Патент на изобретение №2292344
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ПИРИДИЛЦИАНОГУАНИДИНОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ
(57) Реферат:
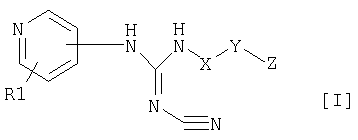
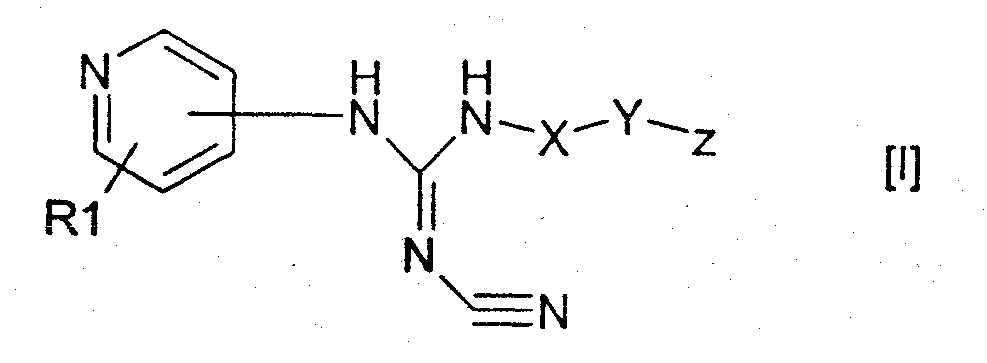
Изобретение относится к новым производным пиридилцианогуанидинов общей формулы (I)
или их фармацевтически приемлемым солям, где R1 обозначает водород; Х обозначает С1-12 углеводородный бирадикал; Y обозначает связь или О; Z обозначает 5-10-членный ароматический гетероциклический радикал, необязательно замещенный гидрокси; при условии, что R1 не присоединен к атому азота в пиридильном цикле. Соединения формулы (I) и их соли обладают антипролиферативной активностью и могут найти применение в медицине. 3 н. и 6 з.п. ф-лы, 1 табл.
Настоящее изобретение относится к новым пиридилцианогуанидиновым лекарственным средствам и фармацевтическим композициям, содержащим их, а также к применению их в производстве медикаментов. Вначале было обнаружено, что пиридилцианогуанидины, такие как пинацидил (N-1,2,2-триметилпропил-N’-циано-N”-(4-пиридил)гуанидин), открывают калиевые каналы, после чего их разрабатывали как противогипертонические средства. Замена боковой цепи пинацидила на более длинные боковые цепи, содержащие арил, приводит к снижению противогипертонической активности таких соединений, однако было обнаружено, что полученные таким образом соединения, с другой стороны, обладают противоопухолевой активностью при пероральном введении на крысиной модели, несущей асцитные опухоли Иошида (Yoshida ascites tumours). Различные классы пиридилцианогуанидинов с антипролиферативной активностью раскрыты, например, в 660823, WO 98/54141, WO 98/54143, WO 98/54144, WO 98/54145, WO 00/61559 и WO 00/61561. Зависимость активности от структуры (SAR – structure-activity relationships) для таких соединений описана в C. Schou et al., Bioorganic and Medicinal Chemistry Letters 7 (24), 1997, pp. 3095-3100, где антипролиферативный эффект ряда пиридилцианогуанидинов анализировали in vitro на различных клеточных линиях рака легких и молочной железы человека, а также на нормальных фибробластах человека. Затем P-J V Hjarnaa et al., Cancer Res. 59, 1999, pp. 5751-5757, опубликовали результаты тестирования конкретного цианогуанидинового соединения, а именно N-(6-(4-хлорфенокси)гексил)-N’-циано-N”-(4-пиридил)гуанидина, в анализах in vitro и in vivo. Данное соединение обладает активностью in vitro, сравнимой с активностью цитостатических агентов, даунорубицина и паклитаксела, но при этом оно демонстрирует гораздо более низкую антипролиферативную активность на нормальных эндотелиальных клетках человека. В анализах in vivo с использованием голых мышей, которым были трансплантированы человеческие опухолевые клетки, соединение демонстрирует значительную противоопухолевую активность, так же как и в отношении опухолевых клеток, устойчивых к традиционным противораковым лекарственным средствам, таким как паклитаксел. Для успешного применения лекарственного средства необходимо разумное соотношение таких факторов, как активность, биодоступность, токсичность, уровень побочных эффектов, растворимость и др., которое имеет место в случае усовершенствованных лекарственных средств на основе цианогуанидинов. Авторы настоящего изобретения обнаружили, что новые пиридилцианогуанидиновые соединения, содержащие гетероциклический радикал, обладают необычайно высокой антипролиферативной активностью. Соответственно, данное изобретение относится к соединениям формулы I
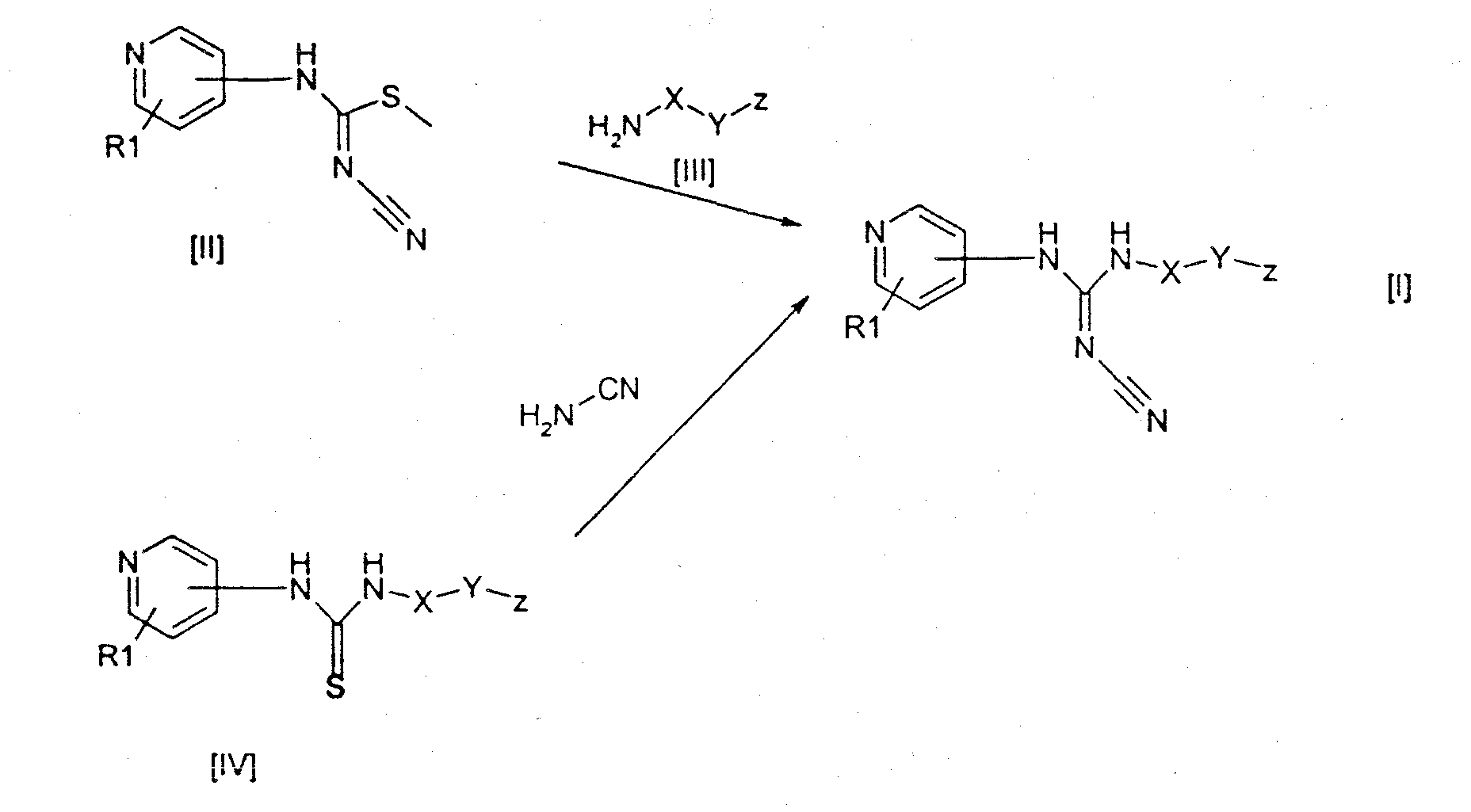
