Патент на изобретение №2293732
|
||||||||||||||||||||||||||
(54) ДИПЕПТИДНИТРИЛЬНЫЕ ИНГИБИТОРЫ КАТЕПСИНА К
(57) Реферат:
Описаны дипептиднитрильные ингибиторы катепсина К, их фармацевтически приемлемые соли или их сложные эфиры, которые применяют для терапевтического или профилактического лечения заболевания или болезненного состояния, в которые вовлечен катепсин К. 2 н. и 1 з.п. ф-лы.
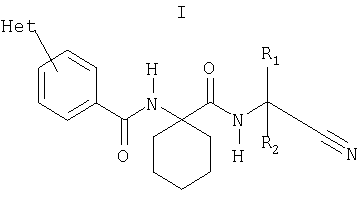
Данное изобретение относится к ингибиторам цистеиновых протеаз, в частности к дипептиднитрильным ингибиторам катепсина К, и к их фармацевтическому применению для лечения или профилактики заболеваний или болезненных состояний, в которые вовлечен катепсин К. Катепсин К является членом семейства цистеиновых катепсиновых ферментов лизосом, например, катепсинов В, К, L и S, которые вовлечены в различные нарушения, включая воспаление, ревматоидный артрит, остеоартрит, остеопороз, опухоли (особенно в инвазию и метастазирование опухолей), ишемическую болезнь сердца, атеросклероз (включая разрушение атеросклеротической бляшки и дестабилизацию), аутоиммунные заболевания, болезни органов дыхания, инфекционные болезни и опосредованные иммунологическим статусом заболевания (включая отторжение трансплантата). Исходящая от заявителей одновременно рассматриваемая заявка на международный патент WO 99/24460 описывает дипептиднитрилы, которые являются ингибиторами цистеиновых катепсинов, и их применение для лечения заболеваний и болезненных состояний, зависимых от цистеиновых катепсинов. В настоящее время получены новые дипептиднитрильные производные, которые являются ингибиторами катепсина К и которые обладают требуемыми свойствами для фармацевтического применения. Соответственно по настоящему изобретению обеспечивается соединение формулы I, или его фармацевтически приемлемая соль, или его сложный эфир
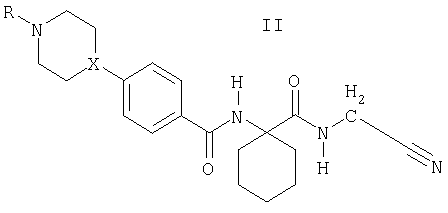
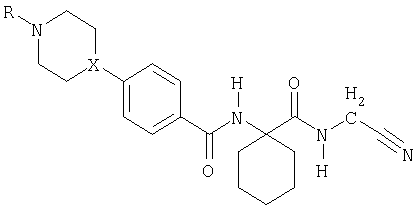
где R1 и R2 означают независимо друг от друга Н или (С1-С7)(низш.)алкил, или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (С3-С8)циклоалкильное кольцо, и Het означает необязательно замещенный азотсодержащий гетероциклический заместитель при условии, что Het не означает 4-пиррол-1-ил. Заместитель Het может быть в положении 2 или 3 фенильного кольца, хотя предпочтительно он находится в положении 4. В настоящем описании термин «азотсодержащий гетероцикл» означает гетероциклическую систему, содержащую по меньшей мере один атом азота, 2-10, предпочтительно 3-7, наиболее предпочтительно 4 или 5 атомов углерода и необязательно один или несколько дополнительных гетероатомов, выбираемых из О, S или предпочтительно N. Заместитель Het может включать ненасыщенный, например, ароматический азотсодержащий гетероцикл; хотя предпочтительно включает насыщенный азотсодержащий гетероцикл. Особо предпочтительными азотсодержащими гетероциклами являются пиперазинил, предпочтительно пиперазин-1-ил, или пиперидинил, предпочтительно пиперидин-4-ил. Het может быть замещен одним или несколькими заместителями, например, до 5 заместителями, независимо друг от друга выбранными из галоида, гидрокси, амино, нитро, необязательно замещенного (С1-С4)алкила (например, алкила, замещенного гидрокси, алкилокси, амино, необязательно замещенного алкиламино, необязательно замещенного диалкиламино, арила или гетероциклила), (С1-С4)алкокси. Предпочтительно Het замещен по атому азота, наиболее предпочтительно монозамещен по атому азота. Предпочтительными заместителями для Het являются (С1-С7)(низш.)алкил, (С1-С7)(низш.)алкокси(С1-С7)(низш.)алкил, (С5-С10)арил(С1-С7)(низш.)алкил или (С3-С8)циклоалкил. R1 и R2 как (С1-С7)(низш.)алкил предпочтительно являются одинаковыми, например, метилом, или R1 и R2 вместе с атомом углерода, к которому они присоединены, предпочтительно образуют (С3-С8)циклоалкильное кольцо, например, циклопропильное кольцо. Наиболее предпочтительно как R1, так и R2 означают Н. Таким образом, в особенно предпочтительных вариантах воплощения изобретение обеспечивает соединение формулы II, или его фармацевтически приемлемую соль, или его сложный эфир
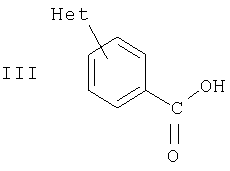
где Х означает СН или N, и R означает Н, (С1-С7)(низш.)алкил, (С1-С7)(низш.)алкокси(С1-С7)(низш.)алкил, (С5-С10)арил(С1-С7)(низш.)алкил, или (С3-С8)циклоалкил. Так, особыми примерами R как (С1-С7)(низш.)алкила являются метил, этил, н-пропил или изопропил. Особым примером R как (С1-С7)(низш.)алкокси(С1-С7)(низш.)алкила является метоксиэтил. Особым примером R как (С5-С10)арил(С1-С7)(низш.)алкила является бензил. Особым примером R как (С3-С8)циклоалкила является циклопентил. Примерами особых соединений формулы II являются: N-[1-(цианометилкарбамоил)циклогексил]-4-(пиперазин-1-ил)бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(4-метилпиперазин-1-ил)-бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(4-этилпиперазин-1-ил)бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-[4-(1-пропил)пиперазин-1-ил]бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(4-изопропилпиперазин-1-ил)бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(4-бензилпиперазин-1-ил)бензамид; N-[1-(цианометилкарбамоил)пиклогексил]-4-[4-(2-метоксиэтил)пиперазин-1-ил]-бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(1-пропилпиперидин-4-ил)бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-[1-(2-метоксиэтил)пиперидин-4-ил]-бензамид; N-[1-(пианометилкарбамоил)циклогексил]-4-(1-изопропилпиперидин-4-ил)бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(1-циклопентилпиперидин-4-ил)бензамид; N-[1-(цианометилкарбамоил)циклогексил]-4-(1-метилпиперидин-4-ил)бензамид и N-[1-(цианометилкарбамоил)циклогексил]-4-(пиперидин-4-ил)бензамид. Соединения формулы I и II и особые соединения, приведенные выше, впредь упоминаются как соединения по изобретению. Соединения по изобретению могут быть получены конденсацией соответствующего Het-замещенного производного бензойной кислоты с цианометиламидом 1-аминоциклогексанкарбоновой кислоты. Например, производное бензойной кислоты, предпочтительно в форме гидрохлорида, смешивают с цианометиламидом 1-аминоциклогексанкарбоновой кислоты, например, в присутствии НОВТ (1-гидроксибензотриазола), водорастворимого карбодиимида (WSCD) и триэтиламина в растворе, например, в ДМФА, и перемешивают, например, в течение ночи при комнатной температуре. Продукт может быть получен, например, упариванием растворителя, затем промыванием водным раствором карбоната натрия, предпочтительно в слабощелочных условиях, затем экстракцией с помощью растворителя, например, этилацетата, высушиванием экстракта, например, над сульфатом натрия, упариванием растворителя и фильтрацией. Могут применяться альтернативные реагенты и методики; например, как описано ниже в примерах. Таким образом, в дальнейшем аспекте изобретение обеспечивает способ получения соединений формулы I, который включает конденсацию соответствующего Het-замещенного производного бензойной кислоты формулы III
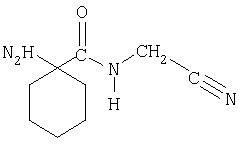
с цианометиламидом 1-аминоциклогексанкарбоновой кислоты.
