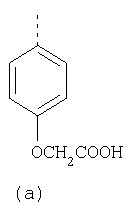Патент на изобретение №2292342
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОЛЕКАРСТВ ИНГИБИТОРОВ ПРОТОННОГО НАСОСА
(57) Реферат:
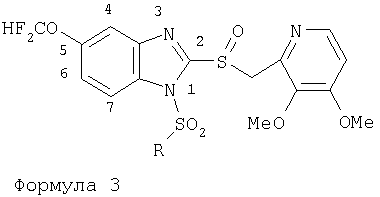
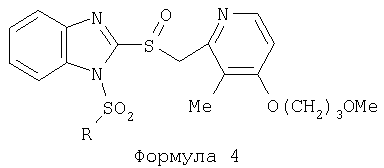
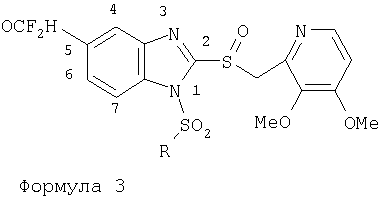
Изобретение относится к новым соединениям Формулы 1, Формулы 2, Формулы 3 или Формулы 4
или изомерам соединений Формул 2 и 3, где значения символов определены в формуле изобретения, и к их фармацевтически приемлемым солям, обладающим улучшенной растворимостью в водных средах и улучшенной биодоступностью, где указанные соединения обладают ингибирующей активностью в отношении секреции желудочной кислоты. 3 н. и 32 з.п. ф-лы, 4 табл.
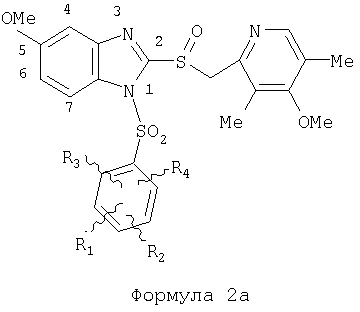
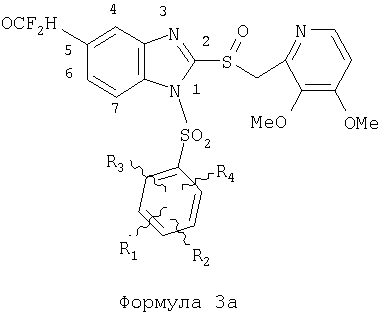
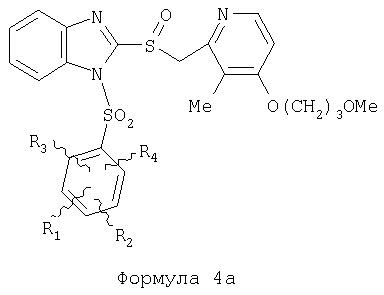
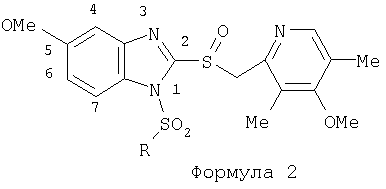
Предпосылки создания изобретения Область изобретения Настоящее изобретение направлено на усовершенствованные пролекарства ингибиторов протонного насоса, которые могут использоваться в качестве противоязвенных средств. Более конкретно, настоящее изобретение направлено на пролекарства, которые медленно гидролизуются с образованием ингибиторов протонного насоса бензимидазольного типа, экзогенно или эндогенно ингибирующих секрецию желудочной кислоты, которые имеют лучшую растворимость в физиологических жидкостях и могут быть использованы для предупреждения и лечения воспалительных желудочно-кишечных заболеваний у млекопитающих, включая человека. Краткое описание уровня техники Производные бензимидазола, предназначенные для ингибирования секреции желудочного сока, раскрыты в патентах США 4045563; 4255431; 4628098; 4686230; 4758579; 4965269; 5021433; 5430042, 5708017, 6093734 и 6599167. В общих чертах, ингибиторы секреции желудочной кислоты бензимидазольного типа действуют посредством перегруппировки с образованием тиофильного фрагмента, который затем ковалентно связывается с желудочной Н,К-АТФазой, ферментом, участвующим в конечной стадии выделения протона в париетальной клетке, и тем самым ингибирует данный фермент. Соединения, ингибирующие желудочный фермент Н,К-АТФазу, большей частью известны как “ингибиторы протонного насоса” (ИПН). Некоторые бензимидазолы, ингибирующие желудочный фермент Н,К-АТФазу, широко используются в качестве лекарственных препаратов при лечении человека и известны под такими наименованиями как LANSOPRAZOLE (лансопразол) (патент США 4628098), OMEPRAZOLE (омепразол) (патенты США 4255431 и 5693818), PANTOPRAZOLE (пантопразол) (патент США 4758579) и RABEPRAZOLE (рабепразол) (патент США 5045552). Заболевания, при лечении которых используются ингибиторы протонного насоса, и в частности четыре упомянутых выше лекарственных препарата, включают наряду с прочими пептическую язву, изжогу, рефлюкс-эзофагит, эрозивный эзофагит, неязвенную диспепсию, инфекцию Helicobacter pylori, ларингит и астму. Несмотря на то что лекарственные препараты типа ингибиторов протонного насоса представляют собой значительный прогресс в области медицины человека и в ветеринарии, они все же не лишены