|
(21), (22) Заявка: 2005139586/04, 19.12.2005
(24) Дата начала отсчета срока действия патента:
19.12.2005
(46) Опубликовано: 20.03.2007
(15) Информация о коррекции:
Версия коррекции № 1 (
Изобретение относится к гуминовым веществам и может использоваться для изучения их физико-химических свойств, взаимодействия с биологическими объектами и распределения в тканях биологических объектов.
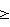
Сопоставление с другими методами
14
Наиболее близким к заявленному является способ введения трития в гуминовые и гуминоподобные вещества взаимодействием этих веществ с содержащим тритий агентом. Здесь делалась попытка ввести тритий в ГВ торфа восстановлением -СНО групп с помощью КВ3H4
Была поставлена задача создания способа введения трития в гуминовые и гуминоподобные вещества так, что равномерность распределения тритиевой метки и ее активность была бы более высокой.
Поставленная задача была решена настоящим изобретением.
В способе получения меченых тритием гуминовых и гуминоподобных веществ взаимодействием гуминовых и гуминоподобных веществ с содержащим тритий агентом согласно изобретению в качестве содержащего тритий агента используют атомарный тритий, полученный при термической каталитической диссоциации трития на вольфрамовой проволоке при давлении 0,3-0,6 Па, перед взаимодействием гуминовые и гуминоподобные вещества наносят на стенки реакционного сосуда лиофилизацией их водных растворов, а термическую диссоциацию производят импульсами 5-10 с при температуре вольфрамовой проволоки 1900-2000 К.
Таким образом, предлагается способ введения трития в гуминовые и гуминоподобные вещества, в котором при обработке атомарным тритием, полученным при термической каталитической диссоциации трития на вольфраме при низком (0.5 Па) давлении, подвергаются не индивидуальные химические соединения, а готовые препараты, нанесенные на стенки реакционного сосуда лиофилизацией их водных растворов. Время обработки ограничивается короткими импульсами (10 с), температура вольфрамовой проволоки ограничивается диапазоном 1900-2000 К. Выбранные условия введения трития обеспечивают равномерное замещение водорода на тритий в любых доступных положениях обрабатываемых молекул.
Важным этапом получения меченого препарата является стадия удаления лабильной метки, проводить которую предлагается в фосфатном буфере с концентрацией 0.028 М при рН 6.8 с помощью диализа через мембраны соответствующего размера (MWCO от 500 до 12000 Да) в течение не менее 24 суток при 4°С. Подбором размера пор мембраны минимизируются потери препарата во время диализа и сохраняется молекулярно-массовое распределение.
На заключительном этапе приготовления меченого образца проводится анализ распределения метки по его компонентам с помощью эксклюзионной хроматографии. Контроль равномерности введения тритиевой метки осуществляется с помощью сравнения профилей выхода с колонки исходного и меченого препаратов по УФ-поглощению при 254 нм, а также по профилю радиоактивности меченого препарата.
Получаемые таким способом препараты ГВ предлагается использовать при изучении их взаимодействия с различными биологическими объектами.
ПРИМЕРЫ
Пример 1. 1 мл водного раствора ГК угля с концентрацией 0.26 мг/мл равномерно распределили на стенках реакционного сосуда объемом 600 см3 и заморозили с помощью жидкого азота. Воду удалили лиофилизацией под вакуумом, собирая ее в ловушке с жидким азотом. Сосуд с мишенью присоединили к специальной вакуумной системе, удалили воздух до остаточного давления 0.001 Topp. Затем охладили стенки реакционного сосуда жидким азотом и продолжили вакуумирование до остаточного давления 0.00005 Topp. С помощью специального натекателя из палладия в реакционный сосуд напустили водород-тритиевую смесь с содержанием трития 35% до давления 0.004 Topp. Нагрели вольфрамовую проволоку, находящуюся в центре реакционного сосуда, до температуры 2000 К с помощью переменного электрического тока. Время нагревания проволоки составило 10 с. Остаточный газ откачали с помощью диффузионного насоса уже без охлаждения реакционного сосуда жидким азотом.
Обработанную тритием мишень ГК угля растворили в 2 мл 0.028 М фосфатного буфера рН 6.8. Его радиоактивность оказалась равной 258 МБк. Раствор поместили в пластиковую пробирку и закрыли диализной мембраной MWCO 2000 Да. Диализную очистку препарата проводили в течение 28 суток против 1 л фосфатного буфера с его периодической заменой и измерением радиоактивности. Радиоактивность раствора меченого препарата составила 63 МБк.
