Патент на изобретение №2292871
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ИНЪЕЦИРУЕМЫЕ КОМПОЗИЦИИ ДЛЯ КОНТРОЛИРУЕМОЙ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИ АКТИВНОГО СОЕДИНЕНИЯ
(57) Реферат:
Настоящее изобретение относится к композициям и способам пролонгирования значений времени высвобождения и снижения токсичности фармакологически активных соединений. Соединения содержат соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворимый в воде растворитель, объединенные вместе с образованием инъецируемой композиции. Липофильный противоион может представлять собой насыщенную или ненасыщенную С8-С22-жирную кислоту и, предпочтительно, может представлять собой насыщенную или ненасыщенную С10-С22-жирную кислоту. При инъекции млекопитающему, по меньшей мере, часть композиции выпадает в осадок и высвобождает активное соединение во времени. Таким образом, композиция образует в организме млекопитающего лекарственное депо активного соединения с замедленным высвобождением. Поэтому настоящее изобретение позволяет осуществлять контролируемое введение дозы активного соединения на период времени до 15 суток и даже дольше. В соответствии с настоящим изобретением могут вводиться многие соединения, включая, но, не ограничиваясь ими, тилмикозин, окситетрациклин, метопролол, флуоксетин, рокситромицин или тербинафин. 7 н. и 56 з.п. ф-лы, 1 табл., 10 ил.
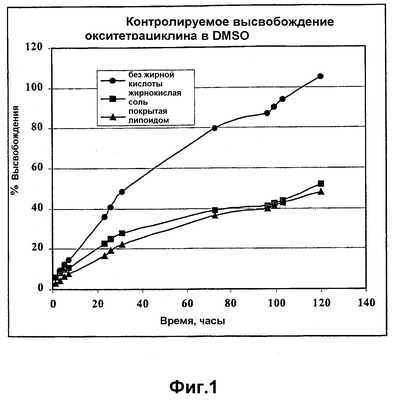
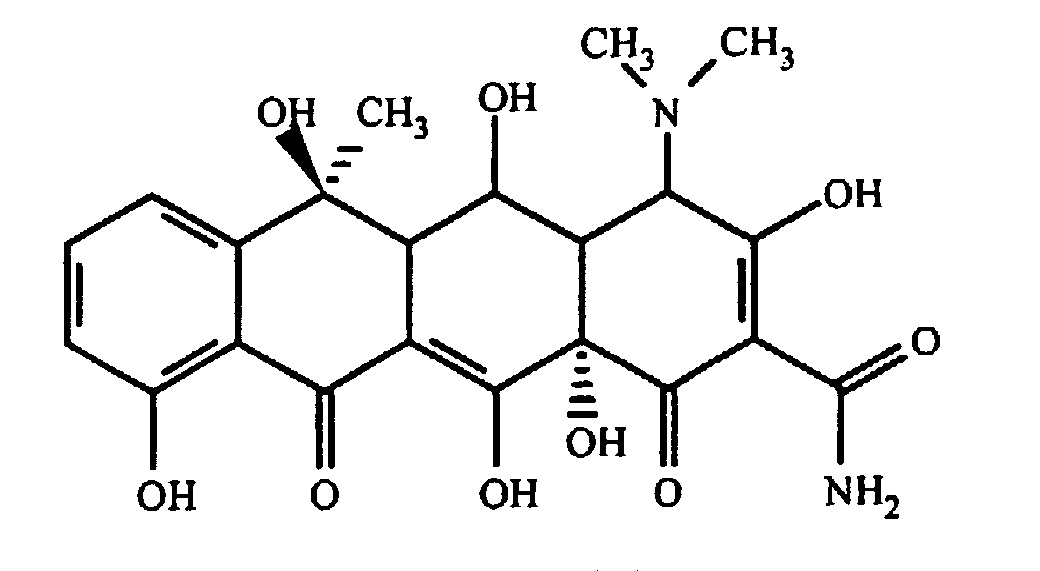
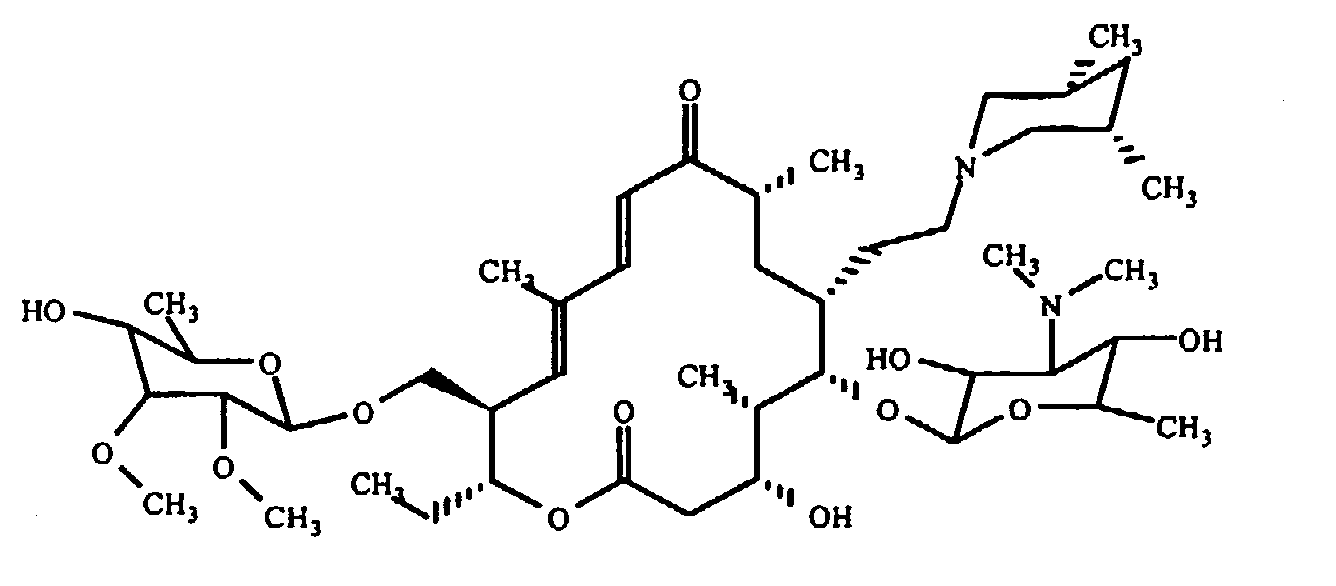
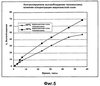
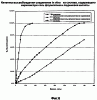
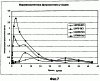
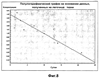
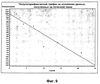
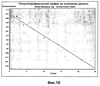
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ Настоящее изобретение относится к способам и композициям для пролонгирования времени высвобождения и снижения токсичности фармакологически активных соединений. ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ Следующее обсуждение предпосылок создания изобретения имеет своей единственной целью помочь читателю в понимании изобретения и не предназначено для описания настоящего изобретения или определения известного уровня техники. Часто для увеличения продолжительности действия инъецированного лекарства или для снижения его токсических эффектов является желательным пролонгирование времени его высвобождения. Составы, которые хорошо растворяются в организме, обычно быстро всасываются и обеспечивают стремительное увеличение содержания доступного лекарства в противоположность к более желательному и постепенному высвобождению фармакологически активного продукта. С целью обеспечения контролируемого и пролонгированного высвобождения фармацевтических соединений было предпринято множество попыток, которые не увенчались успехом в решении всех проблем, связанных с технологией, таких, как достижение пролонгированного времени высвобождения, максимальной стабильности и эффективности, сниженной токсичности, максимальной воспроизводимости в процессе получения и устранение нежелательных физических, биохимических и токсикологических эффектов, вызываемых нежелательными материалами основы. Окситетрациклин является широко применяемым и пригодным антибиотиком для лечения различных инфекций у млекопитающих. В частности, он применяется для лечения и профилактики респираторных инфекций у домашних животных. Повторные введения посредством традиционных средств сопряжены со значительными расходами. Тилмикозин является относящимся к классу макролидов антибиотиком, содержащим две третичных аминогруппы. Он обладает продолжительным биологическим полупериодом существования в тканях, эффективен в отношении широкого спектра микроорганизмов и применяется для лечения респираторных заболеваний у крупного рогатого скота. В повышенных дозах тилмикозин обладает кардиотоксичностью, и его применение на чувствительных видах, таких как кошки, козы, свиньи и лошади, практически полностью прекращено по соображениям безопасности. Коммерческий продукт Микотил® (Eli Lilly & Co., Indianapolis, IN) представляет собой раствор дифосфата и описан в патенте США № 5574020. Этот состав является эффективным для применения у крупного рогатого скота, но антибиотик высвобождается быстро, что выражается в токсичности для многих видов, включая собак и кошек. ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ Заявка на выдачу патента № 60/343625, поданная 19 октября 2001 года, включена в настоящий документ в качестве ссылки во всей своей полноте, включая все графики и чертежи. Настоящее изобретение относится к композициям и способам для пролонгирования времени высвобождения и снижения токсичности фармакологически активных соединений. Соединения включают соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворимый в воде растворитель, объединенные вместе с образованием инъецируемой композиции. Липофильный противоион может представлять собой насыщенную или ненасыщенную C8-C22-жирную кислоту, и, предпочтительно, может представлять собой насыщенную или ненасыщенную C10-C18-жирную кислоту. При инъекции млекопитающему, по крайней мере, часть композиции выпадает в осадок и высвобождает активное соединение во времени. Таким образом, композиция образует в организме млекопитающего лекарственное депо активного соединения с замедленным высвобождением. Поэтому настоящее изобретение позволяет осуществлять контролируемое введение дозы активного соединения на период времени до 15 суток и даже дольше. В предпочтительных вариантах воплощения фармакологически активное соединение может представлять собой тилмикозин, антибиотик, такой как окситетрациклин или доксициклин, или флуоксетин, рокситромицин, турбинафин или метопролол, а липофильный противоион может представлять собой декановую кислоту, лауриновую кислоту, олеиновую кислоту, линолевую кислоту или миристиновую кислоту. В предпочтительных вариантах воплощения фармацевтически приемлемый растворитель является N-метилпирролидоном (NMP). В другом варианте воплощения фармацевтически приемлемый растворитель является пропиленгликолем (например, приблизительно 10%) в глицеринформале, содержащим или не содержащим стабилизаторы. Настоящее изобретение также относится к новым способам введения млекопитающим композиций и составов по настоящему изобретению. Способы обеспечивают композиции активных соединений, применение которых в виде форм, доступных в настоящее время, может приводить к токсическим эффектам у получающего лечение млекопитающего. Таким образом, составы и способы по настоящему изобретению позволяют вводить соединения, широкое применение которых для конкретных видов было ранее невозможно по соображениям безопасности. Данные способы также позволяют пролонгировать время высвобождения соединений и обеспечивают контролируемую дозировку активного соединения для получающего лечение пациента. Способы по настоящему изобретению позволяют вводить получающему лечение млекопитающему фармакологически активное соединение в фармацевтически эффективном количестве на период времени от 4 до 15 суток, включая любое конкретное количество суток