Цианометиламид 1-аминоциклогексанкарбоновой кислоты может быть получен конденсацией 1-аминоциклогексанкарбоновой кислоты, как правило, с соответствующей защитой по аминогруппе, например, FMOC(9-флуоренилметоксикарбонил)-1-аминоциклогексанкарбоновой кислоты, с 2-аминоацетонитрилом. Например, FMOC-1-аминоциклогексанкарбоновую кислоту, например, с НОВТ и WSCD прибавляют к раствору 2-аминоацетонитрила и триэтиламина в ДМФА, смесь перемешивают при 25°С в течение ночи. Цианометиламид 1-аминоциклогексанкарбоновой кислоты можно выделить, как описано в примерах. FMOC-1-аминоциклогексанкарбоновая кислота может быть получена, как описано в примерах. Соединения по изобретению получают или в свободной форме, или в форме соли, если присутствуют солеобразующие группы. Соединения по изобретению, содержащие основные группы, могут быть превращены в кислотно-аддитивные соли, особенно в фармацевтически приемлемые соли. Они образуются, например, с неорганическими кислотами, такими, как минеральные кислоты, например, серная кислота, фосфорная или галоидоводородная кислота, или с органическими карбоновыми кислотами, такими, как (С1-С5)алканкарбоновыми кислотами, которые, например, являются незамещенными или замещенными галоидом, например, уксусная кислота, такими, как насыщенные или ненасыщенные дикарбоновые кислоты, например, щавелевая, янтарная, малеиновая или фумаровая кислота, такими, как гидроксикарбоновые кислоты, например, гликолевая, молочная, яблочная, винная или лимонная кислота, такими, как аминокислоты, например, аспарагиновая или глутаминовая кислота, или с органическими сульфоновыми кислотами, такими, как (С1-С4)алкилсульфоновые кислоты (например, метансульфоновая кислота) или арилсульфоновыми кислотами, которые являются незамещенными или замещенными (например, галоидом). Предпочтительными являются соли, образованные с хлористоводородной кислотой, метансульфоновой кислотой и яблочной кислотой. Принимая во внимание тесную взаимосвязь между соединениями в свободной форме и соединениями в форме их солей, когда бы ни упоминалось соединение в данном контексте, соответствующая соль также подразумевается, при условии, что такое является возможным или уместным при данных обстоятельствах. Соединения, включая их соли, могут также быть получены в форме их гидратов или включать другие растворители, используемые для кристаллизации. Соединения по изобретению обладают ценными фармакологическими свойствами при применении у млекопитающих и особенно полезны в качестве ингибиторов катепсина К. Ингибиторные свойства соединения по изобретению в отношении катепсина К могут быть продемонстрированы in vitro путем измерения ингибирования, например, рекомбинантного человеческого катепсина К. Анализ in vitro проводят следующим образом. Для катепсина К: анализ проводят в 96-луночных планшетах для микротитрования при температуре окружающей среды, используя рекомбинантный человеческий катепсин К. Ингибирование катепсина К анализируют при постоянных концентрациях фермента (0,16 нМ) и субстрата (54 мМ Z(N-карбобензокси)-Phe-Arg-AMCA(7-амино-4-метоксикумарин-3-уксусная кислота)), Peptide Institute Inc., Osaka, Japan) в 100 мМ фосфатном буфере, рН 7,0, содержащем 2 мМ дитиотреит, 20 мМ твин-80 и 1 мМ EDTA (этилендиаминтетрауксусную кислоту). Катепсин К проинкубировали с игибиторами в течение 30 мин, реакцию начинали прибавлением субстрата. Через 30 мин инкубации реакцию останавливали добавлением Е-64 (транс-эпоксисукцинил-L-лейциламидо-(4-гуанидино)бутана) (2 мМ), и интенсивность флуоресценции считывали на аппарате для прочтения многолуночных планшетов при длинах волн возбуждения и излучения 360 и 460 нм соответственно. Соединения по изобретению обычно имеют Кi (константу ингибирования) человеческого катепсина К менее чем примерно 5 нМ, предпочтительно примерно 5 нМ или меньше, например, примерно 1 нМ. Принимая во внимание их активность в качестве ингибиторов катепсина К, соединения по изобретению особенно полезны для млекопитающих как агенты для лечения и профилактики заболеваний и болезненных состояний, включающих повышенные уровни катепсина К. Такие болезни охватывают заболевания, включающие заражение организмами, такими, как Pneumocystis carinii, Trypsanoma cruzi, Trypsanoma brucei, Crithidia fusiculata, а также паразитарные заболевания, такие, как шистосомоз и малярия, опухоли (инвазия опухолей и метастазы опухолей), и другие заболевания, такие, как метахроматическая лейкодистрофия, мышечная дистрофия, атрофия мышечной ткани и подобные заболевания. Катепсин К вовлечен в заболевания, связанные с чрезмерной потерей костной ткани, и, следовательно, соединения по изобретению могут быть применимы для лечения и профилактики таких заболеваний, включая остеопороз, болезни десен, такие, как гингивит и периодонтит, болезнь Педжета, гиперкальциемия при злокачественных новообразованиях, т.е. индуцированная опухолью гиперкальциемия, и метаболическая болезнь костной ткани. Соединения по изобретению могут также применяться для лечения и профилактики заболеваний, связанных с чрезмерной деградацией хрящей или скелета, включая остеоартриты и ревматоидные артриты, а также некоторые опухолевые заболевания, вызывающие экспрессию высоких уровней протеолитических ферментов и деградацию скелета. Соединения по изобретению показаны также для предотвращения или лечения ишемической болезни сердца, атеросклероза (включая разрушение атеросклеротической бляшки и дестабилизацию), аутоиммунных заболеваний, болезней органов дыхания, инфекционных болезней и опосредованных иммунологическим статусом заболеваний (включая отторжение трансплантата). Соединения по изобретению особенно показаны для предупреждения или лечения остеопороза различного генеза (например, возникшего в юношеском возрасте, в климактерическом периоде, в постклимактерическом периоде, вызванного старческим возрастом или кортикостероидной терапией или бездеятельностью). Благоприятные воздействия оценивают в фармакологических тестах in vitro и in vivo, известных, как правило, специалистам и приведенных в контексте в качестве иллюстрации. Упомянутые выше свойства демонстрируются в тестах in vitro и in vivo с преимущественным использованием млекопитающих, например, крыс, мышей, собак, кроликов, обезьян, или изолированных органов и тканей, а также ферментных препаратов млекопитающих или природных, или