Для оценки идентичности исходного и меченого препарата, очищенного от лабильной метки, провели их гель-хроматографическое фракционирование на хроматографической системе Abimed, включающей в себя: ВЭЖХ насос, автосамплер, стеклянную колонку ( 15 мм, L=25 см), спектрофотометрический УФ-детектор, коллектор фракций, плату АЦП для регистрации аналитического сигнала и регистрирующий компьютер. Колонку заполняли гелем “Toyopearl” TSK HW-55S (Toso-Haas, Япония) с диапазоном фракционирования 1000-200000 Да по полидекстранам. В качестве подвижной фазы использовали 0.028 М фосфатный буфер с рН 6.8. Объем элюируемой пробы составлял 1 мл, скорость элюирования 1 мл/мин. 15 мм, L=25 см), спектрофотометрический УФ-детектор, коллектор фракций, плату АЦП для регистрации аналитического сигнала и регистрирующий компьютер. Колонку заполняли гелем “Toyopearl” TSK HW-55S (Toso-Haas, Япония) с диапазоном фракционирования 1000-200000 Да по полидекстранам. В качестве подвижной фазы использовали 0.028 М фосфатный буфер с рН 6.8. Объем элюируемой пробы составлял 1 мл, скорость элюирования 1 мл/мин.
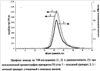
Профили УФ-поглощения исходного и меченого препаратов практически совпали (см. чертеж). Одновременно с гель-хроматографическим анализом проводили его фракционирование, собирая фракции по 1.5 мл. Радиоактивность собранных фракций измеряли с помощью жидкостного сцинтилляционного счета. Полученный профиль по радиоактивности совпал с профилем по УФ-поглощению (см. чертеж). В препарате отсутствовали радиоактивные продукты со временем удержания, отличным от исходного препарата. В результате проведенного анализа было найдено, что раствор содержал 0.25 мг ГК угля. Удельная радиоактивность препарата оказалась равной 254 ГБк/г.
Пример 2. Подготовку препарата ГК угля к введению трития провели так же, как в Примере 1. С помощью специального натекателя из палладия в реакционный сосуд напустили водород-тритиевую смесь с содержанием трития 35% до давления 0.004 Topp. Нагрели вольфрамовую проволоку, находящуюся в центре реакционного сосуда, до температуры 2000 К с помощью переменного электрического тока. Время нагревания проволоки составило 10 с. Остаточный газ откачали с помощью диффузионного насоса до давления 0.0005 Topp, затем напустили новую порцию водород-тритиевой смеси. Повторили нагрев вольфрамовой проволоки электрическим током до 2000 К в течение 10 с, после чего откачали остаточный газ уже без охлаждения реакционного сосуда жидким азотом.
Обработанную тритием мишень ГК угля растворили в 2 мл 0.028 М фосфатного буфера рН 6.8. Его радиоактивность оказалась равной 444 МБк. Раствор поместили в пластиковую пробирку и закрыли диализной мембраной MWCO 2000 Да. Диализную очистку препарата проводили так же, как в Примере 1. Радиоактивность раствора меченого препарата составила в конце диализной очистки 15.8 МБк.
Оценка идентичности исходного и меченого препарата с помощью гель-хроматографического анализа была сделана так же, как в Примере 1. Профили по УФ-поглощению исходного и меченого препаратов и профиль по радиоактивности меченого препарата полностью совпали. В результате проведенного анализа было найдено, что раствор содержал 0.23 мг ГК угля. Удельная радиоактивность препарата оказалась равной 69 ГБк/г.
Пример 3. Препарат ГК угля подготовили к введению трития так же, как в Примерах 1 и 2. Водород-тритиевую смесь с содержанием трития 35% напускали до давления 0.004 Topp. Вольфрамовую проволоку нагревали до температуры 2000 К с помощью переменного электрического тока. Время нагревания проволоки составило 10 с. Остаточный газ откачали с помощью диффузионного насоса до давления 0.0005 Topp, затем напустили новую порцию водород-тритиевой смеси. Такую процедуру повторили 4 раза. Суммарное время нагрева вольфрамовой проволоки составило 40 с.
Обработанную тритием мишень ГК угля растворили в 2 мл 0.028 М фосфатного буфера рН 6.8. Его радиоактивность оказалась равной 924 МБк. Раствор поместили в пластиковую пробирку и закрыли диализной мембраной MWCO 2000 Да. Диализную очистку препарата проводили так же, как в Примерах 1 и 2. Радиоактивность раствора меченого препарата составила в конце диализной очистки 44 МБк.