до 15 суток включительно или даже дольше. Точное время зависит от нескольких переменных, которыми можно управлять с целью оптимизации настоящего изобретения для конкретного фармакологически активного соединения или способа применения. Предпочтительно, соединение содержится в обрабатываемой ткани в течение 4-5 суток после инъекции и, более предпочтительно, соединение содержится в обрабатываемой ткани в фармацевтически эффективном количестве в течение 6 суток, или даже 7 суток, после инъекции. В других вариантах воплощения может быть желательным управлять переменными таким образом, чтобы увеличить время высвобождения более чем на 15 суток. В одном аспекте настоящее изобретение относится к композициям для введения фармакологически активных соединений. Композиции могут включать соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворимый в воде растворитель, объединенные вместе в условиях для образования инъецируемой композиции. При инъекции млекопитающему композиция может выпадать в осадок и высвобождать фармакологически активное соединение во времени. В различных вариантах воплощения композиция по настоящему изобретению может включать широкий ряд фармакологически активных соединений, таких, как тилмикозин, окситетрациклин, доксициклин, метопролол, сульфаметазин, триметоприм, неомицин, стрептомицин, гентамицин, дибукаин, бупивакаин, бензокаин, тетракаин, ацепромазин, итраконазол, тетрациклины, сульфонамиды, аминогликозиды или любое фармакологически активное соединение с подходящей растворимостью и подходящими химическими функциональными группами. Липофильный противоион может представлять собой насыщенную или ненасыщенную жирную кислоту с любым конкретным числом атомов углерода от 8 до 22, предпочтительно, C8-C18-жирную кислоту, и, более предпочтительно, C10-C18-жирную кислоту, такую как лауриновая кислота, линолевая кислота, декановая кислота или миристиновая кислота. Также могут применяться другие липофильные противоионы, например, дикарбоновые кислоты, такие как себациновая кислота, полимерные кислоты, такие как липофильные поликарбоновые кислоты, и ароматические кислоты, такие как бензойная кислота. Фармацевтически приемлемый носитель может представлять собой органический растворитель. В предпочтительных вариантах воплощения растворитель может представлять собой пирролидон, N-метилпирролидон, полиэтиленгликоль, пропиленгликоль, глицеринформаль, изосорбиддиметиловый простой эфир, этанол, диметилсульфоксид, тетрагидрофурфуриловый спирт, триацетин, или любую комбинацию указанных соединений, или другой растворитель, для которого показаны сходные подходящие качества, такие как отсутствие токсичности и растворимость в воде. В другом варианте воплощения композиции по настоящему изобретению представляют собой соль фармакологически активного соединения с противоионом поликарбоновой кислоты и фармацевтически приемлемый растворимый в воде растворитель, объединенные вместе в условиях для образования инъецируемой композиции, которая выпадает в осадок при введении в воду при комнатной температуре или выпадает в осадок в физиологических условиях окружающей среды (in vivo). При инъекции млекопитающему композиция высвобождает активное соединение во времени. Под термином «поликарбоновая кислота» подразумевают молекулу, которая содержит, по меньшей мере, две карбоксильные группы. В предпочтительных вариантах воплощения поликарбоновая кислота является полиаспарагиновой кислотой, полиакриловой кислотой, себациновой кислотой, полисебациновой кислотой, полибензойной кислотой или их комбинацией. Под термином «поли» подразумевают два или более. В одном варианте воплощения фармакологически активное соединение может представлять собой окситетрациклин, липофильный противоион может представлять собой лауриновую кислоту, а фармацевтически приемлемый растворитель может представлять собой пропиленгликоль, полиэтиленгликоль, глицеринформаль или подходящую комбинацию указанных соединений. В другом варианте воплощения фармакологически активное соединение может представлять собой тилмикозин, липофильный противоион может представлять собой лауриновую кислоту, а фармацевтически приемлемый растворитель может представлять собой пропиленгликоль, полиэтиленгликоль, глицеринформаль или подходящую комбинацию указанных соединений. В еще одном варианте воплощения композиции могут выпадать в осадок и высвобождать активное соединение во времени при введении или инъекции в водную окружающую среду. При инъекции композиции также могут образовывать в организме млекопитающего лекарственное депо, которое высвобождает соединение во времени. В другом аспекте настоящее изобретение относится к способам введения млекопитающему фармакологически активного соединения. Данные способы могут включать приготовление композиции из соли фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемого растворимого в воде растворителя, объединенных вместе в условиях для образования инъецируемого состава, и инъекцию композиции млекопитающему. При инъекции млекопитающему, по меньшей мере, часть композиции выпадает в осадок и высвобождает фармакологически активное соединение во времени. В другом аспекте настоящее изобретение относится к способам пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему. Данные способы могут включать приготовление состава из соли фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемого растворимого в воде растворителя, объединенных вместе в условиях для образования инъецируемого состава, и инъекцию композиции млекопитающему, причем при инъекции млекопитающему часть композиции выпадает в осадок и высвобождает фармакологически активное соединение во времени, пролонгируя тем самым время высвобождения соединения. Поэтому настоящее изобретение может обеспечивать контролируемую дозировку активного соединения для получающего лечение млекопитающего. В еще одном аспекте настоящее изобретение относится к способам приготовления инъецируемого состава для введения млекопитающему фармакологически активного соединения. Данные способы могут включать образование соли фармакологически активного соединения с липофильным противоионом, добавление растворимого в воде фармацевтически приемлемого растворителя, объединение соли и растворителя в условиях для образования инъецируемого состава, где при инъекции млекопитающему, по меньшей мере, часть состава выпадает в осадок и высвобождает фармакологически активное соединение во времени. В другом аспекте настоящее изобретение относится к композициям для введения млекопитающему фармакологически активного соединения. Данные композиции содержат соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворитель, объединенные вместе для образования инъецируемой композиции. При инъекции млекопитающему, по меньшей мере, часть растворенной в растворителе соли фармакологически активного соединения с липофильным противоионом выпадает в осадок in vivo и высвобождает фармакологически активное соединение во времени. Поэтому настоящее изобретение сулит важные преимущества по сравнению с составами, доступными в настоящее время. Настоящее изобретение позволяет осуществлять контролируемое высвобождение