора экспрессии pcDNA 3.1(+)/Zeo млекопитающих (фирма Invitrogen). Количественную шкалу вектора для трансфектирования определяли с использованием набора Qiagen Endo-Free Plasmid Maxi Kit и трансфектировали в клетки COS-7 с использованием FuGene 6 (фирма Roche Molecular), как описано в инструкциях фирмы Roche. Клетки COS-7 культивировали в среде DMEM (фирма Gibco), содержащей 10% фетальную телячью сыворотку, инактивированную нагреванием (фирма Gibco), и гентамицин (фирма Gibco), и через 72 ч после трансфектирования клетки собирали. Клетки центрифугировали, осадок промывали буферным раствором ФСБ (фирма Gibco), снова осаждали и быстро замораживали в сухом льду/этаноле и непосредственно использовали для выделения мембранной фракции. Мембранная фракция Все стадии выделения мембранной фракции проводили при 4°С. Клетки COS-7, трансфектированные простаноидным рецептором, или устойчивые трансфектированные клетки СНО гомогенизировали в буферном растворе для анализа (см. ниже) с использованием гомогенизатора Polytron (фирмы Brinkman) и центрифугировали при 48000 g в течение 30 мин. Осадок ресуспендировали в буферном растворе для анализа и обрабатывали в ультразвуковом дезинтеграторе Branson. Концентрацию белка определяли с использованием набора для определения белка DC Protein Assay (фирма BioRad) по методике, описанной в инструкциях фирмы-производителя. Фракцию хранили при -80°С. Анализ связывания с простаноидным рецептором Для анализа использовали методики анализа конкурентного аффинного связывания ЕР2, ЕР3 и ЕР4, описанные в статье M.Abramovitz и др., The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs, Biochimica et Biophysica Acta, 1483, 285-293 (2000). Анализ связывания проводили в конечном объеме 0,2 мл следующих буферных растворов для анализа: 20 мМ HEPES, 1 мМ ЭДТА и 10 мМ MgCl2 (рН 7,4) (для ЕРЗ) или 10 мМ MES, 10 мМ MnCl2 и 1 мМ ЭДТА (рН до 6,0 с помощью NaOH) (для ЕР2 и ЕР4), + радиоактивный лиганд [3H]-PGE2 (200 Ки/ммоль, NEN). {2,25 нМ в случае ЕРЗ и 2,5 нМ в случае ЕР2}. Реакцию инициировали добавлением мембранного белка (приблизительно 50 мкг/в реакционной смеси для ЕР3, 100 мкг для ЕР2 и ЕР4). Концентрация диметилсульфоксида (фирма Sigma) составляет постоянную величину 1% (об./об.) во всех образцах, а конечная концентрация исследуемых соединений 100 мкМ – 0,3 нМ. Неспецифическое связывание определяли в присутствии 10 мкМ нерадиоактивного PGE2 (фирма Cayman Chemical). Инкубирование проводили в течение 60 мин при 30°С (ЕР3) или в течение 45 мин при 23°С (ЕР2 и ЕР4). Для завершения инкубирования смесь быстро фильтровали через 96-луночный фильтр Unifilter GF/B (фирма Packard) (предварительно смоченный 10 мМ MES, 0,01% БСА, pH 6,0 для ЕР2) при 4°C с использованием полуавтоматического 96-луночного прибора для сбора клеток Filtermate 196 (фирма Packard). Фильтры промывали 3-4 мл буферного раствора для промывки (20 мМ HEPES, pH 7,4, для ЕР3, 10 мМ MES, 0,01% БСА, pH 6,0, для ЕР2 и ЕР4), сушили в течение по меньшей мере 1 ч при комнатной температуре и остаточную радиоактивность, связанную с индивидуальными фильтрами, измеряли на сцинтилляционном счетчике Packard TopCount Microplate после добавления 37,5 мкл раствора Microscint 20 (фирма Packard). Данные обрабатывали статистическим методом с использованием программного обеспечения Prism версия 3.0 (GraphPad).