где R1 обозначает водород, галоген или один или несколько линейных или разветвленных, насыщенных или ненасыщенных Cl-6 углеводородных радикалов, необязательно замещенных галогеном, гидрокси, циано, нитро, карбокси, алкокси, алкоксикарбонилом, алкилкарбонилом, формилом, амино, аминоалкилом, аминокарбонилом, алкилкарбониламино, сульфо, аминосульфонилом, алкилсульфониламино, гидроксисульфонилокси, дигидроксифосфиноилокси или фосфоно; X обозначает линейный или разветвленный, насыщенный или ненасыщенный Cl-12 углеводородный бирадикал, необязательно замещенный галогеном, гидрокси, циано, нитро, карбокси, алкокси, алкоксикарбонилом, алкилкарбонилом, формилом, амино, аминоалкилом, аминокарбонилом, алкилкарбониламино, сульфо, аминосульфонилом, алкилсульфониламино, гидроксисульфонилокси, дигидроксифосфиноилокси или фосфоно; Y обозначает связь, O, C(O), S, S(O), S(O)2, C(O)O, NH, C(О)NH, OC(O) или NHC(O); Z обозначает ароматический или неароматический гетероциклический радикал, содержащий 5-12 атомов в цикле, необязательно замещенный галогеном, гидрокси, циано, нитро, алкокси, алкоксикарбонилом, алкилкарбонилом, формилом, аминоалкилом, или линейный или разветвленный, насыщенный или ненасыщенный Cl-4 углеводородный радикал, необязательно замещенный галогеном, гидрокси, циано, нитро, алкокси, алкоксикарбонилом, алкилкарбонилом, формилом или аминоалкилом; при условии, что R1 не присоединен к атому азота в пиридильном цикле; а также к их фармацевтически приемлемым солям, сольватам, гидратам, N-оксидам и пролекарственным формам. Данное изобретение также относится к применению соединения формулы I в терапии и к фармацевтическим композициям, включающим соединение формулы I. Данное изобретение также относится к способам лечения или профилактики заболеваний, включающим введение пациенту эффективной дозы соединения формулы I. Кроме того, данное изобретение также относится к применению соединений формулы I в производстве лекарственных препаратов. В контексте настоящего изобретения термин “углеводород” обозначает фрагмент, включающий только водород и углерод, предпочтительно включающий 1-18, например 1-12, например 1-6, атомов углерода. Примеры упомянутого углеводорода включают метан, этан, этен, этин, бутан, бутен, бутин, изобутан, трет-бутан, гексан, 1,3-диметилгексан, октан, октен, нонин, додекан, додецен и др. Соответствующий радикал или бирадикал представляет собой фрагмент, полученный в результате удаления из углеводорода соответственно одного или двух атомов водорода. Термин “гетероциклический радикал” обозначает моноциклические кольца, содержащие 1-3 гетероатома, выбранных из N, O и S, и конденсированные бициклические кольца, включающие 1-4 гетероатома из приведенных выше. Примеры включают тиенил, фурил, пиранил, изобензофуранил, хроменил, пирролил, имидазолил, пиридил, пиримидинил, индолизинил, пуринил, хинолил, нафтиридинил, циннонилил, хроманил, пирролидинил, имидазолидинил, пиперидил, пиперазинил, морфолинил, оксазинил, тетрагидрофуранил, оксазолидинил, тетрагидропиранил, тетразолил, тиазолил, пиразолил, оксазолил, изооксазолил, пиразинил, бензимидазолил и бензофуранил. Термин “галоген” обозначает фтор, хлор, бром и иод. Термин “фармацевтически приемлемая соль” обозначает соли, полученные в результате взаимодействия соединений формулы I, включающих кислые или основные группы, с подходящими основаниями или кислотами соответственно. Примерами таких кислот являются хлористоводородная, бромистоводородная, иодистоводородная, серная, азотная, уксусная, фосфорная, молочная, малеиновая, фталевая, лимонная, пропионовая, бензойная, глутаровая, глюконовая, метансульфоновая, салициловая, янтарная, винная, толуолсульфоновая, сульфаминовая и фумаровая кислоты. Примерами таких оснований являются гидроксид калия, гидроксид натрия, аммиак и амины. Термин “сольват” обозначает частицы, образовавшиеся в результате взаимодействия соединения, в данном случае соединения формулы I, с растворителем, например спиртом, глицерином или водой, где указанные частицы существуют в твердом виде. Если растворителем является вода, сольват называется гидратом. Термин “N-оксид” обозначает, например, пиридил-N-оксидные производные соединения данного изобретения. Такие соединения могут быть получены в результате окисления N пиридила подходящим окисляющим агентом, например 3-хлорпербензойной кислотой, в инертном растворителе, например дихлорметане. Термин “алкил” обозначает монорадикалы, полученные из алканов, предпочтительно включающие 1-8 атомов углерода, такие как метил, этил, пропил, бутил, изобутил, трет-бутил, пентил, гексил и циклогексил. Термин “алкокси” обозначает радикал формулы -OR, где R представляет собой алкил, как определено выше. Термин “алкоксикарбонил” обозначает радикал формулы -C(O)-OR, где R представляет собой алкил, как определено