фармакологически активных соединений с целью уменьшения токсичности, в особенности у небольших животных, таких как собаки и кошки. Оно также сулит преимущество, делая возможным эффективное введение соединений домашним животным, требуя тем самым меньших затрат времени и средств по сравнению с ранее доступными способами введения лекарств. Фармакологически активное соединение доступно в стабильном инъецируемом составе, который при инъекции выпадает в осадок и медленно высвобождает активное соединение в течение длительного периода времени. Представленное выше краткое описание настоящего изобретения не является ограничивающим, а другие признаки и преимущества настоящего изобретения станут очевидными как из последующего подробного описания предпочтительных вариантов воплощения, так и из формулы изобретения. КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ Фиг.1 является графической иллюстрацией, показывающей, что полученный в соответствии с настоящим изобретением окситетрациклин высвобождается в солевой раствор со скоростью, меньшей по сравнению со свободным лекарством. Растворитель представляет собой DMSO, а липофильный противоион является лауриновой кислотой. Фиг.2 является графической иллюстрацией, показывающей, что полученный в соответствии с настоящим изобретением метопролол высвобождается в солевой раствор со скоростью, меньшей по сравнению со свободным лекарством. Жирная кислота и растворитель являются лауриновой кислотой и N-метилпирролидоном. Фиг.3 является графическим описанием, иллюстрирующим изменение скорости высвобождения фармакологически активного соединения (тилмикозин) под действием жирной кислоты с выбранной длиной цепи. Растворитель: N-метилпирролидон; липофильный противоион: декановая кислота и лауриновая кислота. Фиг.4 является графическим описанием, иллюстрирующим влияние растворителя на кинетику высвобождения тилмикозина in vitro. Липофильный противоион: ди(декановая) кислота; содержание тилмикозина в составе равно 100 мг/мл. Использованы следующие сокращения: PEG = полиэтиленгликоль, THFA = тетрагидрофурфуриловый спирт, DMA = диметилацетамид, ISO-DME = изосорбиддиметиловый эфир, DMSO = диметилсульфоксид, NMP = N-метилпирролидон. Фиг.5 является графическим описанием, иллюстрирующим тот факт, что скорость высвобождения фармакологически активного соединения (тилмикозина) зависит от концентрации жирнокислой соли. Липофильный противоион: декановая кислота. Фиг.6 является графическим описанием, иллюстрирующим кинетику высвобождения соединения in vitro из состава, содержащего жирнокислую соль флуоксетина и лауриновой кислоты. Фиг.7 является графическим описанием фармакокинетики флуоксетингидрохлорида (HCl) и жирнокислой соли флуоксетина и лауриновой кислоты (FAS) у кошек. Фиг.8 является полулогарифмическим графиком изменения концентрации тилмикозина в легочной ткани кошек в течение 21 суток. Использовали восемь самцов и восемь самок кошек, а для всех типов тканей применяли дозу, равную 10 мг/кг массы тела. Фиг.9 является полулогарифмическим графиком изменения концентрации тилмикозина в почечной ткани кошек в течение 21 суток. Использовали восемь самцов и восемь самок кошек, а для всех типов тканей применяли дозу, равную 10 мг/кг массы тела. Фиг.10 является полулогарифмическим графиком изменения концентрации тилмикозина в печеночной ткани кошек в течение 21 суток. Использовали восемь самцов и восемь самок кошек, а для всех типов тканей применяли дозу, равную 10 мг/кг массы тела. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Композиции по настоящему изобретению могут быть приготовлены с применением солей фармакологически активных соединений с основными функциональными группами. Они могут быть приготовлены с применением ряда липофильных кислот, насыщенных или ненасыщенных жирных кислот, холевых кислот, фосфатидных кислот, дикарбоновых кислот, таких как себациновая кислота, или любой кислоты, которая при объединении с фармакологически активным соединением приводит полученную соль в состояние, нерастворимое в воде, но растворимое в растворимом в воде растворителе. Под термином «соль» подразумевают два ковалентно несвязанных соединения, химически связанных посредством ионных взаимодействий. Под термином «смешивающийся с водой» подразумевают растворитель, способный к смешиванию с водой в любых соотношениях без разделения на две фазы. Термин «растворимый в воде» подразумевает, что растворитель обладает значительным показателем растворимости в водных растворах, например, триацетин считается растворимым в воде растворителем, так как он растворяется в воде в соотношении приблизительно 1:14. Под термином «липофильный противоион» подразумевают ионную форму жирорастворимой молекулы. Липофильный противоион может, предпочтительно, представлять собой жирную кислоту, но также может представлять собой другую жирорастворимую молекулу. Противоион содержит, по меньшей мере, одну заряженную группу, противоположную по знаку с зарядом другой образующей соль химической группы, обуславливая тем самым ионное взаимодействие между двумя группами. Под терминами «инъецируемый состав» или «инъецируемая композиция» понимают состав или композицию, которые могут быть набраны в шприц и инъецированы млекопитающему подкожно, внутрибрюшинно или внутримышечно, не вызывая побочных эффектов, обусловленных присутствием в композиции твердых материалов. Твердые материалы включают, но не ограничиваются ими, кристаллы, клейкую массу и гель. Термин «фармакологически активные соединения» подразумевает химическое соединение, которое обуславливает фармакологический эффект у получающего лечение млекопитающего. Например, эффект может заключаться в уничтожении или предотвращении роста микроорганизмов или паразитов, уменьшении воспаления или другом фармацевтическом и измеримом эффекте у получающего лечение млекопитающего. Под термином «выпадать в осадок» подразумевают, что соединение образует осадок или твердое вещество. Осадок является нерастворимым твердым веществом, которое образуется в растворе при комнатной температуре in vitro или в физиологическом микроокружении (in vivo). Осадок может принимать различные формы, такие как, например, твердое вещество, кристалл, клейкая масса или гель. Термин «фармацевтически эффективное количество» подразумевает количество, которое вызывает у получающего лечение млекопитающего измеримый и значимый в медицинском плане эффект, который выражается в прогрессе в лечении или профилактике заболевания, или в смягчении или предупреждении состояния, которое являлось поводом для лечения. «Фармацевтически приемлемым растворителем» является жидкость, в которой растворяется соль фармакологически активного соединения и липофильного противоиона и которая пригодна для применения у людей и/или животных без возникновения чрезмерных неблагоприятных побочных эффектов (таких, как токсичность, раздражение и аллергическая реакция), соразмерных с приемлемым отношением польза/риск. Композиции по настоящему изобретению сулят несколько преимуществ. Композиции являются инъецируемыми композициями, которые содержат активное соединение в высоких концентрациях. В предпочтительных вариантах воплощения