Пример 11 Анализ плотности костной массы Соединения по настоящему изобретению испытывали по их действию на костную массу крыс с удаленным яичником. На взрослых самках крыс Sprague-Dawley или Wistar Hanover проводили фиктивную операцию или удаляли яичники по методу Charles River. Согласно методике крыс помещали парами в помещение с контролируемой окружающей средой и проводили акклиматизацию в течение по меньшей мере одной недели. Животные в процессе акклиматизации получали корм парами. Через 20 сут после хирургической операции животным вводили исследуемое соединение подкожным способом один раз в день в течение 5 недель. Анализируемое соединение растворяли в 10% EtOH/солевом растворе или в 20 мМ фосфатном буферном растворе. Перед лечением и при завершении лечения измеряли плотность минеральной составляющей костной ткани (BMD) сканированием на костном денситометре Hologic QRD-4500 с использованием программного обеспечения высокого разрешения. Затем анализировали изображения отдельных участков, представляющих интерес, таких, как вся бедренная кость, головка бедра, диафиз бедра, дистальная бедренная кость, головка большой берцовой кости, проксимальный большеберцовый метафиз, позвонки L2-L4, позвонок 15. Для оценки влияния удаления яичников на костную массу проводили сравнение результатов сканирования ложно оперированных крыс, крыс OVX и крыс, получавших наполнитель, причем сравнение проводили по критерию Стьюдента. Группы OVX сравнивали с использованием однофакторного дисперсионного анализа (ANOA), а затем по критерию Фишера (по младшему значащему разряду) для сравнения каждой группы обработанных соединением животных с контрольной группой, при условии, если общее действие являлось статистически достоверным. Перед указанным выше анализом данные можно упорядочить и провести непараметрический анализ (с использованием критерия суммы рангов Wilcoxon или Kruskal-Wallis). Удаление яичников приводит к значительной потере общей костной массы, в основном из зрелой костной ткани. Общая BMD у оперированных животных снижалась на 5-20% по сравнению с ложно оперированными контрольными животными.
Формула изобретения
1. Производные пирролидона формулы I
где Q означает -СН2– или кислород, В означает -СН2-, -(CH2)2-, -(CH2)2-, -(CH2)4-, (CH2)5-, -СН=СН-, -СН2-СН=СН-, -СН=СН-СН2– или -СН2-СН=СН-СН2-, при условии, что если В означает -СН=СН- или -СН=СН-СН2-, то Q означает -СН2-, Х означает -O-, -S-, -SO- или -SO2– или простую связь, при условии, что если Х означает простую связь, то Q означает кислород, J означает -(CRbRc)n-, где n означает целое число от 1 до 4, a Rb и Rc оба означают водород или один или оба Rb и Rc означают (С1-С4)алкил, а другой из них означает водород, или Rb и Rc вместе с атомом углерода, к которому они присоединены, образуют С2-С5полиметиленовую группу, или -СН2-СН=СН-, А означает -СН2-СН2-, -СН-СН- или -С Z означает -C(O)OR n означает 1, 2, 3 или 4, R1 означает -(СН2)рR7, где R7 означает C1-С6алкил, С3-С8циклоалкил, фуранил, замещенный трифторметилом, фенил или фенил-Y-фенил, где Y означает кислород или простую химическую связь, причем фенил необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, включающей C1-С6алкил, C1-С6алкоксигруппу, галоген и галоген-С1-С6алкил, р означает 0, 1, 2, 3, 4 или 5, R2 означает водород, C1-С6алкил, а R3, R4, R5 и R6 означают водород, или их фармацевтически приемлемые соли или сольваты, индивидуальные изомеры или рацемическая или нерацемическая смесь изомеров. 2. Производные пирролидона по п.1, где R1 означает -(CH2)pR7, a R2 означает водород. 3. Производные пирролидона по п.2, где R7 означает C1-С6алкил или С3-С8циклоалкил. 4. Производные пирролидона по одному из пп.1-3, где p равно 4, а R7 означает метил. 5. Производные пирролидона по п.1, где Z означает COOH. 6. Производные пирролидона по п.1, где R7 означает фенил, необязательно замещенный заместителем, выбранным из группы, включающей C1-С6алкил, трифторметил, галоген, -Y-R9, где Y означает простую химическую связь, а R9 означает фенил. 7. Производные пирролидона по п.1, где p равно 0. 