выше. Термин “алкилкарбонил” обозначает радикал формулы -C(O)-R, где R представляет собой алкил, как определено выше. Термин “аминоалкил” обозначает радикал формулы -R-NR’2, где R представляет собой алкил, как определено выше, и каждый R’ независимо представляет собой алкил, как определено выше, или водород. Термин “аминокарбонил” обозначает радикал формулы -C(O)-NR’2, где каждый R’ независимо представляет собой алкил, как определено выше, или водород. Термин “аминосульфонил” обозначает радикал формулы -S(O)2-NR’2, где каждый R’ независимо представляет собой алкил, как определено выше, или водород. Термин “алкилсульфониламино” обозначает радикал формулы N(R’)S(О)2-R, где R представляет собой алкил, как определено выше, и каждый R’ независимо представляет собой алкил, как определено выше, или водород.Термин “амино” обозначает радикал формулы -NR’2, где каждый R’ независимо представляет собой алкил, как определено выше, или водород. Термин “пролекарство” обозначает производное активного соединения, которое не обладает, или не обязательно обладает, физиологической активностью активного соединения, но которое при введении пролекарственной формы может подвергаться in vivo ферментативному расщеплению, такому как гидролиз, с высвобождением активного соединения. Получение пролекарственной формы соединений настоящего изобретения раскрыто в международной патентной заявке PCT/DK01/00750. В предпочтительном воплощении данного изобретения R1 обозначает водород, галоген, или один или несколько линейных или разветвленных, насыщенных или ненасыщенных C1-6 углеводородных радикалов; X обозначает линейный или разветвленный, насыщенный или ненасыщенный C1-12 углеводородный бирадикал; Y обозначает O или C(O); Z обозначает ароматический или неароматический гетероциклический радикал, содержащий 5-10 атомов в цикле. В следующем предпочтительном воплощении данного изобретения R1 обозначает водород; X обозначает линейный C5-10 углеводородный радикал; Y обозначает O; Z обозначает ароматический или неароматический гетероциклический радикал, содержащий 5-10 атомов в цикле. В другом предпочтительном воплощении Z выбран из группы, состоящей из пиридила, имидазолила, хинолила или пиримидинила. Конкретные примеры соединений, соответствующих формуле I, включают N-[6-(3-пиридилокси)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(1-имидазолил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(2-хинолилокси)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(3-пиридил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(3-хинолил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(5-пиримидинил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин. Более того, было обнаружено, что соединения, выбранные из группы, состоящей из 3-[6-(N-трет-бутоксикарбониламино)-1-гексилокси]-пиридина; гидрохлорид 3-[6-амино-1-гексилокси]пиридина; 3-[6-амино-1-гексилокси]пиридина; 1-[6-(N-трет-бутоксикарбониламино)-1-гексил]имидазола; гидрохлорид 1-(6-амино-1-гексил)имидазола; 1-(6-амино-1-гексил)имидазола; 2-[6-(N-трет-бутоксикарбониламино)-1-гексилокси]-хинолина; 2-(6-амино-1-гексилокси)-хинолина; могут использоваться при получении соединений формулы I. Соединения формулы I могут быть получены путем взаимодействия соединения формулы II, где R1 такой, как определен в формуле I, с соединением формулы III, где X, Y и Z такие, как определены в формуле I, см. схему. Реакцию можно проводить в подходящем растворителе, таком как пиридин, необязательно в присутствии третичного амина, такого как триэтиламин, и катализатора, такого как 4-(N,N-диметиламино)пиридин, и при температуре, находящейся в интервале от комнатной до 100°C. В течение реакции R1, X, Y и Z могут временно содержать подходящие защитные группы. Соединения формул II и III известны из литературы или могут быть получены с помощью методов, хорошо известных специалистам в данной области. В другом варианте осуществления изобретения тиомочевина формулы IV, которая содержит такие же заместители, как и определенные для формулы (I), и при необходимости временно защищенная, взаимодействует с одним или несколькими эквивалентами N,N’-дициклогексилкарбодиимида (DCCD) и цианамида в инертном растворителе, таком как ацетонитрил, при температуре, превышающей комнатную, с образованием соединения формулы I, см. схему. Соединения формулы IV могут быть получены с помощью методов, хорошо известных специалистам в данной области.