полученных, например, рекомбинантным способом. Соединения по изобретению могут применяться in vitro в виде растворов, например, предпочтительно водных растворов или суспензий, либо in vivo, или энтерально, или парентерально, преимущественно перорально, например, в виде суспензии или в виде водного раствора, или в виде твердой капсулы, или таблетки. Дозирование in vitro может изменяться в диапазоне концентраций между примерно 10-5 моль и 10-9 моль. Дозирование in vivo в зависимости от пути введения может меняться в диапазоне 0,1-100 мг/кг. В соответствии с настоящим изобретением было найдено, что соединения по изобретению обладают хорошей биодоступностью, в частности, хорошей пероральной биодоступностью. Так, например, отобранные соединения по изобретению имеют абсолютную пероральную биодоступность 50% или выше, например, 80% или более. Противоартритная эффективность соединений по изобретению при лечении ревматоидного артрита может быть определена с использованием модели, такой же или подобной крысиной модели стимулированного артрита, которая описана ранее (R.E.Esser и др., J. Rheumatology, 1993, 20, 1176.) Эффективность соединений по изобретению в лечении остеоартрита может быть определена с использованием модели, такой же или подобной кроличьей модели частичной латеральной менискэктомии, которая описана ранее (Colombo и др., Arth. Rheum. 1993 26, 875-886). Эффективность соединений на модели может быть оценена количественно с использованием методик гистологических показателей, как описано ранее (O’Byrne и др., Inflamm. Res. 1995, 44, 117-118). Эффективность соединений по изобретению в лечении остеопороза может быть определена с использованием животной модели, такой, как крыса с удаленными яичниками, или других подобных видов, например, кролика или обезьяны, в опыте, в котором исследуемые соединения вводят животному и наличие маркеров резорбции костной ткани измеряют в моче или сыворотке (например, как описано в Osteoporos. Int. (1997) 7:539-543). В соответствии с дальнейшими аспектами изобретение обеспечивает: соединение по изобретению для применения в качестве фармацевтического средства; фармацевтическую композицию, включающую соединение по изобретению в качестве активного ингредиента; способ лечения пациента, страдающего от заболевания либо болезненного состояния или восприимчивого к заболеванию или болезненному состоянию, в которые вовлечен катепсин К, включающий введение пациенту эффективного количества соединения по изобретению, и применение соединения по изобретению для получения лекарственного средства для терапевтического или профилактического лечения заболевания или болезненного состояния, в которые вовлечен катепсин К. Настоящее изобретение относится к способам применения соединений по изобретению и их фармацевтически приемлемых солей или их фармацевтически приемлемых композиций для ингибирования катепсина К у млекопитающих и к лечению зависимых от катепсина К состояний, таких, как описанные в контексте, например, воспаления, остеопороза, ревматоидного артрита и остеоартрита. В частности, настоящее изобретение относится к способу селективного ингибирования активности катепсина К у млекопитающих, который включает введение нуждающемуся в этом млекопитающему эффективного игибирующего катепсин К количества соединения по изобретению. Более конкретно такая связь со способом лечения остеопороза, ревматоидного артрита и воспаления (и других заболеваний, как описано выше) у млекопитающих включает введение нуждающемуся в этом млекопитающему соответственно эффективного количества соединения по изобретению. Подразумевается, что следующие примеры иллюстрируют изобретение и не должны рассматриваться как ограничивающие его. Температуры приведены в °С. Если не указано иначе, все упаривания осуществляли при пониженном давлении, предпочтительно между примерно 15 и 100 мм рт.ст. (=20-133 мбар). Структура конечных продуктов, промежуточных и исходных материалов подтверждены стандартными аналитическими методиками, например, микроанализом и спектроскопическими характеристиками (например, МС, ИК, ЯМР). Использованные сокращения являются общепринятыми в этой области техники. Примеры A. FMOC-1-аминоциклогексанкарбоновая кислота Заглавное соединение получали по общеизвестным методикам из 1-аминоциклогексанкарбоновой кислоты (700 ммоль), FMOC-хлорида (770 ммоль), диизопропилэтиламина (770 ммоль) и 770 мл 1 н. гидроксида натрия в 950 мл диоксана; tпл 180-182°C; Rf=0,21 (CH2Cl2/МеОН, 95:5). В качестве растворителя вместо диоксана можно использовать ацетонитрил. Б. Цианометиламид FMOC-1-аминоциклогексанкарбоновой кислоты Гидрохлорид 2-аминоацетонитрила (564 ммоль) и триэтиламин (564 ммоль) растворяли в ДМФА (1700 мл). Прибавляли FMOC-1-аминоциклогексанкарбоновую кислоту (564 ммоль), HOBt (564 ммоль) и WSCD (564 ммоля), смесь перемешивали при 25°С в течение ночи. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и экстрагировали трижды этилацетатом. Экстракт промывали водой, 10% лимонной кислотой, насыщенным раствором соли, сушили над сульфатом магния и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 167-169°С, Rf=0,27 (н-гексан: этилацетат = 1:1). Альтернативно в реакции конденсации можно использовать ТГФ в качестве растворителя и 1-хлор-3,5-диметокситриазин (CDMT) в качестве активатора наряду с N-метилморфолином (NMM); в этом случае продукт может быть выделен путем добавления изопропилацетата и воды, отделением органической фазы, последующим промыванием с насыщенным раствором соли, частичным упариванием растворителя, отделением кристаллического продукта фильтрованием и высушиванием. В. Цианометиламид 1-аминоциклогексанкарбоновой кислоты Растворяли цианометиламид FMOC-1-аминоциклогексанкарбоновой кислоты (248 ммоль) в ДМФА (200 мл), прибавляли пиперидин (248 мл), смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь выливали в воду (3000 мл) и перемешивали в течение 30 мин. Суспензию фильтровали, фильтрат подкисляли с 4 н. соляной кислотой и затем экстрагировали этилацетатом. Прибавляли 1 н. гидроксид натрия, чтобы сделать водную фазу щелочной, и трижды экстрагировали смесь этилацетатом. Органические фракции сушили над сульфатом натрия, растворитель упаривали. Осадок сушили (вакуум), получая бледно-желтое масло. Rf=0,26 (CH2Cl2:MeOH=95:5). 