8. Производные пирролидона по п.1, где Q означает -СН2-, В означает -СН2-, а Х означает -О- или -S-. 9. Производные пирролидона по п.1, где J означает -(CHRa)3-, один из Ra означает С1-С4алкил, а другие означают водород. 10. Производные пирролидона по п.1, где J означает -СН2-СН=СН-. 11. Производные пирролидона по п.5, где Q означает -СН2-, В означает -СН2-, -(СН2)2-, -(СН2)3– или -(СН2)4-, а Х означает -O-, -S-, -SO- или -SO2-. 12. Производные пирролидона по п.1, где J означает -(CHRa)3-, один из Ra означает С1-С4алкил, а другие означают водород. 13. Производные пирролидона по п.1, где J означает -СН2-СН=СН-. 14. Производные пирролидона по п.1, где J означает -(СН2)3 –. 15. Производные пирролидона по любому из пп.12-14, где p равно 0 или 1. 16. Производные пирролидона по п.11, где А означает СН=СН, a R1 означает пентил. 17. Производные пирролидона по п.11, где А означает СН2-СН2-, а R1 означает пентил. 18. Производные пирролидона по п.1, выбранные из группы, включающей а) метиловый эфир 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]масляной кислоты, б) 4-[(2-{(2R)-2-[(1Е,3S)-3-гидроксиокт-1-енил]-5-оксопирролидин-1-ил}этил)тио]бутановую кислоту, в) {4-[2-[R-(3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутилсульфанил}уксусную кислоту, г) {4-[2-[R-(1E-3S-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутилсульфанил}уксусную кислоту, д) {4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусную кислоту, е) (4-{(R)-2-[(E)-3-гидрокси-3-(5-трифторметилфуран-2-ил)пропенил]-5-оксопирролидин-1-ил}бутилсульфанил)уксусную кислоту, ж) 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этансульфинил} масляную кислоту, з) 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этансульфонил} масляную кислоту, и) {(Z)-4-[(R)-2-((Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1 -ил]бут-2-енилокси}уксусную кислоту, к) {4-[(R)-2-((Е)-(S)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]бутокси}уксусную кислоту, л) (4-{(R)-2-[(S)-(Е)-3-гидрокси-4-(3-трифторметилфенил)бут-1-енил]-5-оксопирролидин-1-ил}бутокси)уксусную кислоту, м) (4-{(R)-2-[(Е)-3-гидрокси-3-(2 н) (4-{(R)-2-[(Е)-3-(4 о) 4-{2-[(R)-2-((Е)-3-гидрокси-3-пентилокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}масляную кислоту, п) 4-{2-[(R)-2-((Е)-3-гидрокси-3-метилокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}масляную кислоту, р) 4-[2-((S)-2-{(R)-3-[3-(3-фторфенокси)фенил]-3-гидроксипропил}-5-оксопирролидин-1-ил)этилсульфанил]масляную кислоту, с) 4-[2-((S)-2-{(R)-3-[3-(4 т) 4-[2-((S)-2-{(S)-3-[3-(4 у) 4-[2-((S)-2-{(R)-3-[3-(3 ф) 4-[2-((S)-2-{(R)-3-[3-(3 х) 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-4-метилмасляную кислоту, ц) 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-3-метилмасляную кислоту, ч) 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-2-метилмасляную кислоту, ш) 4-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}-2-бутеновую кислоту, щ) 1-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанилметил}циклопропил)уксусную кислоту, э) 5-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}уксусную кислоту, ю) 3-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}пропионовую кислоту, я) 5-{2-[(R)-2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-ил]этилсульфанил}пентановую кислоту, аа) 6-[2-((S)-(Е)-3-гидроксиокт-1-енил)-5-оксопирролидин-1-илокси]гексановую кислоту и бб) 3-{3-[2-(3-гидроксиокт-1-инил)-5-оксопирролидин-1-ил]пропилсульфанил}пропионовую кислоту. 19. Производные пирролидона формулы I по п.1, которые являются сольватом, индивидуальным изомером или рацемической или нерацемической смесью соединений по пп.1-18. 20. Фармацевтическая композиция, проявляющая конкурентную аффинность связывания EP1, ЕР2, ЕР3 и ЕР4, содержащая терапевтически эффективное количество соединения формулы I по любому из пп.1-18 в смеси по меньшей мере с одним пригодным носителем, разбавителем или эксципиентом. 21. Производные пирролидона формулы II
где Q, В, X, J, R1, R3, R4, R5 и R6 имеют значения, указанные в п.1, Z означает C(O)OR R10 означает -СН2ОН, -СНО, -CH=CH-C(O)R1.