Фармацевтические композиции В другом аспекте данное изобретение относится к фармацевтическим композициям, содержащим соединение формулы I. Композиции настоящего изобретения, предназначенные как для применения в ветеринарии, так и для лечения человека, включают активные ингредиенты в сочетании с фармацевтически приемлемым носителем(ями) и необязательно с другим терапевтическим ингредиентом(ами). Носитель должен быть “приемлемым” в том смысле, что он должен быть совместимым с другими ингредиентами композиций и не оказывать вредного воздействия на реципиента. Обычно активный ингредиент составляет 0,1-100 мас. % от массы композиции. Как правило, разовая доза композиции содержит от 0,07 мг до 1 г соединения формулы I. Под термином “разовая доза” подразумевают однократную, т. е. единичную дозу, которую можно ввести пациенту и которую можно легко обрабатывать и расфасовывать, при этом она остается физически и химически стабильной разовой дозой, содержащей либо активное вещество как таковое, либо его смесь с твердыми или жидкими фармацевтическими разбавителями или носителями. Композиции включают, например, формы, подходящие для перорального (включая замедленное или регулируемое высвобождение), ректального, парентерального (включая подкожное, интраперитонеальное, внутримышечное, внутрисуставное и внутривенное), чрескожного, офтальмического, местного, назального или буккального введения. Композиции обычно могут быть представлены в виде дозированной лекарственной формы и могут быть получены с помощью любого из хорошо известных в области фармацевтики способов, например, как раскрыто в Remington, The Science and Practice of Pharmacy, 20th ed., 2000. Все способы включают стадию смешивания активного ингредиента с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. В основном композиции получают путем равномерного и тщательного смешивания активного ингредиента с жидким носителем, или мелко размельченным твердым носителем, или с обоими, и затем, при необходимости, формования продукта в целевую композицию. Композиции настоящего изобретения, подходящие для перорального введения, могут находиться в виде дискретных единиц, таких как капсулы, саше, таблетки или пастилки, каждая из которых содержит предварительно определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкости, такой как этанол или глицерин; или в виде эмульсии типа масло в воде или вода в масле. Такими маслами могут быть пищевые масла, такие как, например, хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают синтетические или природные смолы, такие как трагакант, альгинат, гуммиарабик, дкстран, натрий карбоксиметилцеллюлозу, желатин, метилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, карбомеры и поливинилпирролидон. Активные ингредиенты также могут быть введены в виде болюса, электуария или пасты. Таблетки можно получить путем прессования или формования активного ингредиента необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки могут быть получены путем прессования в подходящем устройстве активного ингредиента(ов) в свободнотекучем виде, таком как порошок или гранулы, необязательно в смеси со связующим средством, таким как, например, лактоза, глюкоза, крахмал, желатин, аравийская камедь, трагакантовая камедь, альгинат натрия, карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, полиэтиленгликоль, воски и т.п.; смазывающим средством, таким как, например, олеат натрия, стеарат натрия, бензоат натрия, ацетат натрия, хлорид натрия или т.п.; разрыхлителем, таким как, например, крахмал, метилцеллюлоза, агар, бентонит, кроскармелоза натрия, натриевая соль гликолята крахмала, кросповидон или т.