1Н-ЯМР (d6-ДМСО): 1,05-1,80 (m, 10Н); 4.0 (уш. s, 2H); NH (очень широкий сигнал). Альтернативно на этапе удаления FMOC-защитной группы можно использовать ТГФ вместо ДМФА и диэтиламин вместо пиперидина. Пример 1: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-пиперазин-1-илбензамида А. Метиловый эфир 4-пиперазин-1-илбензойной кислоты Растворяли 1-(4-цианофенил)пиперазин (11 ммоль) в 15 мл смеси концентрированной сульфоновой кислоты и метанола (5 н.) и перемешивали в запаянной трубке при 110°С в течение 3 ч. После упаривания растворителя остаток растворяли в воде и экстрагировали этилацетатом. Прибавление карбоната натрия к водной фазе до рН 9 привело к осаждению белого твердого вещества, которое отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с Rf=0,59 (CH2Cl2:MeOH(+3 н. NH3)=9:1). Б. Гидрохлорид 4-пиперазин-1-илбензойной кислоты Растворяли метиловый эфир 4-пиперазин-1-илбензойной кислоты (17 ммоль) в 6 н. соляной кислоте (25 мл) и нагревали при кипении в течение 3 ч. Смесь охлаждали в бане со льдом до 0-4°С, образовавшееся твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок белого цвета с tпл>240°С. В. 4-(4-FMOC-пиперазин-1-ил)бензойная кислота Растворяли гидрохлорид 4-пиперазин-1-илбензойной кислоты (10,5 ммоль) в 15 мл диоксана и 11,6 мл гидроксида натрия (2 н.) и охлаждали до 0°С. Одновременно прибавляли по каплям FMOC-хлорид (11,6 ммоль) в диоксане (5 мл) и диизопропилэтиламин (11,6 ммоль) в диоксане (5 мл) в течение 20 мин при 0°С, смесь перемешивали в течение 15 мин, затем давали нагреться до комнатной температуры и перемешивали в течение ночи. Смесь разбавляли водой (50 мл) и дважды экстрагировали диэтиловым эфиром. Водную фазу подкисляли водной соляной кислотой (4 н.) при 0-4°С, образовавшееся твердое вещество отделяли фильтрованием, промывали водой и сушили (вакуум). Получали порошок белого цвета с Rf=0,2 (CH2Cl2:MeOH=95:5). Г. N-[1-(Цианометилкарбамоил) циклогексил1-4-(4-FMOC-пиперазин-1-ил)бензамид Растворяли цианометиламид 1-аминоциклогексанкарбоновой кислоты (8,3 ммоль), 4-(4-РМОС-пиперазин-1-ил)бензойную кислоту (8,3 ммоль), НОВТ (8,3 ммоль) и WSCD (8,3 ммоль) в ДМФА (20 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы создать слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью этилацетат/гексан (4:1) в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 192-194°С, Rf=0,26 (CH2Cl2:MeOH=95:5). Д. N-[1-(Цианометилкарбамоил)циклогексил]-4-(пиперазин-1-ил)бензамид Растворяли N-[1-(цианометилкарбамоил)циклогексил]-4-(4-FMOC-пиперазин-1-ил)бензамид (4,4 ммоль) в ДМФА (15 мл), прибавляли пиперидин (4,4 ммоль), смесь перемешивали при комнатной температуре в течение 4 ч. Прибавляли 4 дополнительных капли пиперидина и смесь перемешивали в течение ночи. Реакционную смесь выливали в смесь воды и этилацетата, суспензию фильтровали, фильтрат подкисляли 4 н. соляной кислотой, затем экстрагировали этилацетатом. Прибавляли насыщенный раствор карбоната натрия, чтобы сделать водную фазу основной, смесь трижды экстрагировали этилацетатом. Водную фазу насыщали хлористым натрием и снова трижды экстрагировали этилацетатом. Органические фракции сушили над сульфатом натрия, растворитель упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью CH2Cl2MeOH (+3 н. NH3)=95:5 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 206-210°С, Rf=0,28 (CH2Cl2:MeOH (+3 н. NH3)=9:1). 1Н-ЯМР (d6-ДМСО): 1,15-1,35 (m, 1H); 1,4-1,6 (m, 5H); 1,65-1,8 (m, 2H); 2,05-2,15 (m, 2H); 2,8 (m, 4H); 3,15 (m, 4H); 4,0 (d, 2H), 6,95 (d, 2H); 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). Пример 2: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-(метилпиперазин-1-ил)бензамида А. Метиловый эфир 4-(4-метилпиперазин-1-ил)бензойной кислоты Метиловый эфир 4-фторбензойной кислоты (34 ммоль), 1-метилпиперазин (75 ммоль) и карбонат калия (34 ммоль) суспендировали в ацетонитриле (30 мл) и перемешивали при кипении в течение трех дней. После упаривания растворителя остаток растворяли в воде и трижды экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью СН2Cl2:МеОН=95:5 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок бледно-желтого цвета с tпл 117-119°С, Rf=0,20 (CH2Cl2:MeOH=95:5). Б. Гидрохлорид 4-(4-метилпиперазин-1-ил)бензойной кислоты Растворяли метиловый эфир 4-(4-метилпиперазин-1-ил)бензойной кислоты (8,5 ммоль) в 4 н. соляной кислоте (15 мл) и нагревали при кипении в течение 8 ч. Смесь охлаждали в бане со льдом до 0-4°С, разбавляли 5 мл ацетона, твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок белого цвета с tпл>270°C, Rf=0,11 (CH2Cl2:MeOH=9:1). В. N-[1-(Цианометилкарбамоил)циклогексил]-4-(4-метилпиперазин-1-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (1,38 ммоль), гидрохлорид 4-(4-метилпиперазин-1-ил)бензойной кислоты (1,38 ммоль), НОВТ (1,38 ммоль), WSCD (1,38 ммоль) и триэтиламин (1,38 ммоль) растворяли в ДМФА (5 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 218-220°С, Rf=0,19 (CH2Cl2:MeOH=9:1). 1H-ЯМР (d6-ДМСО): 1,15-1,35 (m, 1H); 1,4-1,6 (m, 5H); 1,65-1,8 (m, 2H); 2,05-2,15 (m, 2H); 2,2 (s, 3Н); 2,4 (m, 4H); 3,2 (m, 4H); 4,0 (d, 2H), 6,95 (d, 2H); 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). Пример 3: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-(4-этилпиперазин-1-ил)бензамида А. Метиловый эфир 4-(4-этилпиперазин-1-ил)бензойной кислоты Метиловый эфир 4-фторбензойной кислоты (53 ммоль), 1-этилпиперазин (44 ммоль) и карбонат калия (44 ммоль) суспендировали в диметилацетамиде (50 мл) и перемешивали при кипении в течение ночи. После упаривания растворителя остаток растворяли в воде и трижды экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали коричневатый порошок с tпл 102-104°C, Rf=0,22 (CH2Cl2:MeOH=95:5). Б. Гидрохлорид 4-(4-этилпиперазин-1-ил)бензойной кислоты Метиловый эфир 4-(4-этилпиперазин-1-ил)бензойной кислоты (15 ммоль) растворяли в 4 н. соляной кислоте (35 мл) и нагревали при кипении в течение 8 ч. Смесь охлаждали в бане со льдом до 0-4°С, образовавшееся твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок серого цвета с tпл>270°С, Rf=0,08 (CH2Cl2:MeOH=9:1). В. N-[1-(Цианометилкарбамоил)циклогексил1-4-(4-этилпиперазин-1-ил)-бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (0,9 ммоль), гидрохлорид 4-(4-этилпиперазин-1-ил)бензойной кислоты (0,9 ммоль), НОВТ (0,9 ммоль), WSCD (0,9 ммоль) и триэтиламин (0,9 ммоль) растворяли в ДМФА (5 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью СН2Cl2:МеОН (+3 