PC4A – Регистрация договора об уступке патента СССР или патента Российской Федерации на изобретение
Прежний патентообладатель:
(73) Патентообладатель:
Договор № РД0038886 зарегистрирован 25.07.2008
Извещение опубликовано: 10.09.2008 БИ: 25/2008
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
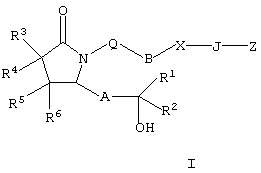
 С-;
С-; , где R
, где R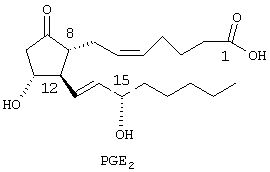
 -ФНО, см., например, К.К.Meja и др., Characterization ofprostanoid receptor(s) on human blood monocytes at which prostaglandin E2 inhibits lipopolysaccharide-induced tumor necrosis factor-alpha generation, Br. J.Pharmacol., 122, 149-157 (1997), и A.Eigler и др., Anti-inflammatory activities of cAMP-elevating agents: enhancement of IL-10 synthesis and concurrent suppression of TNF production, J. Leukoc. BioL, 63, 101-107 (1998), или лечение глаукомы, см., например, M.Takamatsu и др., Localization of Prostaglandin E Receptor Subtypes in the Ciliary Body of Mouse Eye, Exp. Eye Res., 70, 623-628 (2000), и D.F.Woodward и др., Molecular Characterization and Ocular Hypotensive Properties of the Prostanoid EP2 Receptor, J. Ocul. Pharmacol. Ther., 11, 447 (1995).
-ФНО, см., например, К.К.Meja и др., Characterization ofprostanoid receptor(s) on human blood monocytes at which prostaglandin E2 inhibits lipopolysaccharide-induced tumor necrosis factor-alpha generation, Br. J.Pharmacol., 122, 149-157 (1997), и A.Eigler и др., Anti-inflammatory activities of cAMP-elevating agents: enhancement of IL-10 synthesis and concurrent suppression of TNF production, J. Leukoc. BioL, 63, 101-107 (1998), или лечение глаукомы, см., например, M.Takamatsu и др., Localization of Prostaglandin E Receptor Subtypes in the Ciliary Body of Mouse Eye, Exp. Eye Res., 70, 623-628 (2000), и D.F.Woodward и др., Molecular Characterization and Ocular Hypotensive Properties of the Prostanoid EP2 Receptor, J. Ocul. Pharmacol. Ther., 11, 447 (1995).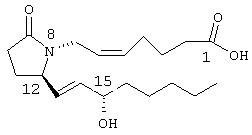
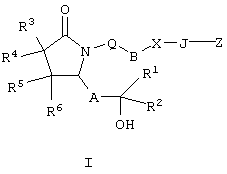
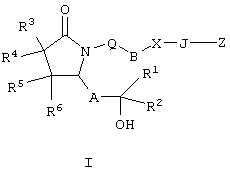
 , версия 4.0, компьютерной системы института Бельштейна для формирования систематической номенклатуры IUPAC.