композиция может содержать фармакологически активное соединение в диапазоне 10-60% (масс./объем.). Но специалисту в данной области техники должно быть понятно, что этот диапазон может варьироваться в широких пределах в зависимости от растворимости или нерастворимости фармакологически активного соединения, выбранного липофильного противоиона, выбранного растворителя, возможности применения конечного продукта для инъекций и любых других уместных требований для конкретного применения. Активное соединение может содержаться в таком малом количестве, как 10% или 5%, или даже 1%, но все еще обладать нужным эффектом. Сходным образом содержание активного соединения может составлять 70% или даже более, если этого требует необходимость. Экзотических носителей или наполнителей не требуется. Композиции легко подвергаются фильтрации, упрощая тем самым процесс производства. Считается, что исключение воды из состава придает составам большую стабильность и ингибирует рост микроорганизмов. Описанные здесь способы приготовления композиций являются простыми, а введение в соответствии с настоящим изобретением должно приводить к более мягким реакциям в месте инъекции вследствие нейтрализации фармакологически активного соединения. Настоящее изобретение обеспечивает способность модулировать скорость высвобождения и время высвобождения фармакологически активного соединения. Скорость высвобождения можно модулировать путем варьирования липофильности и молекулярной массы противоиона, применяемого для получения соли. Например, соли лауриновой кислоты и тилмикозина обычно высвобождаются медленнее, чем деканоаты. Кроме того, более высокие концентрации соли в составе обычно приводят к более низким скоростям высвобождения. Деканоат тилмикозина высвобождается более медленно из состава, содержащего 60% жирнокислой соли тилмикозина, чем из состава, содержащего 30% жирнокислой соли тилмикозина. Сходным образом, как объясняется здесь, для увеличения или уменьшения времени высвобождения активного соединения до требуемого значения может применяться управление другими переменными, такими, как выбор липофильного противоиона, выбор растворителя, концентрация соли и другими. Как правило, это может быть желательным для солей, основанных на молярном отношении заряженных групп. Но каждый может с успехом получать нерастворимые соли путем применения геми-соли или другим путем, варьируя от соотношения 1:1. Фармацевтически приемлемый растворитель может представлять собой смешивающийся с водой растворитель или растворимый в воде растворитель, и предпочтительно представляет собой смешивающийся с водой растворитель. Также могут применяться смеси растворимого и/или смешивающегося с водой растворителей. Специалисту в данной области техники должно быть понятно, что для оптимизации результата конкретного применения могут быть смешаны различные растворимые в воде растворители. Например, для получения оптимального растворителя могут быть получены смеси полиэтиленгликоля, пропиленгликоля и глицеринформаля в различных соотношениях. В некоторых вариантах воплощения подходящий растворитель может быть получен путем смешивания в приблизительно равных количествах. В других вариантах воплощения составы по настоящему изобретению, содержащие соль фармакологически активного соединения с липофильным противоионом, могут быть объединены с не включенным в состав соли активным соединением для получения более высокой начальной дозы активного соединения. Не желая быть связанным какой-либо конкретной теорией, инъецируемые композиции могут быть приготовлены, когда полученную соль фармакологически активного средства с липофильным противоионом объединяют с подходящим для парентерального введения органическим растворителем. Считается, что при инъекции данного состава млекопитающему растворитель может диффундировать из места инъекции, тогда как тканевые жидкости диффундируют по направлению к месту инъекции, что приводит к выпадению в осадок фармакологически активного соединения у получающего лечение млекопитающего. Осадок может принимать различные формы, например, твердое вещество, кристаллы, клейкую массу или гель. Таким образом, должна существовать концентрация активного соединения, которая высвобождает фармацевтически эффективное количество в течение желаемого периода времени. Осадок может действовать в качестве лекарственного депо в организме млекопитающего, что приводит к высвобождению соединения в течение периода времени. Время высвобождения может составлять, по меньшей мере, 3 суток, по меньшей мере, 4 суток, по меньшей мере, 5 суток, по меньшей мере, 6 суток, по меньшей мере, 7 суток, или любое количество суток до 15 суток включительно, или даже дольше, если это требуется. Под термином «лекарственное депо» подразумевают концентрацию осадка фармакологически активного соединения в организме получающего лечение млекопитающего, которая высвобождает фармацевтически эффективное количество активного соединения во времени. Было показано, что длина цепи жирной кислоты, конкретные сочетания жирных кислот, процентное соотношение в составе фармакологически активного соединения и липофильного противоиона и выбор фармацевтически приемлемого растворителя влияют на кинетику высвобождения фармакологически активного соединения. Поэтому кинетика высвобождения фармакологически активного соединения может подходящим образом и легко управляться путем манипулирования и этими, и другими переменными. Также было показано, что составы устойчивы к стерилизации путем автоклавирования. Специалисту в данной области техники следует понимать, что настоящее изобретение может применяться в отношении многих фармакологически активных соединений, обладающих подходящей растворимостью и подходящими химическими функциональными группами. Поэтому предполагается, что настоящее изобретение может применяться в отношении широкого ряда фармакологически активных соединений, таких как лекарственные препараты, лекарственные средства, питательные вещества или другие желаемые соединения для введения млекопитающему. Специалисту в данной области техники следует понимать, что могут быть желательны некоторые модификации представленных здесь способов, которые основываются на конкретных характеристиках применяемого фармакологически активного соединения. В следующих не ограничивающих примерах представлены дополнительные применения настоящего изобретения, и они предназначены исключительно для иллюстрации в качестве примеров. Пример 1: Окситетрациклин
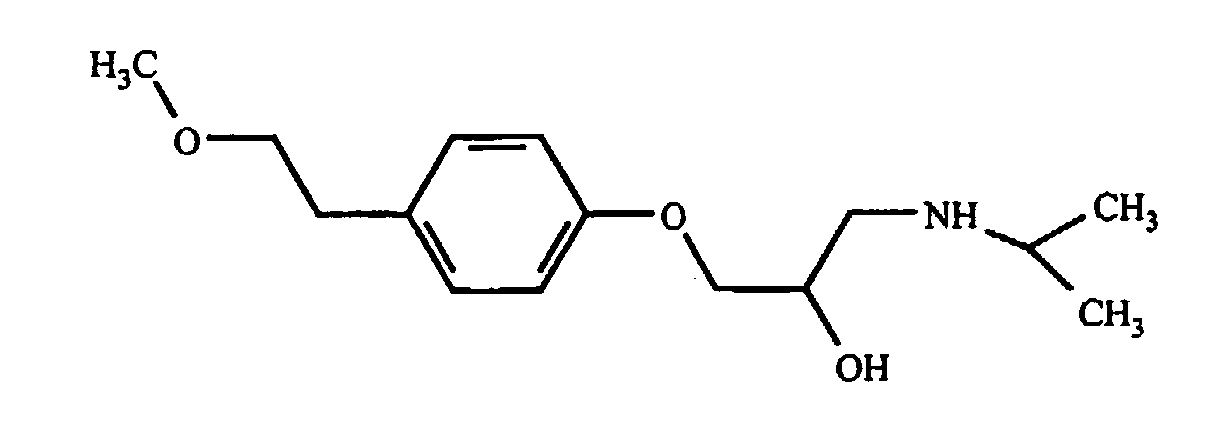
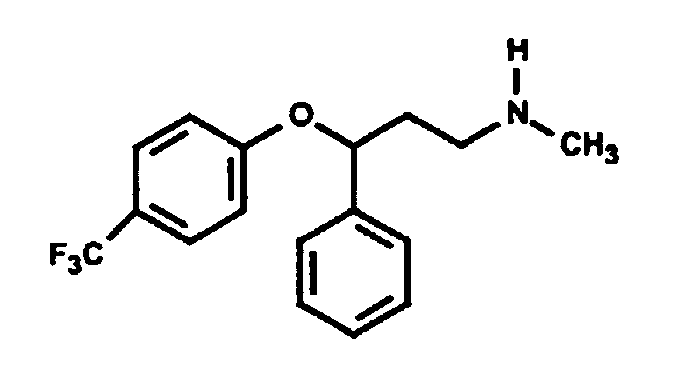