Оценка идентичности исходного и меченого препарата с помощью гель-хроматографического анализа была сделана так же, как в Примерах 1 и 2. Профили по УФ-поглощению исходного и меченого препаратов и профиль по радиоактивности меченого препарата полностью совпали. В результате проведенного анализа было найдено, что раствор содержал 0.22 мг ГК угля. Удельная радиоактивность препарата оказалась равной 200 ГБк/г.
Пример 4. 1 мл водного раствора ГК угля с концентрацией 0.3 мг/мл равномерно распределили на стенках реакционного сосуда объемом 400 см3 и заморозили с помощью жидкого азота. Дальнейшая подготовка препарата к введению тритиевой метки осуществлялась аналогично Примеру 1. С помощью специального натекателя из палладия в реакционный сосуд напустили водород-тритиевую смесь с содержанием трития 43% до давления 0.004 Торр. Нагрели вольфрамовую проволоку, находящуюся в центре реакционного сосуда, до температуры 1880 К с помощью переменного электрического тока. Время нагревания проволоки составило 10 с. Остаточный газ откачали с помощью диффузионного насоса уже без охлаждения реакционного сосуда жидким азотом.
Мишень растворили в 1,5 мл 0,028 М фосфатного буфера рН 6,8. Его радиоактивность оказалась равной 178 МБк. Удаление лабильной метки проводили в течение 25 суток через мембрану MWCO 2000 Да. Хроматографический анализ проводили аналогично описанию в Примере 1. Профили по УФ-поглощению исходного и меченого препаратов и профиль по радиоактивности меченого препарата полностью совпали. Удельная радиоактивность препарата оказалась равной 153 ГБк/г.
Примеры 5-6. Препараты ГК угля готовили к введению тритиевой метки аналогично Примеру 4. Нагревание вольфрамовой проволоки осуществляли до 1750 и 1610 К в течение 10 с. Радиоактивность раствора после растворения мишени составила 60 и 39 МБк. После удаления лабильной метки в течение 25 дней проводили хроматографический анализ аналогично Примеру 1. Профили по УФ-поглощению исходного и меченого препаратов и профиль по радиоактивности меченого препарата полностью совпали. Удельная радиоактивность препаратов оказалась равной 42 и 17 ГБк/г.
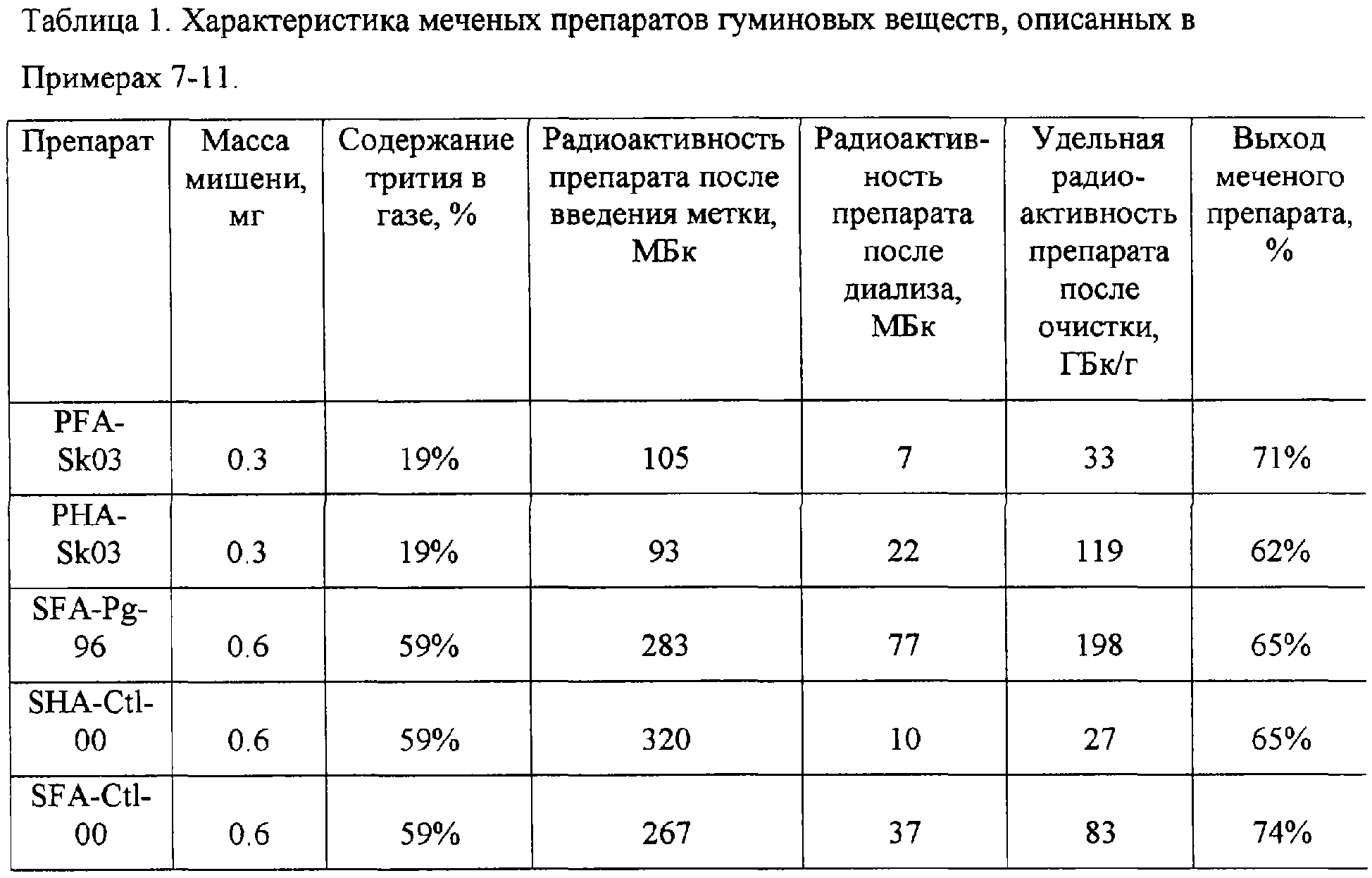
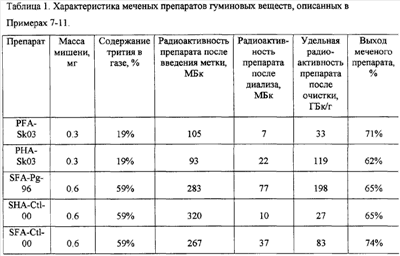