, версия 4.0, компьютерной системы института Бельштейна для формирования систематической номенклатуры IUPAC.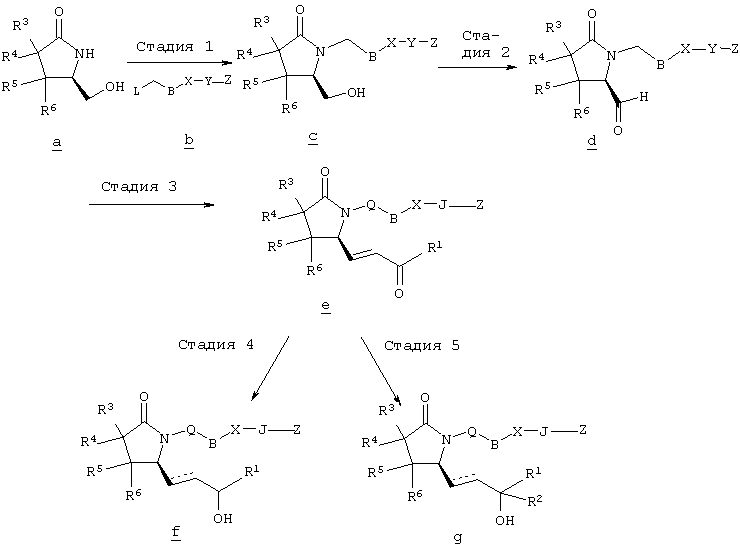
 -кетофосфонатом общей формулы R1-С(O)-СН2РО(ОСН3)2 (соединение k, получение см. на схеме В) и третичным амином в присутствии основания, такого, например, как гидрид натрия, трет-бутоксид калия, гексаметилдисилазид калия или хлорид лития, в инертном растворителе из группы простых эфиров, таких, как тетрагидрофуран, 1,2-диметоксиэтан или трет-бутилметиловый эфир, получают кетон общей формулы е.
-кетофосфонатом общей формулы R1-С(O)-СН2РО(ОСН3)2 (соединение k, получение см. на схеме В) и третичным амином в присутствии основания, такого, например, как гидрид натрия, трет-бутоксид калия, гексаметилдисилазид калия или хлорид лития, в инертном растворителе из группы простых эфиров, таких, как тетрагидрофуран, 1,2-диметоксиэтан или трет-бутилметиловый эфир, получают кетон общей формулы е.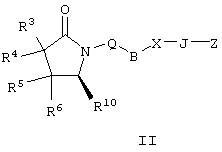
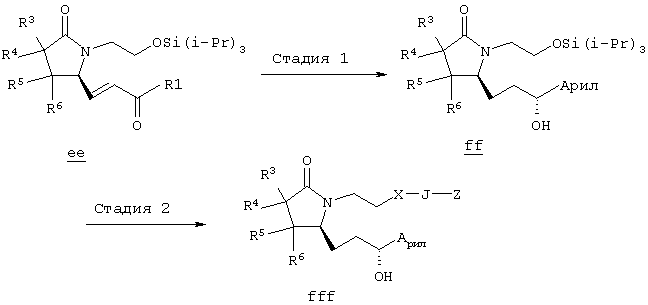
 -тиобутиролактоном, в инертном растворителе из группы простых эфиров, такого, как тетрагидрофуран или 1,2-диметоксиэтан, с образованием соединения fff, в котором Х означает S. При необходимости сложноэфирную группу Z гидролизуют с образованием -С(O)ОН по вышеуказанным методикам.