н. NH3)=93:7 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета. 1Н-ЯМР (d6-ДМСО): 1,0 (t, 3Н), 1,15-1,35 (m, 1H); 1,4-1,6 (m, 5H); 1,65-1,8 (m, 2H); 2,05-2,15 (m, 2H); 2,35 (q, 2H); 2,45 (m, 4H); 3,2 (m, 4H); 4,0 (d, 2H), 6,95 (d, 2H); 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). Пример 4: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-[4-(1-пропил)пиперазин-1-ил]бензамида А. Метиловый эфир 4-[4-(1-пропил)пиперазин-1-ил]бензойной кислоты Метиловый эфир 4-фторбензойной кислоты (165 ммоль), дигидробромид 1-(1-пропил)пиперазина (138 ммоль) и карбонат калия (690 ммоль) суспендировали в диметилацетамиде (320 мл) и перемешивали при кипении в течение ночи. После упаривания растворителя остаток растворяли в воде и трижды экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали коричневатый порошок с tпл 99-101°С, Rf=0,23 (CH2Cl2:MeOH=95:5). В описанной выше методике вместо карбоната калия можно использовать карбонат цезия. Б. Гидрохлорид 4-[4-(1-пропил)пиперазин-1-ил]бензойной кислоты Метиловый эфир 4-[4-(1-пропил)пиперазин-1-ил]бензойной кислоты (38 ммоль) растворяли в 4 н. соляной кислоте (60 мл) и нагревали при кипении в течение 7 ч. Смесь охлаждали в бане со льдом до 0-4°С, образовавшееся твердое вещество отделяли фильтрованием, промывали холодной водой и сушили (вакуум). Получали белесый порошок с tпл>270°C, Rf=0,19 (CH2Cl2:MeOH=9:1). Альтернативно 4-[4-(1-пропил)пиперазин-1-ил]бензойная кислота может быть получена в виде внутренней соли с уксусной кислотой. Например, метиловый эфир 4-[4-(1-пропил)пиперазин-1-ил]бензойной кислоты суспендировали в смеси вода/метанол при 70°С и гидролизовали путем добавления 1 экв. гидроксида натрия; раствор осветляли фильтрованием, продукт осаждали добавлением 1 экв. уксусной кислоты, отделяли фильтрованием и сушили. В. N-[-[1-(Цианометилкарбамоил)циклогексил]-4-[4-(1-пропил)пиперазин-1-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (22 ммоль), гидрохлорид 4-[4-(1-пропил)пиперазин-1-ил]бензойной кислоты (22 ммоль), НОВТ (22 ммоль), WSCD (22 ммоль) и триэтиламин (22 ммоль) растворяли в ДМФА (50 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью (CH2Cl2:MeOH=9:1) в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 216-218°С, Rf=0,34 (CH2Cl2:MeOH=9:1). 1Н-ЯМР (d6-ДМСО): 0,85 (t, 3Н), 1,2-1,3 (m, 1H); 1.4-1,6 (m, 7Н); 1,65-1,8 (m, 2Н); 2,05-2,15 (m, 2Н); 2,25 (t, 2Н); 2,45 (m, 4Н); 3,2 (m, 4H); 4,0 (d, 2H), 6,95 (d, 2H); 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). По альтернативной методике внутреннюю соль уксусной кислоты с 4-[4-(1-пропил)пиперазин-1-ил)бензойной кислотой в ацетонитриле обрабатывали НОВТ, NMM (нитрозометилмочевиной) и диизопропилкарбодиимидом (DICI), после перемешивания в течение 1 ч при 40°С прибавляли раствор цианометиламида 1-аминоциклогексанкарбоновой кислоты в ацетонитриле. По завершении реакции продукт осаждали добавлением воды к реакционной смеси, фильтровали и последующим соупариванием с этанолом окончательно высушивали продукт. Пример 5: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-(4-изопропилпиперазин-1-ил)бензамида А. Метиловый эфир 4-[4-изопропилпиперазин-1-ил]бензойной кислоты Трис(дибензилиденацетон)дипалладий (0,05 ммоль), (2-дициклогексилфосфанилбифенил-2-ил)диметиламин (0,1 ммоль) и карбонат калия (4,6 ммоля) суспендировали в 1,2-диметоксиэтане (10 мл) в атмосфере N2, не содержащей кислорода. Прибавляли метиловый эфир 4-бромбензойной кислоты (3,3 ммоль) и 1-изопропилпиперазин (3,9 ммоль), перемешиваемую смесь нагревали при кипении в течение 28 ч. После охлаждения растворитель упаривали, к остатку прибавляли воду, затем трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью CH2Cl2:MeOH=95:5 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок бледно-коричневого цвета с Rf=0,23 (CH2Cl2:MeOH=95:5). Б. Гидрохлорид 4-(4-изопропилпиперазин-1-ил)бензойной кислоты Метиловый эфир 4-(4-изопропилпиперазин-1-ил)бензойной кислоты (0,9 ммоль) растворяли в 4 н. соляной кислоте (2 мл) и нагревали при кипении в течение 7 ч. Смесь охлаждали в бане со льдом до 0-4°С и прибавляли ацетон. Образовавшееся твердое вещество отделяли фильтрованием, промывали холодным ацетоном и сушили (вакуум). Получали порошок бледно-коричневого цвета с tпл>270°C, Rf=0,08 (CH2Cl2:MeOH=9:1). В. N-[1-(Цианометилкарбамоил)циклогексил]-4-(4-изопропилпиперазин-1-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (0,6 ммоль), гидрохлорид 4-(4-изопропилшшеразин-1-ил)бензойной кислоты (0,6 ммоль), НОВТ (0,6 ммоль), WSCD (0,6 ммоль) и триэтиламин (0,6 ммоль) растворяли в ДМФА (2 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в смеси этилацетат/диэтиловый эфир, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 218-220°C, Rf=0,28 (CH2Cl2:MeOH=9:1). 1Н-ЯМР (d6-ДМСО): 1,0 (d, 6Н), 1,2-1,3 (m, 1H); 1,4-1,6 (m, 5H); 1,65-1,8 (m, 2Н); 2,05-2,15 (m, 2H); 2.45 (m, 4Н); 2,65 (m, 1H); 3,2 (m, 4H); 4,0 (d, 2H), 6,95 (d, 2Н); 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). Пример 6: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-(4-бензилпиперазин-1-ил)бензамида А. Метиловый эфир 4-(4-бензилпиперазин-1-ил)бензойной кислоты Суспендировали трис(дибензилиденацетон)дипалладий (0,03 ммоль), (2-дициклогексилфосфанилбифенил-2-ил)диметиламин (0,9 ммоль) и трет.-бутилат натрия (6,5 ммоль) в толуоле (20 мл) в атмосфере без кислорода (N2). Прибавляли метиловый эфир 4-бромбензойной кислоты (4,65 ммоль) и 1-(бензил)пиперазин (5,6 ммоль), перемешиваемую смесь нагревали при кипении в течение 4 ч. После охлаждения прибавляли смесь этилацетата и диэтилового эфира, смесь фильтровали. Затем растворитель упаривали, остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 105-107°C, Rf=0,67 (СН2Cl2:МеОН=95:5). Б. Гидрохлорид 4-(4-бензилпиперазин-1-ил)бензойной кислоты Метиловый эфир 4-(4-бензилпиперазин-1-ил)бензойной кислоты (0,84 ммоль) растворяли в 4 н. соляной кислоте (2 мл) и нагревали при кипении в течение 8 ч. Смесь охлаждали в бане со льдом до 0-4°С, образовавшееся твердое вещество отделяли фильтрованием, промывали холодным ацетоном и сушили (вакуум). Получали порошок серого цвета с tпл>270°С, Rf=0,18 (СН2Cl2:МеОН=95:5). В. N-[1-Цианометилкарбамоил)циклогексил]-4-(4-бензилпиперазин-1-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (0,84 ммоль), гидрохлорид 4-[4-(2-пропил)пиперазин-1-ил]бензойной кислоты (0,84 ммоль), НОВТ (0,84 ммоль), WSCD (0,84 ммоль) и триэтиламин (0,84 ммоль) растворяли в ДМФА (2 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в метаноле, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 210-212°C, Rf=0,20 (CH2Cl2:MeOH=95:5). 