Окситетрациклин содержит одну третичную аминогруппу, а гидрохлорид окситетрациклина хорошо растворим в воде. Было обнаружено, что добавление одного моля жирных кислот к одному молю окситетрациклина приводит к образованию соли, которая, по сравнению с исходным окситетрациклином, обладает меньшей растворимостью в воде, но большей растворимостью в N-метилпирролидоне (NMP). Если к содержащему NMP составу добавить воду, то соль выпадает в осадок. Скорость высвобождения окситетрациклина in vitro может быть определена путем герметизации состава в мешке для диализа (Pierce, Rockford, IL), помещения его в резервуар с солевым раствором и измерения содержания лекарства в солевом растворе как функции времени. Состав по настоящему изобретению сравнивали с существующими составами, содержащими окситетрациклин. На фиг.1 показано, что содержащий окситетрациклин состав высвобождается в солевой раствор со скоростью, значительно меньшей по сравнению со свободным лекарством. Содержащую окситетрациклин композицию в соответствии с настоящим изобретением приготавливали путем добавления 0,464 г окситетрациклина и 0,203 г лауриновой кислоты к 3 мл NMP. Смесь перемешивали в течении 60 минут с получением прозрачного раствора. Один миллилитр данного раствора герметично закрывали в мешке для диализа и суспендировали мешок в 150 мл фосфатного солевого буферного раствора с pH 7,4. Через различные интервалы времени отбирали аликвоты и спектрофотометрически определяли концентрацию окситетрациклина. Из результатов, представленных на фиг.1, видно, что окситетрациклин продолжает высвобождаться из мешка путем диффузии в течение более чем 120 часов, причем в данной точке наблюдается высвобождение лишь приблизительно 50% содержащегося окситетрациклина. Пример 2: Метопролол Метопролол является антигипертензивным, антиангинозным и противоаритмическим лекарством со следующей структурной формулой:
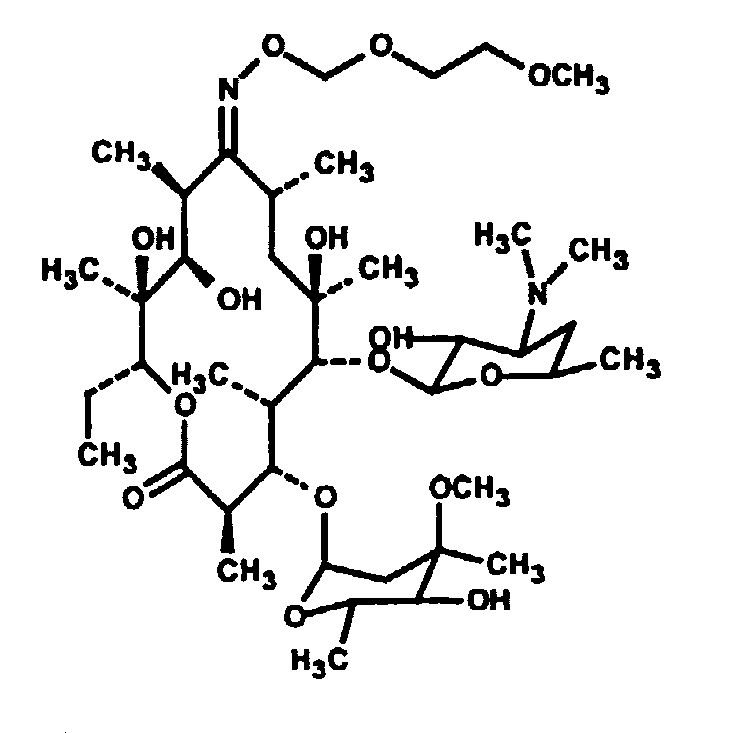
Его сукцинаты и тартраты являются коммерчески доступными под различными торговыми наименованиями. Обе эти соли, как и основная форма лекарства, хорошо растворимы в воде. Основную форму метопролола получали из коммерчески доступного тартрата в соответствии со стандартной методикой. Если аминогруппа метопролола нейтрализована добавлением лауриновой кислоты, полученная соль является плохо растворимой в воде, но хорошо растворимой в фармацевтически приемлемых неводных растворителях. Содержащую метопролол композицию в соответствии с настоящим изобретением приготавливали путем добавления 0,3224 г основания метопролола и 0,2661 г лауриновой кислоты к 2,415 мл NMP. Смесь перемешивали в течение 30 минут с получением прозрачного раствора. Один миллилитр данного раствора герметично закрывали в мешке для диализа и суспендировали мешок в 150 мл фосфатного буферного раствора с pH 7,4. Через различные интервалы времени отбирали аликвоты и спектрофотометрически определяли концентрацию метопролола. Результаты, представленные на фиг.2, показывают, что метопролол продолжает высвобождаться из мешка путем диффузии в течение более чем 48 часов, в то время как контрольный раствор основания метопролола (приготовленный путем растворения 150 мг в 1,124 мл NMP) высвобождается путем диффузии быстро. Пример 2A: Тилмикозин Тилмикозин является антибиотиком из класса макролидов со следующим строением:
Он является эффективным в отношении широкого спектра микроорганизмов и применяется для лечения респираторных инфекций у крупного рогатого скота. Основная форма является умеренно растворимой в водных растворах, в то время как хлоридные и фосфатные соли являются высокорастворимыми. В повышенных дозах тилмикозин обладает кардиотоксичностью, и поэтому не вводится внутривенно. По соображениям безопасности его применение у чувствительных видов, таких как кошки, козы, свиньи и лошади, практически полностью прекращено. Если две аминогруппы тилмикозина нейтрализованы любой из нескольких жирных кислот (таких как, например, декановая C10, лауриновая C12, миристиновая C14, пальмитиновая C16, стеариновая C18, олеиновая C18, элаидиновая C18, линолевая C18 и эруковая C22), полученная соль является плохо растворимой в воде, но хорошо растворимой в фармацевтически приемлемых неводных растворителях. Если содержащие соль составы герметично закрыть в мешках для диализа и поместить в солевой раствор, соль тилмикозина выпадает в осадок, и тилмикозин медленно высвобождается из мешка. Скорость высвобождения зависит от длины цепи жирной кислоты (фиг.3), растворителя (фиг.4) и концентрации жирнокислой соли тилмикозина (фиг.5). Пример 3: Соли тилмикозина in vitro 10 граммов (0,0115 моль) тилмикозина и 0,0253 моль различных карбоновых кислот (таких как, например, декановая, лауриновая, линолевая или миристиновая кислоты, в раздельных пробах) помещали в колбу, доводили добавлением N-метилпирролидона до конечного объема, равного 100 мл, и перемешивали в течение 60 минут с получением прозрачного раствора. Аликвоты (1 мл) данных растворов герметично закрывали в мешке для диализа и суспендировали мешок в колбах, содержащих 150 мл фосфатного солевого буферного раствора с pH 7,4. Было обнаружено, что соль выпадает в осадок в мешке в течение приблизительно 1 часа. Через различные интервалы отбирали аликвоты солевого раствора и определяли концентрацию тилмикозина по методу ВЭЖХ. Результаты, полученные для солей декановой кислоты (C-10) и лауриновой кислоты (C-12) и представленные на фиг.3, показывают, что тилмикозин продолжает высвобождаться из мешка путем диффузии в течение более чем 120 часов. Кислоты с более длинной цепью обуславливают более медленное высвобождение тилмикозина. Микотил® (Eli Lilly, Indianapolis, IN), фосфатная соль тилмикозина является хорошо растворимой и быстро высвобождается из мешка путем диффузии. Пример 4: Соль тилмикозина и ди(декановой) кислоты в различных растворителях Приготавливали растворы соли тилмикозина и ди(декановой) кислоты в различных смешивающихся с водой растворителях, объединяя 10 г (0,0115 моль) тилмикозина и 0,0253 моль декановой кислоты в различных растворителях, до конечного объема, равного 100 мл. Скорости высвобождения in vitro измеряли в соответствии со способом, описанным в примере 1. Результаты, представленные на фиг.4, показывают, что скорость высвобождения зависит от растворителя, но для каждого растворителя наблюдали более низкую скорость высвобождения по сравнению с фосфатной солью (Микотил®), результаты для которой представлены на фиг.3. Пример 5: Влияние концентрации на высвобождение тилмикозина Растворы соли тилмикозина и ди(декановой) кислоты приготавливали путем объединения 30 г (0,0345 моль) или 60 г (0,0690 моль) тилмикозина с 2 эквивалентами декановой кислоты в NMP до конечного объема, равного 100 мл. Скорости высвобождения in vitro измеряли в соответствии со способом, описанным в примере 1, а результаты, представленные на фиг.5, показывают, что более высокие начальные концентрации приводят к более медленной скорости высвобождения. Пример 6: Тилмикозин in vivo Приготавливали составы, содержащие дидеканоат, дилаурат и димиристат тилмикозина в N-метилпирролидоне в концентрации 100 мг/мл, и вводили кошкам подкожно в заднюю часть шеи в дозах, равных 45 или 75 мг/кг массы тела. Полученные ранее данные показывают, что фосфатная соль тилмикозина в концентрации 25 мг/кг является смертельной для кошек. После инъекции у кошек наблюдается гипотермия и летаргический сон, что является показательным для оценки биопригодности лекарства. Было обнаружено, что для составов, содержащих жирные кислоты с длиной цепи более, чем C10, токсичность была существенно меньшей, что согласуется с более медленным высвобождением лекарства из данных составов. Все кошки выжили, а их поведение в течение 3 суток после инъекции было нормальным. Результаты просуммированы в таблице 1. Таблица 1: Содержание тилмикозина в крови в определенные периоды времени Составы, содержащие соль тилмикозина и декановой кислоты, лауриновой кислоты и миристиновой кислоты в N-метилпирролидоне в концентрации 100 мг/мл, инъецировали 9 здоровым взрослым кошкам в указанных дозировках. Полученные концентрации тилмикозина в клетках крови и плазме крови для каждой кошки в отдельности, измеренные через 6 часов и 2 суток, представлены в таблице 1.