Примеры 7-11. Мишени препаратов гуминовых кислот торфа PHA-Sk03, фульфокислот торфа PFA-Sk03, гуминовых кислот почв SHA-Ctl-00, фульфокислот почв SFA-Ctl-00 и SFA-Pg-96 готовили в реакционных сосудах объемом 600 см3 аналогично описанию Примера 1. Для введения трития использовали водород-тритиевую смесь при давлении 0.004 Topp. Время нагревания вольфрамовой проволоки составляло 10 с, температура вольфрамовой проволоки поддерживали равной 2000 К. Растворение мишеней, диализную очистку и хроматографический анализ проводили, как описано ранее. Данные по количеству вещества, взятого на приготовление мишеней, по составу водород-тритиевой смеси и радиоактивности препаратов на разных стадиях очистки приведены в таблице 1. Для всех препаратов получено хорошее совпадение выходящих с колонки профилей по поглощению при 254 нм для исходных и меченых препаратов, а также профиля радиоактивности меченого препарата.
Примеры 12-13. Мишени препаратов гуминоподобного вещества лигфола-16 (продукт гидролиза древесного лигнина) готовили лиофилизацией 1 мл раствора с концентрацией 0,64 г/л на стенках реакционных сосудах объемом 600 см3 и вводили тритиевую метку аналогично описанию в Примере 1. Для удаления метки из лабильных положений использовали два подхода: меченый препарат длительно выдерживали при 4°С и затем проводили его лиофилизацию; меченый препарат очищали с помощью диализа при 4°С, как описано в Примере 1.
При гель-хроматографическом фракционировании исходного и меченых препаратов было найдено, что кривая элюирования меченого препарата, очищенного путем лиофилизации, практически идентична исходному препарату. Как в исходном препарате, так и в меченом аналоге, наблюдается явно выраженное плечо в области низких молекулярных масс, что свидетельствовало о присутствии низкомолекулярной составляющей в данном препарате лигфола. В то же время, профиль элюирования препарата, очищенного с помощью диализа, существенно отличается от исходного. Максимум кривой элюирования диализованного препарата был сдвинут в сторону больших молекулярных масс. При этом практически отсутствовало плечо в области низких молекулярных масс. Кроме того, площадь пика свидетельствовала о значительной (33%) потере препарата при диализе. Указанный характер изменений объясним тем, что низкомолекулярные соединения, входящие в состав препарата, были удалены при диализной очистке. В то же время для обоих меченых препаратов наблюдалось хорошее совпадение профилей их выхода по УФ-поглощению и по радиоактивности, что свидетельствовало о равномерном распределении трития по компонентам препаратов. В таблице 2 приведены основные характеристики меченых препаратов лигфола.