1Н-ЯМР (d6-ДМСО): 1,15-1,35 (m, 1H); 1,4-1,6 (m, 5H); 1,65-1,8 (m, 2H); 2,05-2,15 (m, 2H); 2,45 (m, 4H); 3,2 (m, 4H); 3,5 (s, 2H); 4,0 (d, 2H), 6,9 (d, 2H); 7,2-7,4 (m, 5H), 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). Пример 7: синтез Н-[1-(цианометил1карбамоил)циклогексил1-4-[4-(2-метоксиэтил)пиперазин-1-ил]бензамида А. Метиловый эфир 4-(4-бензилпиперазин-1-ил)бензойной кислоты Метиловый эфир 4-фторбензойной кислоты (200 ммоль), 1-бензилпиперазин (300 ммоль) и карбонат калия (300 ммоль) суспендировали в ацетонитриле (400 мл) и перемешивали при кипении в течение 6 дней. После упаривания растворителя остаток растворяли в воде и трижды экстрагировали диэтиловым эфиром. Экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле с (CH2Cl2 сначала, затем CH2Cl2:MeOH=15:1) в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок с tпл 105-107°С. Б. Метиловый эфир 4-(пиперазин-1-ил)бензойной кислоты Метиловый эфир 4-(4-бензилпиперазин-1-ил)бензойной кислоты (19,4 ммоль) растворяли в метаноле (150 мл) и прибавляли Pd/C (0,6 г). Смесь перемешивали в атмосфере водорода до прекращения его поглощения. Катализатор удаляли фильтрованием, фильтрат упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок с tпл 95-97°C. В. Метиловый эфир [4-(2-метоксиэтил)пиперазин-1-ил]бензойной кислоты Метиловый эфир 4-(пиперазин-1-ил)бензойной кислоты (19 ммоль), 2-бромэтилметиловый эфир (21 ммоль) и карбонат калия суспендировали в ацетонитриле (950 мл) и перемешивали при 80°С в течение 8 ч. После упаривания растворителя остаток растворяли в воде и трижды экстрагировали метиленхлоридом. Экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок с tпл 103-105°С. Г. Гидрохлорид [4-(2-метоксиэтил)пиперазин-1-ил]бензойной кислоты Метиловый эфир [4-(2-метоксиэтил)пиперазин-1-ил]бензойной кислоты (17 ммоль) растворяли в 4 н. соляной кислоте и нагревали при кипении в течение 5 ч. После охлаждения растворитель упаривали, остаток суспендировали в этаноле, твердое вещество отделяли фильтрованием, промывали диэтиловым эфиром и сушили (вакуум). Получали порошок с tпл>270°С, Rf=0,35 (CH2Cl2:MeOH=9:1). Д. N-[1-(Цианометилкарбамоил)циклогексил]-4-[4-(2-метоксиэтил)пиперазин-1-ил]бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (1,0 ммоль), гидрохлорид [4-(2-метоксиэтил)пиперазин-1-ил]бензойной кислоты (1,0 ммоль), НОВТ (1,0 ммоль), WSCD (1,0 ммоль) и триэтиламин (1,0 ммоль) растворяли в ДМФА (4 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью СН2Cl2/МеОН (92,5:7,5) в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 166-168°C, Rf=0,37 (CH2Cl2:MeOH=9:1). 1Н-ЯМР (d6-ДМСО): 1,15-1,35 (m, 1H); 1,4-1,6 (m, 5H); 1,65-1,8 (m. 2H); 2,05-2,15 (m, 2H); 2,45 (m, 6H); 3,2 (m, 7H); 3,45 (t, 2H); 4,0 (d, 2H), 6,95 (d, 2H); 7,65 (s, 1H); 7,75 (d, 2H), 8,15 (m, 1H). Пример 8: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-(1-пропилпиперидин-4-ил)бензамид А. 1-(4-Фенилпиперидин-1-ил)этанон Растворяли 4-фенилпиперидин (87 ммоль) и пиридин (96 ммоль) в безводном метиленхлориде (100 мл) и к перемешиваемому раствору прибавляли по каплям при 10°С ацетилхлорид (96 ммоль) в метиленхлориде (40 мл). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Смесь трижды экстрагировали водой, водную фазу снова экстрагировали метиленхлоридом. Объединенные органические фазы сушили над сульфатом натрия и упаривали. Получали бледно-коричневое масло с Rf=0,13 (этилацетат/гексан, 1:1). Б. 4-Пиперидин-4-илбензойная кислота Растворяли 1-(4-фенилпиперидин-1-ил)этанон (84 ммоль) в метиленхлориде (250 мл) и прибавляли по каплям оксалилхлорид (336 ммоль) при -20 до -10°С. После добавления оксалилхлорида прибавляли порциями треххлористый алюминий (260 ммоль) при -10°С. Смесь перемешивали при -10°С в течение 3 ч. Охлаждающую баню убирали и смесь перемешивали в течение ночи при комнатной температуре. Реакционную массу выливали на смесь лед/вода (600 мл) и трижды экстрагировали метиленхлоридом. Объединенные органические фазы промывали водой, сушили над сульфатом натрия и упаривали. Остаток растворяли в водном растворе гидроксида натрия (2 н., 250 мл) и прибавляли при 0°С 6 н. соляную кислоту, чтобы подкислить раствор. Образовавшийся осадок отделяли фильтрованием и промывали водой. Твердое вещество суспендировали в 6 н. соляной кислоте, смесь нагревали при кипении в течение 18 ч. После охлаждения до комнатной температуры растворитель удаляли, остаток суспендировали в этаноле. Твердое вещество отделяли фильтрованием и сушили. Получали порошок коричневого цвета с tпл>270°C. В. Метиловый эфир 4-пиперидин-4-илбензойной кислоты Растворяли 4-пиперидин-4-илбензойную кислоту (47 ммоль) в метаноле (300 мл) и прибавляли 1 мл концентрированной сульфоновой кислоты. Смесь нагревали при кипении в течение ночи. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить основные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Получали порошок коричневого цвета с Rf=0,18 (CH2Cl2:MeOH=9:1). Г. Метиловый эфир 4-(1-пропилпиперидин-4-ил)бензойной кислоты Метиловый эфир 4-пиперидин-4-илбензойной кислоты (28 ммоль), этилизопропиламин (31 ммоль) и 1-йодпропан (42 ммоль) растворяли в 1,2-диметоксиэтане (100 мл) и смесь нагревали при 70°С в течение ночи. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить основные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле с CH2Cl2:MeOH=9:1 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок бледно-коричневого цвета с Rf=0,35 (CH2Cl2:MeOH=9:1). Д. Гидрохлорид 4-(1-пропилпиперидин-4-ил)бензойной кислоты Метиловый эфир 4-(1-пропилпиперидин-4-ил)бензойной кислоты (32 ммоль) растворяли в 4 н. соляной кислоте (45 мл) и нагревали при кипении в течение 7 ч. Смесь охлаждали в бане со льдом до 0-4°С, образовавшееся твердое вещество отделяли фильтрованием, промывали холодным ацетоном и сушили (вакуум). Получали порошок коричневого цвета с tпл>2700C, Rf=0,08 (CH2Cl2:MeOH=9:1). Е. N-[1-(Цианометилкарбамоил)циклогексил]-4-(1-пропилпиперидин-4-ил)-бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (23 ммоль), гидрохлорид 4-(1-пропилпиперидин-4-ил)бензойной кислоты (23 ммоль), НОВТ (23 ммоль), WSCD (23 ммоль) и триэтиламин (23 ммоль) растворяли в ДМФА (50 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить основные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 198-200°C, Rf=0,34 (CH2Cl2:MeOH=9:1). 