Содержание солей тилмикозина также было исследовано в тканях. Жирнокислую соль тилмикозина и лауриновой кислоты в 10% пропиленгликоле в глицеринформале в концентрации 100 мг/мл вводили кошкам подкожно в дозе 10 мг/кг массы тела и определяли биораспределение в организме кошек. Описанные здесь способы разрабатывали для определения и количественного анализа тилмикозина в различных тканях и сыворотке крови кошек, в особенности в печеночной, почечной и легочной ткани и сыворотке крови кошек. Специалисту в данной области техники следует понимать, что возможны различные вариации описанных здесь способов без выхода за рамки настоящего изобретения. Образцы почечной, печеночной и легочной ткани отбирали на 2, 3, 4, 7, 14 и 21 сутки после инъекции животным содержащего тилмикозин состава в дозе, равной 10 мг/кг массы тела. Результаты представлены на фиг.8, 9 и 10. Было обнаружено, что для кошек маркерной тканью является почечная ткань, тогда как для коров и свиней маркерной является печеночная ткань. Содержание тилмикозина в почках было соответственно выше, чем в печени со значениями Cmax (почки: 13,8 мкг/г; печень: 7,3 мкг/г), которые достигались в обеих тканях приблизительно через 48 часов. Для легких значение Cmax составляло 7,5 мкг/г и наблюдалось приблизительно через 48 часов после инъекции дозы. Определяемые количества тилмикозина содержались в тканях в течение 21 суток исследования. Для концентраций, равных 1 мкг/г ткани, получали значение эффективности экстракции лекарства, равное 98%. Было определено, что предел обнаружения тилмикозина в различных тканях кошек составляет 0,032 мкг/г. Для сыворотки крови кошек было получено значение эффективности экстракции, равное 95%, в диапазоне концентраций от 0,15 до 6 мкг/мл после обогащения. Было определено, что предел обнаружения тилмикозина составляет 0,16 мкг/мл с линейным характером роста в диапазоне от 0,15 до 6 мкг/мл. ПОЛУЧЕНИЕ ОБРАЗЦОВ ТКАНЕЙ Образцы тканей получали путем измельчения при помощи ножниц или скальпеля на бумажном полотенце. В каждую пробирку добавляли 10 мл метанола и по отдельности гомогенизировали образцы в течение 10-15 минут. Образцы обрабатывали ультразвуком на льду в течение 1 минуты и центрифугировали при 10000 об/мин в течение 30 минут при 4°С. Метанольный экстракт декантировали в свежие пробирки для центрифуги и ресуспендировали образцы ткани в пробирках с 10 мл метанола и 5,0 мл 100 мМ фосфата. Пробирки перемешивали встряхиванием и центрифугировали при 10000 об/мин в течение 30 минут при 4°С. Экстракт декантировали в свежие пробирки для центрифуги и центрифугировали при 5000 об/мин в течение 10 минут при 4оС. Метанольный экстракт добавляли к 70 мл воды в колбе и перемешивали вращением в колбе. Каждую серию экстрактов подвергали экстракции в твердой фазе (SPE) на картридже C18 Sep-Pak Plus® (Waters, Milford, MA) с применением вакуумного коллектора или гидростатического насоса для пропускания образцов через картриджи. После полной загрузки образца каждый картридж для SPE промывали 10 мл воды, а затем 10 мл 25% ацетонитрила/воды со скоростью потока менее чем 5 мл/мин. Картриджи для SPE отсоединяли от устройства и полностью высушивали в высоком вакууме (26 дюймов рт. ст.) в вакуумном эксикаторе в течение 10 минут. Анализируемое вещество элюировали из картриджей для SPE добавлением 5% уксусной кислоты/метанола. Отбирали только первые 2,0 мл элюата. Волюметрические колбы переворачивали и перемешивали, и хранили в течение ночи при 4°С. Элюаты образцов фильтровали через 0,22 мкм PVDF-фильтр и анализировали по методу ВЭЖХ на колонке SphereClone® 5 мкм phenyl column (Phenomenex, Torrance, CA). Пример 7: Флуоксетин
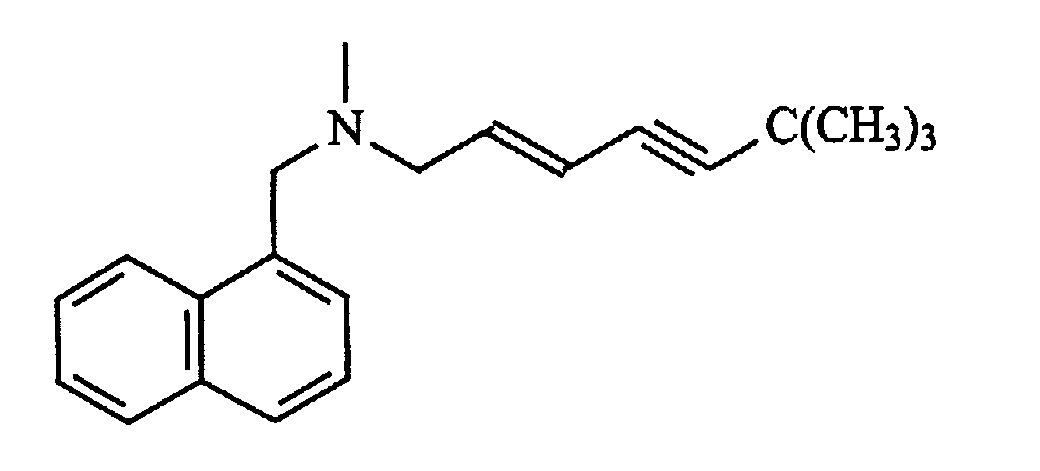
Флуоксетин является избирательным ингибитором повторного поглощения серотонина и широко применяется для лечения психических расстройств, таких как навязчиво-компульсивное расстройство у людей. Было показано, что флуоксетин эффективен для лечения агрессивного поведения и связанного с разлучением беспокойства у собак и поведенческого мечения мочой у кошек. Составы, содержащие флуоксетин в концентрации 100 и 150 мг/мл, приготавливали в виде соли лауриновой кислоты в 10% пропиленгликоле в глицеринформале при отношении лекарство:жирная кислота 1:1. Кинетику высвобождения in vitro изучали для обоих составов в концентрации 100 мг/мл с применением методики оценки диализа, описанной в примере 1, результаты измерений представлены на фиг.6. В качестве контроля в эксперименте применяли основание флуоксетина в 10% пропиленгликоле в глицеринформале в концентрации 100 мг/мл. В то время как содержащий основание флуоксетина состав высвобождается в течение приблизительно 160 ч, для состава, содержащего соль лауриновой кислоты (лекарство:лауриновая кислота, 1:1), время высвобождения составляет 700 ч. Состав, содержащий жирнокислую соль с отношением флуоксетин:лауриновая кислота (1:1,1) в концентрации 150 мг/мл, инъецировали кошкам подкожно в дозах, равных 20 и 30 мг/кг. В то же самое время двум кошкам в течение 28 суток перорально вводили состав в дозе 1 мг/кг/сутки. Образцы сыворотки крови отбирали до 42-х суток и анализировали их на присутствие флуоксетина по методу ВЭЖХ на колонке C18, элюируя 35% ацетонитрилом в фосфатном буфере, pH 2,7. Результаты показывают, что однократная подкожная инъекция состава, содержащего жирнокислую соль, в течение 42 суток обеспечивает концентрацию лекарства, сравнимую с ежедневной дозой для перорального приема (фиг.7). В виде прозрачного раствора приготавливали состав соли флуоксетина и линолевой кислоты, растворенной в 10% пропиленгликоле в глицеринформале. Было обнаружено, что эта соль также выпадает в осадок в воде. Пример 8: Рокситромицин Рокситромицин является антибиотиком из класса макролидов со следующим строением:
Он является эффективным в отношении широкого спектра микроорганизмов и применяется для лечения респираторных заболеваний у крупного рогатого скота. Аминогруппу рокситромицина нейтрализуют добавлением линолевой кислоты в 10% пропиленгликоле в глицеринформале, что приводит к получению прозрачного раствора при концентрации 200 мг/мл. Как и в случае с тилмикозином, соль выпадает в осадок при введении раствора в воду. Пример 9: Тербинафин Тербинафин является противогрибковым средством со строением, представленным ниже:
Тербинафин является специфическим ингибитором сквален-эпоксидазы, ключевого фермента в синтезе эргостерола грибками. Аминогруппу тербинафина нейтрализуют добавлением линолевой кислоты в 10% пропиленгликоле в глицеринформале, что приводит к получению прозрачного раствора при концентрации 150 мг/мл. Полученная соль является плохо растворимой в воде и выпадает в осадок при введении раствора в воду. Хотя настоящее изобретение было описано и рассмотрено на примерах достаточно подробно, для того, чтобы специалист в данной области техники мог осуществить и применить его, различные альтернативы, модификации и усовершенствования будут очевидными, не выходя при этом за пределы существа и объема изобретения. Специалист в данной области техники с легкостью поймет, что настоящее изобретение хорошо приспособлено для достижения целей и получения как упомянутых результатов и преимуществ, так и присущих им преимуществ. Его модификации и другие применения будут очевидны специалисту в данной области техники. Данные модификации подпадают под объем настоящего изобретения и определяются объемом притязаний в соответствии с формулой изобретения. Для специалиста в данной области техники очевидно, что в настоящем изобретении могут быть сделаны различные замены или модификации, не выходящие при этом за пределы существа и объема изобретения. Все патенты и публикации, упомянутые в описании изобретения, являются показательными на уровне среднего специалиста в области техники, к которой относится настоящее изобретение. Настоящее изобретение, иллюстративно описанное здесь, подходящим образом может применяться на практике в отсутствие любого элемента или элементов, ограничения или ограничений, которые не были здесь конкретно раскрыты. Использованные термины и выражения применены с целью описания, а не с целью ограничения, и применение данных терминов и выражений не имеет целью исключить любые эквиваленты указанных и описанных признаков или их частей, но подразумевается, что внутри объема заявленного изобретения возможны различные изменения. Поэтому следует понимать, что, хотя настоящее изобретение было подробно раскрыто с помощью предпочтительных вариантов воплощения и необязательных признаков, специалист в данной области техники может прибегнуть к модификации и вариации раскрытых здесь понятий, и что такие модификации и вариации подпадают под объем настоящего изобретения, определенный прилагаемой формулой изобретения. Кроме того, если признаки и аспекты настоящего изобретения описываются с помощью формул Маркуша, специалисту в данной области техники будет понятно, что тем самым изобретение также включает в себя каждый отдельный член или подгруппу членов формулы Маркуша. Например, если X определен как выбранный из группы, состоящей из брома, хлора и йода, то пункты, в которых X является бромом, и пункты, в которых X является бромом и хлором, полностью описаны. Другие варианты воплощения представлены в следующей формуле изобретения.
Формула изобретения
1. Инъецируемая композиция для пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающая: соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворитель, объединенные вместе с образованием инъецируемой композиции, которая выпадает в осадок при инъекции в воду, и где при инъекции млекопитающему композиция высвобождает эффективное количество активного соединения во времени. 2. Композиция по п.1, где фармацевтически приемлемый растворитель является смешивающимся с водой растворителем. 3. Композиция по п.1, где фармакологически активное соединение является антибиотиком. 4. Композиция по п.1, где фармакологически активное соединение является тилмикозином, окситетрациклином, доксициклином, флуоксетином, рокситромицином, тербинафином или метопрололом. 5. Композиция по п.1, где фармакологически активное соединение выбрано из группы, состоящей из триметоприма, неомицина, стрептомицина, гентамицина, дибукаина, бупивакаина, бензокаина, тетракаина, ацепромазина, итраконазола, тетрациклинов, сульфонамидов и аминогликозидов. 6. Композиция по п.1, где липофильный противоион является С10-С22-насыщенной или ненасыщенной жирной кислотой. 7. Композиция по п.1, где липофильный противоион является C10-C18-насыщенной или ненасыщенной жирной кислотой. 8. Композиция по п.7, где жирная кислота выбрана из группы, состоящей из лауриновой кислоты, декановой кислоты, миристиновой кислоты, олеиновой кислоты и линолевой кислоты. 9. Композиция по п.1, где липофильный противоион является поликарбоновой кислотой. 10. Композиция по п.9, где поликарбоновая кислота выбрана из группы, состоящей из себациновой кислоты, полисебациновой кислоты, полиаспарагиновой кислоты, полиакриловой кислоты и полибензойной кислоты. 11. Композиция по п.1, где фармацевтически приемлемый растворитель выбран по отдельности или в комбинации из группы, состоящей из пирролидона, N-метилпирролидона, полиэтиленгликоля, пропиленгликоля, глицеринформаля, изосорбиддиметилового простого эфира, этанола, диметилсульфоксида и тетрагидрофурфурилового спирта. 12. Композиция по п.1, где фармацевтически приемлемый растворитель содержит 10% пропиленгликоль в глицеринформале, со стабилизаторами или без них. 13. Композиция по п.1, где фармацевтически приемлемый растворитель является триацетином. 14. Композиция по п.1, где фармакологически активное соединение является окситетрациклином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбран по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 15. Композиция по п.1, где фармакологически активное соединение является тилмикозином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбран по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 16. Способ пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающий: приготовление композиции, включающей: соль фармакологически активного соединения и липофильного противоиона и фармацевтически приемлемый растворитель, объединенные вместе с образованием инъецируемого состава, и инъекцию композиции млекопитающему, где при инъекции млекопитающему, по меньшей мере, часть композиции выпадает в осадок и высвобождает эффективное количество активного соединения во времени. 17. Способ по п.16, где фармацевтически приемлемый растворитель является смешивающимся с водой растворителем. 18. Способ по п.16, где фармакологически активное соединение является антибиотиком. 19. Способ по п.18, где антибиотик является тилмикозином, тетрациклином или доксициклином. 20. Способ по п.18, где антибиотик выбирают из группы, состоящей из триметоприма, неомицина, стрептомицина, гентамицина, тетрациклинов, сульфонамидов и аминогликозидов. 21. Способ по п.16, где липофильный противоион является жирной кислотой. 22. Способ по п.21, где жирная кислота является С10-С22-жирной кислотой. 23. Способ по п.22, где жирную кислоту выбирают из группы, состоящей из лауриновой кислоты, декановой кислоты, миристиновой кислоты, олеиновой кислоты и линолевой кислоты. 24. Способ по п.16, где липофильный противоион является себациновой кислотой. 25. Способ по п.16, где фармацевтически приемлемый растворитель выбирают из группы, состоящей из пирролидона, N-метилпирролидона, полиэтиленгликоля, пропиленгликоля, глицеринформаля, изосорбиддиметилового эфира, этанола, диметилсульфоксида и тетрагидрофурфурилового спирта. 26. Способ по п.16, где фармацевтически приемлемый растворитель является триацетином. 