недостатков. Недостатки используемых в настоящее время лекарственных препаратов типа ингибиторов протонного насоса (ИПН) проще всего можно объяснить при более подробном описании способа их действия, заболеваний или состояний, при лечении которых они используются, и условий их применения. Итак, кислотозависимые заболевания включают, не ограничиваясь перечнем, эрозивный эзофагит, рефлюкс-эзофагит, желудочную и дуоденальную язву, неязвенную диспепсию, инфекцию Helicobacter pylori. Обычная терапия всех этих заболеваний, за исключением инфекции, вызванной бактерией Н.pylori, включает лечение препаратами, подавляющими секрецию кислоты, одним из типов которых являются упомянутые выше ингибиторы протонного насоса. Используемые в настоящее время ингибиторы протонного насоса представляют собой пиридилметилсульфинилбензимидазолы (или соединения, имеющие аналогичную структуру) с установленной величиной рКа от 4,0 до 5,0. Механизм их действия заключается в аккумуляции в кислой области париетальной клетки (секреторном канальце, рН около 1,0) и последующем, катализируемым ионами водорода, превращении в реакционноспособные тиофильные соединения, способные ингибировать желудочный фермент АТФазу, приводя к эффективному ингибированию желудочной секреции со значительным терапевтическим индексом. Благодаря такому механизму используемые в настоящее время лекарственные препараты типа ИПН нуждаются в специальной желудочной защите для того, чтобы оставаться активными при дуоденальной абсорбции. По этой причине, а также из-за легкости разложения в кислой среде желудка лекарственные препараты типа ИПН, предназначенные для перорального приема, покрывают энтеросолюбильной оболочкой. Необходимость в энтеросолюбильном покрытии является недостатком ввиду его высокой стоимости и чувствительности к влаге. Из-за потребности аккумулирования в кислой области париетальной клетки для эффективного действия лекарственных препаратов типа ИПН необходима секреция кислоты. Было установлено, что период полувыведения из плазмы таких препаратов составляет от 60 до 90 минут. Все кислотные насосы являются неактивными в течение этого времени, вернее, лишь около 75%, в среднем, являются активными в течение времени, которое лекарственный препарат находится в крови после перорального введения. В ходе медицинских исследований также было установлено, что при повседневном пероральном одноразовом приеме максимальное ингибирование стимулированного высвобождения кислоты составляет приблизительно 66%. Это происходит благодаря сочетанию короткого периода полувыведения вещества из плазмы, ограниченного числа кислотных насосов, активных в момент присутствия вещества, и обороту кислотных насосов. В повседневной практике зачастую невозможно при помощи вечернего перорального приема препарата надлежащим образом контролировать секрецию кислоты в ночное время, поскольку к моменту секреции кислоты, происходящей после полуночи, препарат исчезает из плазмы. Для терапии кислотозависимых заболеваний и лечения инфекции Н.pylori (в сочетании с антибиотиками), а также для ослабления симптомов неязвенной диспепсии идеальным было бы полное ингибирование секреции