п., или диспергирующим средством, таким как полисорбат 80. Формованные таблетки могут быть получены путем формования в подходящем устройстве смеси порошкообразного активного ингредиента и подходящего носителя, увлаженного инертным жидким разбавителем. Композиции для ректального введения могут быть получены в виде свечей, в которых соединение настоящего изобретения смешано с низкоплавкими растворимыми или нерастворимыми в воде твердыми веществами, такими как масло какао, гидрированные растительные масла, полиэтиленгликоль или сложные эфиры жирных кислот и полиэтиленгликолей, тогда как эликсиры могут быть получены с использованием миристилпальмитата. Композиции, подходящие для парентерального введения, обычно включают стерильные масляные или водные препараты активных ингредиентов, которые предпочтительно являются изотоническими по отношению к крови реципиента, например изотонический солевой раствор, изотонический раствор глюкозы или буферный раствор. Как правило, композицию можно стерилизовать, например, фильтрацией через фильтр, задерживающий бактерии, добавлением к композиции стерилизующего средства, облучением или нагреванием композиции. Липосомальные композиции, как раскрыто, например, в Encyclopedia of Pharmaceutical Technology, vol. 9,1994, также подходят для парентерального введения. Альтернативно соединение формулы I может быть представлено в виде стерильного твердого препарата, например лиофилизированного порошка, который легко растворяется в стерильном растворителе непосредственно перед применением. Чрескожные композиции могут быть представлены в виде пластыря или аппликатора. Композиции, подходящие для офтальмического введения, могут быть представлены в виде стерильного водного препарата активных ингредиентов, которые могут находиться в микрокристаллическом виде, например в виде суспензии микрокристаллов в воде. Для офтальмического введения активного ингредиента также можно использовать липосомные композиции или биоразрушаемые полимерные системы, например, как раскрыто в Encyclopedia of Pharmaceutical Technology. vol. 2,1989. Композиции, подходящие для местного или офтальмического введения, включают жидкие или полужидкие препараты, такие как линименты, лосьоны, гели, аппликации (applicants), эмульсии типа масло в воде или вода в масле, такие как кремы, мази или пасты; или растворы или суспензии, такие как капли. Композиции, подходящие для назального или буккального введения, включают порошкообразные композиции, композиции, вводимые без помощи вспомогательных средств (selfpropelling), и композиции в виде спрея, такие как аэрозоли и распылители. В добавление к вышеуказанным ингредиентам композиции на основе соединения формулы I могут включать один или несколько дополнительных ингредиентов, таких как разбавители, буферы, ароматизирующие средства, красители, поверхностно-активные вещества, загустители, консерванты, например метилгидроксибензоат (включая антиоксиданты), эмульгирующие средства и т.п. При системном лечении с использованием настоящего изобретения вводят суточные дозы соединения формулы I, составляющие 0,001-500 мг на килограмм массы тела, предпочтительно 0,002-100 мг/кг массы тела млекопитающего, например 0,003-20 мг/кг или 0,003-5 мг/кг, как правило, в соответствии с суточной дозой для взрослого человека, составляющей от 0,01 до 37000 мг. Однако настоящее изобретение также предоставляет соединения и композиции, предназначенные для введения с более длинными интервалами, например, каждую неделю, каждые три недели или каждый месяц. При местном лечении дерматологических нарушений вводят мази, кремы или лосьоны, содержащие 0,1-750 мг/г, предпочтительно 0,1-500 мг/г, например 0,1-200 мг/г, соединения формулы I. При местном применении в офтальмологии вводят мази, капли или гели, содержащие 0,1-750 мг/г, предпочтительно 0,1-500 мг/г, например 0,1-200 мг/г соединения формулы I. Пероральные композиции получают предпочтительно в виде таблеток, капсул или капель, содержащих 0,07-1000 мг, предпочтительно 0,1-500 мг, соединения формулы I в дозированной лекарственной форме. Было обнаружено, что производные цианогуанидина способны модулировать активность I NF Роль активированного NF Комплекс I Как описано выше, регулирование уровня активированного NF Апоптоз представляет собой генетически кодируемую программу клеточной гибели, характеризующуюся “активным решением” клетки умереть, принятым на основе информации, полученной из окружающей среды, истории ее развития и т.