Пример 14. Определение фактора бионакопления ГК угля бактериями. Для определения фактора бионакопления ГК угля бактериями тетрациклин-устойчивый штамм грамотрицательной бактерии Escherichia coli наращивали путем глубинного культивирования при 37°С в среде М9 (Na2HPO4×12H2O 15.1 г/л, КН2PO4 3.0 г/л, NaCl 0.5 г/л, NH4Cl 1.0 г/л, MgSO4×12H2O 0.5 г/л, CaCl2 5.6 мг/л, глюкоза 0.25 г/л) в течение 10-14 часов. Автоклавирование сред проводили при 120°С в течение 30 мин. Стерилизацию растворов глюкозы, солей кальция и магния и препарата ГК угля проводили путем фильтрования через мембранный фильтр с размером пор 20 мкм; указанные компоненты добавляли в среду раздельно. В качестве бактериостатического агента использовали тетрациклин в концентрации 30 мг/л, его добавляли в среду непосредственно перед инокуляцией. рН среды устанавливали 7.5 с использованием NaOH.
В питательную среду добавляли немеченый препарат ГК угля в концентрациях 5-50 мг/л и меченый препарат ГК угля для создания удельной радиоактивности растворов 1500-2000 Бк/мл. Затем суспензию бактерий центрифугировали при 5000 об/мин в течение 30 мин и в супернатанте определяли равновесную концентрацию ГК угля. Далее супернатант заменяли эквивалентным объемом среды М9, бактерии ресуспендировали и в полученной суспензии определяли количество ГК угля, сорбированного бактериями.
Рост бактерий контролировали по поглощению при 600 нм (OD600). Сначала для серии суспензий с различным содержанием бактерий измеряли OD600 и сырой вес бактерий, который определяли путем взвешивания осадка, образующегося при центрифугировании суспензии бактерий при 5000 об/мин в течение 30 мин. Затем рассчитывали зависимость сырого веса бактерий от OD600:
Вес бактерий (г/л)=2.5167×OD600+3.2158.
Далее вес бактерий рассчитывали по полученной зависимости.
На основании полученных данных строили изотермы сорбции ГК угля бактериями и рассчитывали фактор бионакопления, как тангенс угла наклона изотермы. Полученный фактор бионакопления составил 3.2 л/кг.
Пример 15. Определение количества ГК угля, поступившего в клетки бактерий. Для определения количества ГК угля, поступившего внутрь клеток бактерий, проводили наращивание бактерий Е.coli, как описано в Примере 14. Отделение бактерий от культуральной среды осуществляли путем центрифугирования при 5000 об/мин в течение 30 мин. Супернатант заменяли эквивалентным объемом среды М9, осадок бактерий ресуспендировали и по удельной радиоактивности супернатанта определяли количество ГК угля, сорбированного бактериями. Затем в суспензии проводили лизис бактериальных клеток путем добавления хлороформа. Суспензию с разрушенными клетками вновь подвергали центрифугированию при 5000 об/мин в течение 30 мин, отделяя таким образом дебрис от внутреннего содержимого клеток. На основании удельной радиоактивности супернатанта рассчитывали количество ГК угля, поступившего в клетки. Оказалось, что в клетки бактерий поступает 50% от общего количества препарата, сорбируемого клетками.
Формула изобретения
Способ получения меченных тритием гуминовых и гуминоподобных веществ взаимодействием гуминовых и гуминоподобных веществ с содержащим тритий агентом, отличающийся тем, что в качестве содержащего тритий агента используют атомарный тритий, полученный при термической каталитической диссоциации трития на вольфрамовой проволоке при давлении 0,3-0,6 Па, перед взаимодействием гуминовые и гуминоподобные вещества наносят на стенки реакционного сосуда лиофилизацией их водных растворов, а термическую диссоциацию производят импульсами 5-10 с при температуре вольфрамовой проволоки 1900-2000 К.
РИСУНКИ
TH4A – Переиздание описания изобретения к патенту Российской Федерации
Причина переиздания: Коррекция библиографических данных в реферате на английском языке
Извещение опубликовано: 10.01.2008 БИ: 01/2008
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 20.12.2008
Извещение опубликовано: 27.11.2009 БИ: 33/2009
NF4A – Восстановление действия патента СССР или патента Российской Федерации на изобретение
Дата, с которой действие патента восстановлено: 10.12.2009
Извещение опубликовано: 10.12.2009 БИ: 34/2009
|