кислоты. При помощи используемых в настоящее время лекарственных препаратов типа ИПН это достижимо лишь при внутривенной инфузии; в случае препарата омепразол требуются внутривенные инфузии по 8 мг/час. Очевидно, что существует необходимость в препарате или препаратах, действующих по механизму лекарственных препаратов типа ИПН, но способных достигать или приближаться к полному ингибированию секреции кислоты при пероральном применении. Из-за неполного и продолжающегося менее 24 часов ингибирования секреции кислоты при пероральном введении лекарственых форм современных препаратов типа ИПН лечение желудочных и дуоденальных изъязвлений занимает от 4 до 8 недель. Это происходит несмотря на то, что время воспроизведения поверхностных клеток пищевода, желудка и двенадцатиперстной кишки составляет приблизительно 72 часа. Несомненно, что столь длительное лечение существующими препаратами обусловлено неадекватным подавлением кислоты и вызванным кислотой разрушением. Все вышесказанное подчеркивает необходимость создания препарата или препаратов, действующих по механизму лекарственных средств типа ИПН, способных достигать или приближаться к полному ингибированию секреции кислоты при пероральном применении. В качестве дополнительных предпосылок, имеющих отношение к настоящему изобретению, заявители отмечают концепцию пролекарства, хорошо известную в данной области. Вообще говоря, пролекарства по существу являются производными лекарственных соединений, которые превращаются в физиологически активные соединения после введения в организм. Превращение может протекать самостоятельно, как, например, гидролиз в физиологической среде, либо быть катализируемым ферментами. Из значительного объема научной литературы, посвященной общим сведениям о пролекарствах, можно привести следующие примеры: Design of Prodrugs (Создание пролекарств) (Bundgaard H. ed.) 1985 Elsevier Science Publishers B.V. (Biomedical Division), Chapter 1; Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities (Создание пролекарств: Биообратимые производные для различных функциональных групп и химических объектов) (Hans Bundgaard); Bundgaard et al. Int. J. of Pharmaceutics 22 (1984) 45-56 (Elsevier); Bundgaard et al. Int. J. of Pharmaceutics 29 (1986) 19-28 (Elsevier); Bundgaard et al. J. Med. Chem. 32 (1989) 2503-2507 Chem. Abstracts 93, 137935y (Bundgaard et al.); Chem. Abstracts 95, 138493f (Bundgaard et al.); Chem. Abstracts 95, 138592n (Bundgaardet al.); Chem. Abstracts 110, 57664p (Alminger et al.); Chem. Abstracts 115, 64029s (Buur et al.); Chem. Abstracts 115, 189582y (Hansen et al.); Chem. Abstracts 117, 14347q (Bundgaard et al.); Chem. Abstracts 117, 55790x (Jensen et al.), и Chem. Abstracts 123, 17593b (Thomsen et al.). Насколько известно настоящим заявителям, в данное время не используют пролекарства ингибиторов протонного насоса. Однако отдельные патенты США описывают соединения, которые могут действовать как пролекарства некоторых ингибиторов протонного насоса. В частности, Патент США 4686230 (Rainer et al.) описывает производные пиридилметилсульфинилбензимидазолов, содержащие при одном из атомов азота бензимидазольного