1Н-ЯМР (d6-ДМСО): 0,85 (t, 3Н); 1,2-1,3 (m, 1H); 1.4-1,6 (m, 7H); 1,6-1,8 (m. 6Н); 1,9-2,0 (m, 2H); 2,05-2,15 (m, 2Н); 2,25 (t, 2H); 2,55 (m, 1H); 2,95 (d, 2H); 4,0 (d, 2H), 7,35 (d, 2H); 7,8 (d, 2H), 7,9 (s, 1H); 8.15 (m, 1H). Пример 9: синтез N-[1-(цианометилкарбамоил)циклогексил]-4-[1-(2-метоксиэтил)пиперидин-4-ил]бензамида А. Метиловый эфир 4-карбоксифенилборной кислоты Растворяли 4-карбоксифенилборную кислоту (300 ммоль) в метаноле (400 мл) и к перемешиваемому раствору прибавляли 1,5 мл концентрированной соляной кислоты. Реакционную смесь нагревали при кипении в течение 30 ч. Растворитель упаривали, образующийся остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 201-205°C, Rf=0,28 (CH2Cl2:MeOH=95:5). Данный порошок, являющийся смесью метилового эфира 4-карбоксифенилборной кислоты и димерного ангидрида метилового эфира 4-карбоксифенилборной кислоты, использовали без дальнейшей очистки. Б. Метиловый эфир 4-пиридин-4-илбензойной кислоты Метиловый эфир 4-карбоксибензилборной кислоты (248 ммоль) из раздела А, 4-бромпиридин (248 ммоль), тетракис(трифенилфосфин)палладий (2,5 ммоль) и карбонат калия (744 ммоль) суспендировали в 1,2-диметоксиэтане (1100 мл). Перемешиваемую смесь нагревали при кипении в течение 8 ч. После охлаждения растворитель упаривали, к остатку прибавляли воду и затем трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок светло-коричневого цвета с tпл 99-101°C, Rf=0,39 (CH2Cl2:MeOH=95:5). В. 4-(4-Метоксикарбонилфенил)-1-(2-метоксиэтил)пиридинийбромид Метиловый эфир 4-пиридин-4-илбензойной кислоты (70 ммоль) и 2-бромэтилметиловый эфир (28 мл) нагревали в течение 1 ч при 110°С. После охлаждения реакционную смесь суспендировали в ацетоне, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок бледно-коричневого цвета с tпл 170-171°C, Rf=0,22 (CH2Cl2:MeOH=9:1). Г. Метиловый эфир 4-[1-(2-метоксиэтил)пиперидин-4-ил[бензойной кислоты Суспендировали 4-(4-метоксикарбонилфенил)-1-(2-метоксиэтил)пиридинийбромид (67 ммоль) в метаноле (250 мл) и прибавляли оксид платины (1,2 г). Смесь перемешивали в атмосфере водорода при нормальном давлении до прекращения поглощения водорода. Катализатор удаляли фильтрованием, фильтрат упаривали. Остаток растворяли в метиленхлориде и экстрагировали водным раствором карбоната натрия. Органическую фазу сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью CH2Cl2:MeOH=9:1 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Получали масло бледно-желтого цвета с Rf=0,22 (CH2Cl2:MeOH=95:5). Д. Гидрохлорид 4-[1-(2-метоксиэтил)пиперидин-4-ил]бензойной кислоты Метиловый эфир 4-[1-(2-метоксиэтил)пиперидин-4-ил]бензойной кислоты (47 ммоль) растворяли в 4 н. соляной кислоте (80 мл) и нагревали при кипении в течение 12 ч. После охлаждения растворитель упаривали, остаток суспендировали в ацетоне, твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок белого цвета с tпл>270°С. Е. N-[1-(Цианометилкарбамоил)циклогексил]-4-[1-(2-метоксиэтил)пиперидин-4-ил]бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (107 ммоль), гидрохлорид 4-[1-(2-метоксиэтилпиперидин-4-ил]бензойной кислоты (107 ммоль), НОВТ (107 ммоль), WSCD (107 ммоль) и триэтиламин (107 ммоль) растворяли в ДМФА (250 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить слабоосновные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью СН2Cl2:МеОН (с 2 н. NH3)=9:1 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в смеси диэтиловый эфир/этилацетат, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 160-162°C, Rf=0,42 (СН2Cl2:МеОН (с 3 н. HN3)=9:1). 1Н-ЯМР (d6-ДМСО): 1,2-1,3 (m, 1H); 1,4-1,6 (m, 5H); 1,6-1,8 (m, 6H); 2,0-2.2 (m, 4Н); 2,45 (m, 2H); 2,55 (m, 1H); 2,95 (уш. d, 2H); 3,2 (s, 3Н); 3,4 (dd, 2H); 4,0 (d, 2H); 7,35 (d, 2H); 7.8 (d, 2H); 7,9 (s, 1H); 8,15 (m, 1H). Пример 10: N-[1-(цианометилкарбамоил)циклогексил]-4-(1-изопропилпиперидин-4-ил)бензамид А. 1-Изопропил-4-(4-метоксикарбонилфенил)пиридинийбромид Метиловый эфир 4-пиридин-4-илбензойной кислоты (2,3 ммоль) и 2-йодпропан (1,0 мл) нагревали в течение 24 ч при 90°С. После охлаждения растворитель упаривали, остаток суспендировали в ацетоне, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок бледно-желтого цвета с tпл 187-189°C, Rf=0,27 (CH2Cl2:MeOH=9:1). Б. Гидройодид метилового эфира 4-(1-изопропилпиперидин-4-ил)бензойной кислоты Суспендировали 1-изопропил-4-(4-метоксикарбонилфенил)пиридинийбромид (1,9 ммоль) в метаноле (10 мл) и прибавляли оксид платины 980 мг). Смесь перемешивали в атмосфере водорода при нормальном давлении до прекращения поглощения водорода. Катализатор удаляли фильтрованием, фильтрат упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 219-224°C, Rf=0,41 (CH2Cl2:MeOH=9:1). В. Гидрохлорид 4-(1-изопропилпиперидин-4-ил)бензойной кислоты Растворяли гидройодид метилового эфира 4-(1-изопропилпиперидин-4-ил)бензойной кислоты в 4 н. соляной кислоте (5 мл) и нагревали при кипении в течение 10 ч. После охлаждения растворитель упаривали, остаток суспендировали в ацетоне, твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок серо-коричневого цвета с tпл>270°C. Г. N-[1-(Цианометилкарбамоил)циклогексил-4-(1-изопропилпиперидин-4-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (0,95 ммоль), гидрохлорид 4-(1-изопропилпиперидин-4-ил]бензойной кислоты (0,95 ммоль), НОВТ (0,95 ммоль), WSCD (0,95 ммоль) и триэтиламин (0,95 ммоль) растворяли в ДМФА (5 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить основные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 214-216°С, Rf=0,38 (CH2Cl2:MeOH (с 3 н. NH3)=9:1). 