п. В отличие от клеток, подвергающихся некрозу, клетки, вступающие в апоптоз, часто способны выжить, но предпочитают умереть для блага всего организма. Апоптоз также отличается от некроза тем, что некроз часто связан с травмой ткани и повреждением клетки, тогда как клетки, следующие курсу апоптоза, разрушаются изнутри контролируемым образом [Tran, Science and Medicine, 6, 18-27, 1999; Williams, Trends Cell Biol., 2, 263-267, 1992]. В предпочтительном воплощении данное изобретение предоставляет фармацевтические композиции, включающие соединение формулы I в сочетании с одним или несколькими другими фармакологически активными соединениями, использующимися для лечения пролиферативных заболеваний. Примеры соединений, использующихся для лечения пролиферативных заболеваний, которые можно применять вместе с соединениями настоящего изобретения, включают производные S-триазина, такие как алтретамин; ферменты, такие как аспарагиназа; антибиотические средства, такие как блеомицин, дактиномицин, даунорубицин, доксорубицин, идарубицин, митомицин, эпирубицин и пликамицин; алкилирующие средства, такие как бусульфан, карбоплатин, кармустин, хлорамбуцил, цисплатин, циклофосфамид, дакарбазин, ифосфамид, ломустин, мехлоретамин, мелфалан, прокарбазин и тиотепа; антиметаболиты, такие как кладрибин, цитарабин, флоксуридин, флударабин, фторурацил, гидроксимочевина, меркаптопурин, метотрексат, гемцитабин, пентостатин и тиогуанин; антимитотические средства, такие как этопозид, паклитаксел, тенипозид, винбластин, винорелбин и винкристин; гормональные средства, например ингибиторы ароматазы, такие как аминоглютетимид, кортикостероиды, такие как дексаметазон и преднизон, а также фактор, высвобождающий лютеинизирующий гормон (LH-RH – luteinizing hormone releasing hormone); антиэстрогены, такие как тамоксифен, форместан и летрозол; антиандрогены, такие как флутамин; модуляторы биологического ответа, например лимфокины, такие как альдеслейкин и другие интерлейкины; интерферон, такой как интерферон- В особенности, такие антипролиферативные соединения, как паклитаксел, фторурацил, этопозид, циклофоспамид, цисплатин, карбоплатин, винкристин, гемцитабин, винорелбин, хлорамбуцил, доксорубицин, мелфалан и сеокальцитол, успешно используются в комбинированных композициях настоящего изобретения. Предполагается, что комбинированная композиция настоящего изобретения может быть предоставлена в виде смесей соединений или в виде отдельных соединений, предназначенных для одновременного или последовательного введения. Выбор временных интервалов в режиме последовательного введения может быть сделан опытным врачом или ветеринаром. В частности, пролиферативные заболевания или состояния, подлежащие лечению с помощью способа настоящего изобретения, включают ряд раковых и опухолевых заболеваний или состояний, таких как лейкемия, острая миелоидная лейкемия, хроническая миелоидная лейкемия, хроническая лимфатическая лейкемия, миелодисплазия, множественная миелома, болезнь Ходжкина или не-ходжкинская лимфома, мелкоклеточная или немелкоклеточная карцинома легкого, рак желудка, кишечника или ободочной и прямой кишки, рак простаты, яичника или молочной железы, рак головы, мозга или шеи, рак мочевого тракта, почек или рак мочевого пузыря, злокачественная меланома, рак печени, рак матки или поджелудочной железы. Данное изобретение также относится к применению соединения формулы I, необязательно вместе с другими противоопухолевыми соединениями, описанными выше, в производстве лекарственных средств. В особенности, если предполагается использовать упомянутое лекарственное средство для лечения пролиферативных заболеваний, например раков, таких, как описанные выше. СПОСОБЫ ПОЛУЧЕНИЯ Значения химических сдвигов для спектров 1H ядерного магнитного резонанса (ЯМР) (300 МГц) и 13C ЯМР (75,6 МГц) приводят по отношению к внутренним стандартам триметилсилану ( Способ получения 1 3-[6-(N-трет-бутоксикарбониламино)-1-гексилокси]пиридин 3-Гидроксипиридин (262 мг) добавляют к суспензии 60% гидрида натрия (128 мг) в N,N-диметилформамиде (5 мл) и смесь перемешивают 30 минут при 60°C. После охлаждения на льду добавляют по каплям раствор N-(трет-бутоксикарбонил)-6-бромгексиламина (770 мг) (Helv. Chim. Acta 76 891 (1993) в N,N-диметилформамиде (3 мл) и перемешивание продолжают в течение ночи при комнатной температуре. Добавляют воду и лед и смесь экстрагируют три раза диэтиловым эфиром. Органические фазы промывают насыщенным раствором хлорида натрия, сушат и упаривают, получая желтое масло, которое затем очищают хроматографией на силикагеле с использованием диэтилового эфира в качестве элюента, получая целевое соединение в виде бесцветного масла. 