-тиобутиролактоном, в инертном растворителе из группы простых эфиров, такого, как тетрагидрофуран или 1,2-диметоксиэтан, с образованием соединения fff, в котором Х означает S. При необходимости сложноэфирную группу Z гидролизуют с образованием -С(O)ОН по вышеуказанным методикам.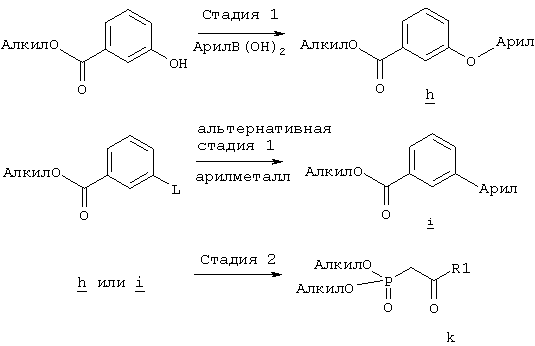
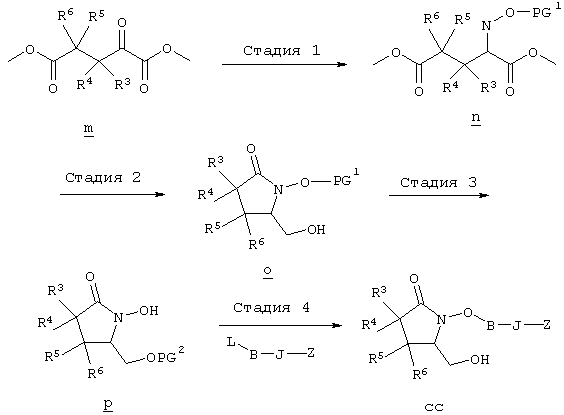
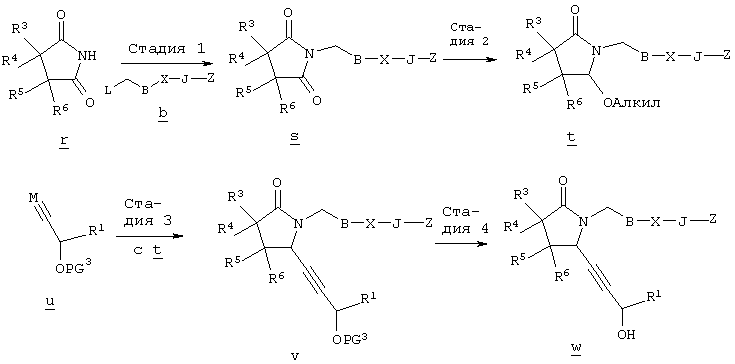
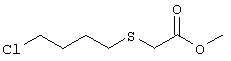
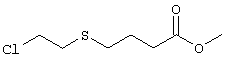
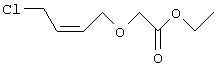
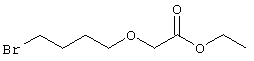
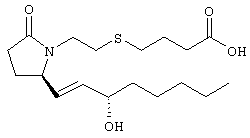
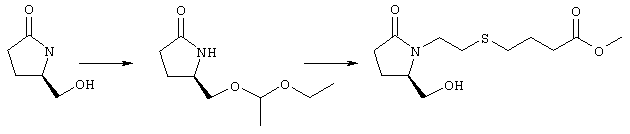
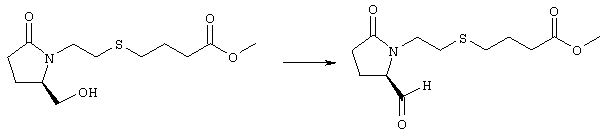
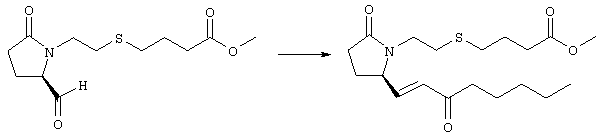
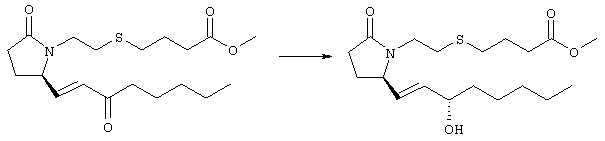
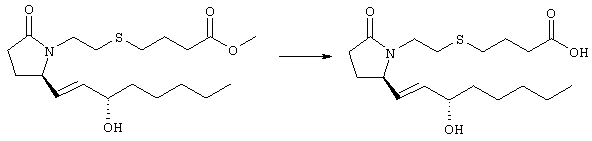
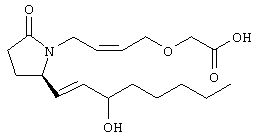
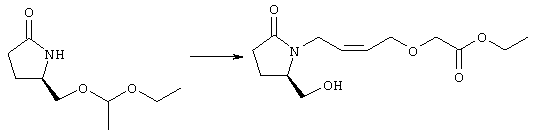