1Н-ЯМР (d6-ДМСО): 0,95 (d, 6Н); 1,2-1,3 (m, 1H); 1,4-1,8 (m, 11H); 2,05-2,25 (m, 4H); 2,55 (m, 1H); 2,7 (m, 1H); 2,85 (d, 2H); 4,0 (d, 2H), 7,35 (d, 2H); 7,8 (d, 2Н), 7,9 (s, 1H); 8,15 (m, 1Н). Пример 11: N-[1-(цианометилкарбамоил)циклогексил]-4-(1-циклопентилпиперидин-4-ил)бензамид А. 1-Циклопентил-4-(4-метоксикарбонилфенил)пиридинийбромид Метиловый эфир 4-пиридин-4-илбензойной кислоты (2,35 ммоль) и 1-йодциклопентан (1,0 мл) нагревали в течение 4 ч при 110°С. Прибавляли йодциклопентан (0,5 мл) и нагревали смесь в течение еще 4 ч при 120°С. После охлаждения растворитель упаривали, остаток суспендировали в ацетоне, твердое вещество отделяли фильтрованием и сушили (вакуум). Твердый остаток очищали флэш-хроматографией на силикагеле со смесью CH2Cl2:MeOH=9:1 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок желтого цвета с tпл 183-185°С, Rf=0,35 (CH2Cl2:MeOH=9:1). Б. Гидройодид метилового эфира 4-(1-циклопентилпиперидин-4-ил)бензойной кислоты Суспендировали 1-циклопентил-4-(4-метоксикарбонилфенил)пиридинийбромид (1,27 ммоль) в метаноле (8 мл) и прибавляли оксид платины (50 мг). Смесь перемешивали в атмосфере водорода при нормальном давлении до прекращения поглощения водорода. Катализатор удаляли фильтрованием, фильтрат упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали белесый порошок с tпл 204-210°C, Rf=0,27 (CH2Cl2:MeOH=95:5). В. Гидрохлорид 4-(1-пиклопентилпиперидин-4-ил)бензойной кислоты Гидрохлорид метилового эфира 4-(1-циклопентилпиперидин-4-ил)бензойной кислоты (1,06 ммоль) растворяли в 4 н. соляной кислоте (5 мл) и нагревали при кипении в течение 10 ч. После охлаждения растворитель упаривали, остаток суспендировали в ацетоне, твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок серо-коричневого цвета с tпл>270°С. Г. N-[1-(Цианометилкарбамоил)циклогексил]-4-(1-циклопентилпиперидин-4-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (0,74 ммоль), гидрохлорид 4-(1-циклопентилпиперидин-4-ил)бензойной кислоты (0,74 ммоль), НОВТ (0,74 ммоль), WSCD (0,74 ммоль) и триэтиламин (0,74 ммоль) растворяли в ДМФА (5 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить основные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в диэтиловом эфире, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 233-234°C, Rf=0,34 (СН2Cl2:МеОН (с 3 н. NH3)=9:1). 1Н-ЯМР (d6-ДМСО): 1,2-1,85 (m, 20H); 1,9-2,15 (m, 4H); 2,4-2,6 (m, 2H); 3,05 (d, 2H); 4,0 (d, 2H), 7,35 (d, 2H); 7,8 (d, 2H), 7,9 (s, 1H); 8.15 (m, 1H). Пример 12: N-[1-(цианометилкарбамоил)циклогексил]-4-(1-метилпиперидин-4-ил)бензамид А. 4-Фенил-1-метилпиперидин Суспендировали 4-фенилпиперидин (12,4 ммоль), параформальдегид (24,8 ммоль) и тетраизопропоксититан (12,4 ммоль) в 1,2-диметоксиэтане (20 мл), нагревали до 60°С в течение 30 мин и перемешивали при комнатной температуре дополнительно 1 ч. Порциями прибавляли натрийборгидрид (12,4 ммоль), смесь перемешивали при комнатной температуре в течение 2 ч и при 60°С в течение дополнительных 3 ч. После охлаждения растворитель упаривали, остаток растворяли в смеси водного аммиака (60 мл) и этилацетата и осторожно фильтровали. Смесь трижды экстрагировали этилацетатом, объединенные органические фазы сушили над сульфатом натрия и упаривали. Получали масло бледно-коричневого цвета. Б. Метиловый эфир 4-(1-метилпиперидин-4-ил)бензойной кислоты Растворяли 4-фенил-1-метилпиперидин (9,9 ммоль) в метиленхлориде (60 мл) и прибавляли по каплям оксалилхлорид (39,6 ммоля) при -20 до -10°С. После добавления оксалилхлорида прибавляли порциями при -10°С треххлористый алюминий. Смесь перемешивали при -10°С в течение 1,5 ч. Затем охлаждающую баню убирали, смесь перемешивали при комнатной температуре дополнительные 2 ч. Смесь снова охлаждали до 0°С и по каплям прибавляли метанол (30 мл). По завершении прибавления метанола охлаждающую баню удаляли и смесь перемешивали при комнатной температуре в течение ночи. Реакционную массу выливали в смесь водного карбоната натрия (чтобы обеспечить основные условия) и этилацетата, суспензию осторожно фильтровали. Фильтрат трижды экстрагировали этилацетатом, объединенные экстракты сушили над безводным сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле со смесью CH2Cl2:MeOH=9:1 в качестве мобильной фазы. Фракции, содержащие продукт, объединяли и упаривали. Получали масло желтого цвета с Rf=0,29 (CH2Cl2:MeOH=9:1). В. Гидрохлорид 4-(1-метилпиперидин-4-ил)бензойной кислоты Метиловый эфир 4-(1-метилпиперидин-4-ил)бензойной кислоты (4,55 ммоль) растворяли в 4 н. соляной кислоте (10 мл) и нагревали при кипении в течение 8 ч. После охлаждения растворитель упаривали, остаток суспендировали в ацетоне, твердое вещество отделяли фильтрованием, промывали ацетоном и сушили (вакуум). Получали порошок бледно-коричневого цвета с tпл>270°С. Г. N-[1-(Цианометилкарбамоил)циклогексил]-4-(1-метилпиперидин-4-ил)бензамид Цианометиламид 1-аминоциклогексанкарбоновой кислоты (0,98 моль), гидрохлорид 4-(1-метилпиперидин-4-ил)бензойной кислоты (0,98 ммоль), НОВТ (0,98 ммоль), WSCD (0,98 ммоль) и триэтиламин (0,98 ммоль) растворяли в ДМФА (5 мл) и перемешивали в течение ночи при комнатной температуре. После упаривания растворителя остаток растворяли в смеси воды и карбоната натрия (чтобы обеспечить основные условия) и трижды экстрагировали этилацетатом. Объединенный экстракт сушили над сульфатом натрия и упаривали. Остаток суспендировали в смеси диэтиловый эфир/пентан, твердое вещество отделяли фильтрованием и сушили (вакуум). Получали порошок белого цвета с tпл 215-217°С, Rf=0,34 (СН2Cl2:МеОН (с 3 н. NH3)=9:1). 1Н-ЯМР (d6-ДМСО): 1,2-1,3 (m, 1Н); 1,4-1,8 (m, 11H); 1,85-2,0 (m, 2H); 2,05-2,2 (m, 5H); 2,55 (m, 1H); 2,95 (d, 2H); 4,0 (d, 2H), 7,35 (d, 2H); 7.8 (d, 2H), 7,9 (s, 1Н); 8,15 (m, 1H). Подобным образом получали N-[1-(цианометилкарбамоил)циклогексил]-4-(пиперидин-4-ил)бензамид, по существу так, как описано выше в примере 12; например, исключая этап А и начальную синтетическую методику на этапе Б, используя 4-фенил пиперидин в качестве исходного вещества.
Формула изобретения
1. Способ лечения пациента, страдающего от заболевания либо болезненного состояния или чувствительного к таковым, в которое вовлечен катепсин К, включающий введение пациенту эффективного количества соединения формулы II или его фармацевтически приемлемой соли, или его сложного эфира
где Х означает CH или N, R означает (С1-С7)алкил, (С1-С7)алкокси(С1-С7)алкил, (С5-С10)арил(С1-С7)алкил или (С3-С8)цилкоалкил. 2. Применение соединения формулы II или его фармацевтически приемлемой соли, или его сложного эфира формулы
где X означает СН или N, и R означает (С1-С7)алкил, (С1-С7)алкокси(С1-С7)алкил, (C5-C10)арил(C1-С7)алкил или (С3-С8)цилкоалкил, для получения лекарственного средства для терапевтического или профилактического лечения заболевания или болезненного состояния, в которое вовлечен катепсин К. 3. Применение по п.2, в котором заболеванием или болезненным состоянием, в которое вовлечен катепсин К, является остеопороз.
|
||||||||||||||||||||||||||