1H ЯМР (CDCl3) Способ получения 2 3-[6-амино-1-гексилокси]пиридина гидрохлорид 3-[6-(N-трет-Бутоксикарбониламино)-1-гексилокси]пиридин (180 мг) обрабатывают большим избытком хлористого водорода в диэтиловом эфире при перемешивании в течение 45 минут, после чего упаривают в вакууме. Растирают с диэтиловым эфиром, декантируют и упаривают, получая указанное в заголовке соединение в виде бесцветного порошка. 1H ЯМР (ДМСО) Способ получения 3 3-[6-амино-1-гексилокси]пиридин Раствор гидрохлорида 3-[6-амино-1-гексилокси]пиридина в воде сильно подщелачивают гидроксидом натрия и дважды экстрагируют хлороформом. Органическую фазу сушат и упаривают в вакууме, получая масло, которое используют на следующей стадии без дополнительной очистки. Способ получения 4 1-[6-(N-трет-Бутоксикарбониламино)-1-гексил]имидазол Имидазол (70 мг) и 1,4M метоксид натрия (1 мл) добавляют к N,N-диметилформамиду (3 мл) и смесь перемешивают при комнатной температуре в течение 30 минут. Добавляют раствор N-(трет-бутоксикарбонил)-6-бромгексиламина (280 мг) в N,N-диметилформамиде (1 мл) и после нагревания при 80-90°C в течение 30 минут перемешивание продолжают в течение ночи при комнатной температуре. После упаривания в вакууме перемешивают с ацетоном и фильтруют, получая фильтрат, который после упаривания очищают хроматографией на силикагеле, используя этилацетат/метанол (4: 1) в качестве элюента, и получают указанное в заголовке соединение в виде бесцветного масла. 1H ЯМР (CDCl3) Способ получения 5 1-(6-Амино-1-гексил)имидазола гидрохлорид Получают, как описано в способе получения 2, но 3-[6-(N-трет-бутоксикарбониламино)-1-гексилокси]пиридин заменяют на 1-[6-(N-трет-бутоксикарбониламино)-1-гексил]имидазол. Бесцветные кристаллы. 1H ЯМР (ДМСО) Способ получения 6 1-(6-Амино-1-гексил)имидазол Получают, как описано в способе получения 3, но гидрохлорид 3-[6-амино-1-гексилокси]-пиридина заменяют на гидрохлорид 1-(6-амино-1-гексил)имидазола. Бесцветное масло. Способ получения 7 2-[6(N-трет-Бутоксикарбониламино)-1-гексилокси]хинолин Данное соединение получают, как описано в способе получения 1, но 3-гидроксипиридин заменяют на 2-гидроксихинолин. Неочищенный продукт очищают хроматографией на силикагеле с этилацетатом в качестве элюента, получая целевое соединение в виде бесцветного масла. 1H ЯМР (CDCl3) Способ получения 8 2-(6-Амино-1-гексилокси)хинолин 2-[6-(N-трет-бутоксикарбониламино)-1-гексилокси]хинолин (480 мг) обрабатывают большим избытком хлористого водорода в диэтиловом эфире при перемешивании в течение 1 часа при комнатной температуре. Кристаллический продукт выделяют фильтрацией и повторно растворяют в воде, после чего раствор сильно подщелачивают гидроксидом натрия и дважды экстрагируют хлороформом. Органическую фазу сушат над карбонатом калия, фильтруют и упаривают, получая указанное в заголовке соединение в виде бесцветного масла. 1H ЯМР (CDCl3) Пример 1 N-[6-(3-пиридилокси)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин Смесь 3-[6-амино-1-гексилокси]пиридина (150 мг), S-метил-N-циано-N’-4-пиридилизотиомочевины (123 мг), триэтиламина (0,18 мл), 4-(N,N-диметиламино)пиридина (3,5 мг) и пиридина (5 мл) перемешивают в течение ночи при 60°C. После охлаждения до комнатной температуры пиридин удаляют, дважды упаривая в вакууме с толуолом, и остаток распределяют между водой и этилацетатом. Органическую фазу сушат и упаривают, получая неочищенный продукт, который очищают хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/метанол/водный аммиак (40:10:2,5). Чистые фракции объединяют и упаривают, получая указанное в заголовке соединение, которое кристаллизуют из этилацетата 1H ЯМР (ДМСО) Пример 2 N-[6-(1-имидазолил)-1-гексилl-N’-циано-N”-(4-пиридил)гуанидин Данное соединение получают, как описано в примере 1, но 3-[6-амино-1-гексилокси]пиридин заменяют на 1-(6-амино-1-гексил)имидазол 1H ЯМР (ДМСО) Пример 3 N-[6-(2-Хинолилокси)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин Получают, как описано в примере 1, но 3-[6-амино-1-гексилокси]пиридин заменяют на 2-(6-амино-l-гексилокси)хинолин. 