27. Способ по п.16, где фармакологически активное соединение является окситетрациклином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбирают по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 28. Способ по п.16, где фармакологически активное соединение является тилмикозином, липофильный противоион является декановой кислотой, а фармацевтически приемлемый растворитель выбирают по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 29. Способ пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающий: приготовление состава, включающего соль фармакологически активного соединения и липофильного противоиона и фармацевтически приемлемый растворитель, объединенные вместе с образованием инъецируемого состава, и инъекцию состава млекопитающему, где при инъекции млекопитающему, по меньшей мере, часть состава выпадает в осадок и высвобождает эффективное количество активного соединения во времени, пролонгируя тем самым время высвобождения фармакологически активного соединения. 30. Способ по п.29, где фармацевтически приемлемый растворитель является смешивающимся с водой растворителем. 31. Способ по п.29, где липофильный противоион является C10-C18-жирной кислотой. 32. Способ по п.31, где антибиотик является тилмикозином, окситетрациклином или доксициклином. 33. Способ по п.32, где жирную кислоту выбирают из группы, состоящей из лауриновой кислоты, декановой кислоты и миристиновой кислоты. 34. Способ по п.29, где липофильный противоион является себациновой кислотой. 35. Способ по п.29, где фармакологически активное соединение является окситетрациклином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбирают по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 36. Способ по п.29, где фармакологически активное соединение является тилмикозином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбирают по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 37. Способ приготовления инъецируемого состава для пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающий: получение соли фармакологически активного соединения и липофильного противоиона, получение фармацевтически приемлемого растворителя, объединение соли и растворителя с образованием инъецируемого состава, где при инъекции млекопитающему, по меньшей мере, часть состава выпадает в осадок и высвобождает эффективное количество активного соединения во времени. 38. Способ по п.37, где фармацевтически приемлемый растворитель является смешивающимся с водой растворителем. 39. Способ по п.37, где фармакологически активное соединение является антибиотиком. 40. Способ по п.37, где антибиотик является тилмикозином, окситетрациклином или доксициклином. 41. Способ по п.37, где липофильный противоион является C10-C18-жирной кислотой. 42. Способ по п.41, где жирную кислоту выбирают из группы, состоящей из лауриновой кислоты, декановой кислоты и миристиновой кислоты. 43. Способ по п.37, где липофильный противоион является себациновой кислотой. 44. Способ по п.37, где фармацевтически приемлемый растворитель выбирают из группы, состоящей из пирролидона, N-метилпирролидона, полиэтиленгликоля, пропиленгликоля, глицеринформаля, изосорбиддиметилового эфира, этанола, диметилсульфоксида и тетрагидрофурфурилового спирта. 45. Способ по п.37, где фармацевтически приемлемый растворитель является триацетином. 46. Способ по п.37, где фармакологически активное соединение является окситетрациклином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбирают по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 47. Способ по п.37, где фармакологически активное соединение является тилмикозином, липофильный противоион является лауриновой кислотой, а фармацевтически приемлемый растворитель выбирают по отдельности или в комбинации из группы, состоящей из полиэтиленгликоля, пропиленгликоля и глицеринформаля. 48. Инъецируемая композиция для пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающая: соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворитель, объединенные вместе с образованием инъецируемой композиции, и где при инъекции в водную окружающую среду, по меньшей мере, часть композиции выпадает в осадок и высвобождает эффективное количество активного соединения во времени. 49. Композиция по п.48, где фармацевтически приемлемый растворитель является смешивающимся с водой. 50. Композиция по п.48, где антибиотик является тилмикозином, окситетрациклином или доксициклином. 51. Композиция по п.48, где фармакологически активное соединение выбрано из группы, состоящей из триметоприма, неомицина, стрептомицина, гентамицина, дибукаина, бупивакаина, бензокаина, тетракаина, ацепромазина, итраконазола, тетрациклинов, сульфонамидов и аминогликозидов. 52. Композиция по п.48, где липофильный противоион является C10-C18-жирной кислотой. 53. Композиция по п.52, где жирная кислота выбрана из группы, состоящей из лауриновой кислоты, декановой кислоты и миристиновой кислоты. 54. Композиция по п.48, где липофильный противоион является себациновой кислотой. 55. Композиция по п.48, где фармацевтически приемлемый растворитель выбран по отдельности или в комбинации из группы, состоящей из пирролидона, N-метилпирролидона, полиэтиленгликоля, пропиленгликоля, глицеринформаля, изосорбиддиметилового эфира, этанола, диметилсульфоксида и тетрагидрофурфурилового спирта. 56. Композиция по п.48, где фармацевтически приемлемый растворитель является триацетином. 57. Инъецируемая композиция для пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающая: соль фармакологически активного соединения с липофильным противоионом и фармацевтически приемлемый растворитель, объединенные вместе с образованием инъецируемой композиции, и где, по меньшей мере, часть фармацевтически активного соединения растворяется в растворителе и выпадает в осадок in vivo. 58. Инъецируемая композиция для пролонгирования времени высвобождения фармакологически активного соединения, введенного млекопитающему, включающая: соль фармакологически активного соединения с противоионом поликарбоновой кислоты и фармацевтически приемлемый растворитель, объединенные вместе с образованием инъецируемой композиции, которая выпадает в осадок при инъекции в воду, и где при инъекции млекопитающему композиция высвобождает эффективное количество активного соединения во времени. 59. Композиция по п.58, где поликарбоновая кислота выбрана из группы, состоящей из полиаспарагиновой кислоты, полиакриловой кислоты, себациновой кислоты, полисебациновой кислоты и полибензойной кислоты. 60. Композиция по п.58, где фармацевтически приемлемый растворитель является смешивающимся с водой растворителем. 61. Композиция по п.58, где фармакологически активное соединение является антибиотиком. 62. Композиция по п.58, где фармакологически активное соединение является тилмикозином, окситетрациклином, доксициклином, флуоксетином, рокситромицином, тербинафином или метопрололом. 63. Композиция по п.58, где фармакологически активное соединение выбрано из группы, состоящей из триметоприма, неомицина, стрептомицина, гентамицина, дибукаина, бупивакаина, бензокаина, тетракаина, ацепромазина, итраконазола, тетрациклинов, сульфонамидов и аминогликозидов.
РИСУНКИ
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||