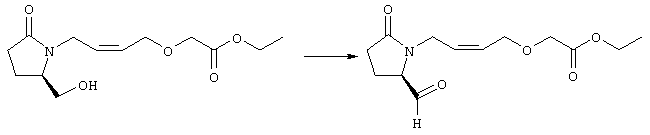
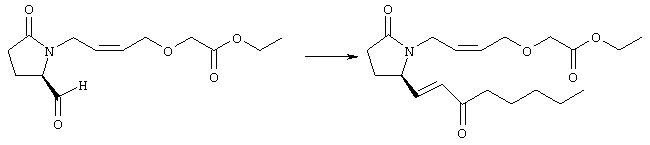
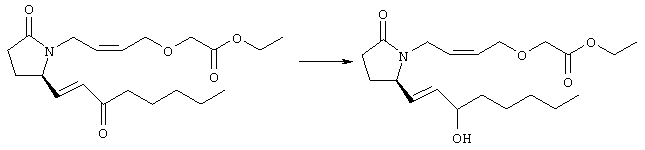
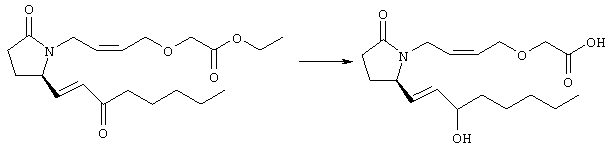
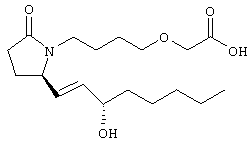
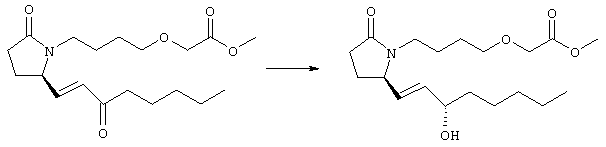
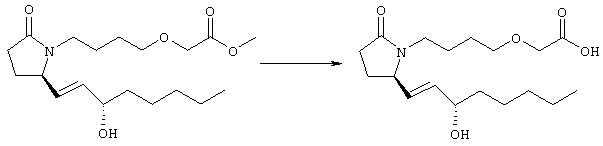
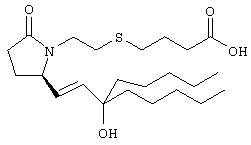
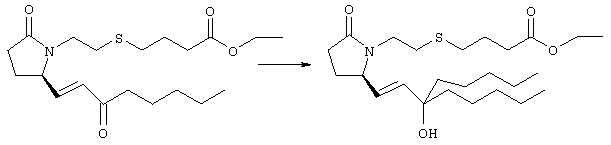
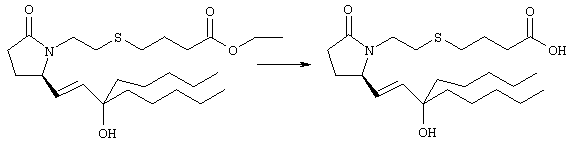
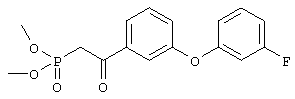
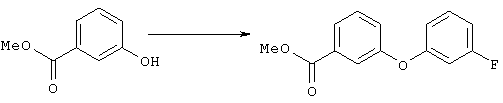
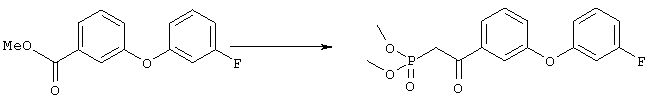
 7,78 (t, J 0,6, 0,9, 7,8 Гц, 1Н), 7,63 (t, J 2,1 Гц, 1Н), 7,48 (t, J 8,1 Гц, 1Н), 7,32-7,26 (m, 2H), 6,90-6,78 (m, 2H), 6,70 (dt, J 2,4, 9,9 Гц, 1Н), 3,80 (d, J 11,2 Гц, 6Н), 3,61 (d, J 22,6 Гц, 2H).
7,78 (t, J 0,6, 0,9, 7,8 Гц, 1Н), 7,63 (t, J 2,1 Гц, 1Н), 7,48 (t, J 8,1 Гц, 1Н), 7,32-7,26 (m, 2H), 6,90-6,78 (m, 2H), 6,70 (dt, J 2,4, 9,9 Гц, 1Н), 3,80 (d, J 11,2 Гц, 6Н), 3,61 (d, J 22,6 Гц, 2H).