1H ЯМР (ДМСО) Пример 4 N-[6-(3-Пиридил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин Получают, как описано в примере 1, но 3-[6-амино-1-гексилокси]пиридин заменяют на 3-(6-амино-1-гексил)пиридин. 13C ЯМР (ДМСО) Пример 5 N-[6-(3-хинолил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин Получают, как описано в примере 1, но 3-[6-амино-1-гексилокси]пиридин заменяют на 3-(6-амино-1-гексил)хинолин. 13C ЯМР (ДМСО) Пример 6 N-[6-(5-пиримидинил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин Получают, как описано в примере 1, но 3-[6-амино-1-гексилокси]пиридин заменяют на 5-(6-амино-1-гексил)пиримидин. 13C ЯМР (ДМСО) Пример 7 Фармацевтическая композиция в форме таблетки 0,1 мг соединения примера 1, 300 мг глюкозы, 6,2 мг лактозы, 0,6 мг стеарата магния, 30 мг крахмала тщательно перемешивают и прессуют в подходящем устройстве для формования таблетки. Фармакологическая активность Испытания проводились путем оценки пролиферации клеток мелкоклеточной карциномы легких человека (NYH). Методика и техника эксперимента соответствуют традиционной, применяемой в аналогичных исследованиях. Результаты испытаний in vitro оценки пролиферации клеток мелкоклеточной карциномы легких человека (NYH).
Как видно из таблицы, указанные соединения способны ингибировать пролиферацию раковых клеток in vitro при низких концентрациях.
Формула изобретения
1. Соединение формулы I
где R1 обозначает водород; Х обозначает линейный или разветвленный, насыщенный или ненасыщенный С1-12 углеводородный бирадикал; Y обозначает связь или О; Z обозначает 5-10-членный ароматический гетероциклический радикал, необязательно замещенный гидрокси; при условии, что R1 не присоединен к атому азота в пиридильном цикле; а также его фармацевтически приемлемые соли. 2. Соединение по п.1, где R1 обозначает водород; Х обозначает линейный C4-12 углеводородный бирадикал; Y обозначает связь или О; Z обозначает 5-10-членный ароматический гетероциклический радикал с 1-3 атомами азота. 3. Соединение по п.1, где R1 обозначает водород; X обозначает линейный насыщенный С5-10 углеводородный бирадикал; Y обозначает О; Z обозначает 5-10-членный ароматический гетероциклический радикал с 1-3 атомами азота. 4. Соединение по п.1, где Z обозначает пиридил, имидазолил, хинолил или пиримидинил. 5. Соединение по п.1, выбранное из группы, состоящей из N-[6-(3-пиридилокси)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(1-имидазолил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(2-хинолилокси)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(3-пиридил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(3-хинолил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин; N-[6-(5-пиримидинил)-1-гексил]-N’-циано-N”-(4-пиридил)гуанидин. 6. Соединение по любому из пп.1-5 для изготовления лекарственного средства для лечения пролиферативных заболеваний. 7. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая соединение по любому из пп.1-5, вместе с фармацевтически приемлемыми эксципиентами. 8. Композиция по п.7 в виде разовой лекарственной формы. 9. Применение соединения по любому из пп.1-5 в производстве лекарственного средства для лечения пролиферативных заболеваний.
|
||||||||||||||||||||||||||
 B киназы (далее используется аббревиатура IKK). Модулируя активность IKK в клетках можно регулировать уровень активированного NF
B киназы (далее используется аббревиатура IKK). Модулируя активность IKK в клетках можно регулировать уровень активированного NF и I
и I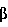 экспрессируются в большинстве клеток, где они связываются с Rel белками p65, т. е. NF
экспрессируются в большинстве клеток, где они связываются с Rel белками p65, т. е. NF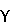 , с общей молекулярной массой 900 кДа. IKK
, с общей молекулярной массой 900 кДа. IKK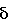 =0,00), хлороформу (
=0,00), хлороформу (