кольца группу, обозначенную как R5. Предполагается, что группа R5 будет отщепляться в условиях физиологической среды или под воздействием фермента с образованием соответствующего соединения со свободной связью N-H (см. раздел 3 Патента США 4686230). Патенты США 5021433 (Alminger et al.), 4045563 (Berntsson et al.) и 4965269 (Brändström et al.) также описывают пиридилметилсульфинилбензимидазолы, в которых один из атомов азота бензимидазольного фрагмента содержит заместитель, способный отщепляться в физиологических или ферментных условиях. Патент США 4045563 (Berntsson et al.) описывает производные N-алкоксикарбонилбензимидазола. В публикации Sih. et al. Journal of Medicinal Chemistry, 1991, vol.34, pp.1049-1062, дано описание N-ацилоксиалкильных, N-алкоксикарбонильных, N-аминоэтильных и N-алкоксиалкильных производных бензимидазолсульфоксида в качестве пролекарств ингибиторов протонного насоса. Согласно этой статье такие пролекарства проявляли лучшую химическую стабильность в твердом состоянии и в водных растворах, но обладали такой же или меньшей активностью, чем соответствующие исходные соединения, содержащие в имидазольной части свободную группу N-H. В указанной публикации не приводятся данные о длительности ингибирующей активности таких пролекарств. В патенте США 6093734 и публикации РСТ WO 00109498 (опубликованной 24 февраля 2000 г.) описаны пролекарства ингибиторов протонного насоса, содержащие замещенный арилсульфонильный фрагмент, присоединенный к одному из атомов азота бензимидазольного кольца ингибиторов протонного насоса, имеющие структуру, идентичную структуре лекарственных препаратов – ингибиторов протонного насоса, известных под названиями лансопразол, омепразол, пантопразол и рабепразол. В публикации РСТ WO 02/30920 описаны бензимидазольные соединения, заявленные как ингибирующие секрецию желудочной кислоты и обладающие действием против Н.pylori. В публикации РСТ WO 02/00166 описаны соединения, заявленные как высвобождающие окись азота (NO) производные ингибиторов протонного насоса с бензимидазольной структурой. Настоящее изобретение является еще одним шагом в данной области, поскольку представляет пролекарства ингибиторов протонного насоса, обладающие лучшей растворимостью в физиологических средах и лучшим проникновением в клетку, и представляет доказательства применимости пролекарств изобретения в качестве пролекарств ингибиторов протонного насоса с улучшенной эффективностью при терапии кислотозависимых заболеваний за счет пролонгирования присутствия ингибиторов протонного насоса в организме. СУЩНОСТЬ ИЗОБРЕТЕНИЯ Настоящее изобретение относится к соединениям Формулы 1, Формулы 2, Формулы 3 или Формулы 4
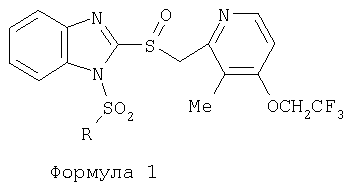
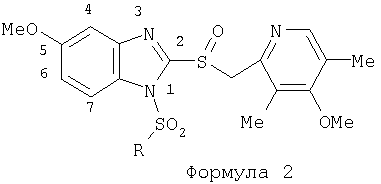
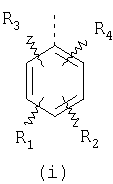
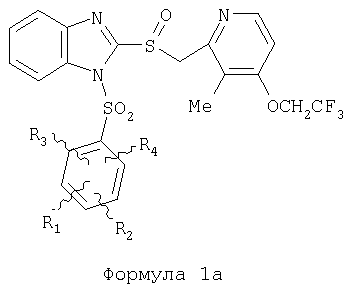
или изомерам соединений Формул 2 и 3, в которых группы ОСН3 и HF2CO, соответственно, находятся в 6 положении бензимидазольного кольца, где R представляет собой группу (i)
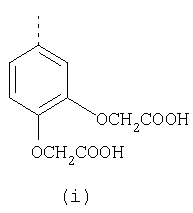
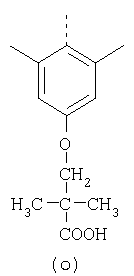
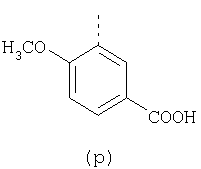
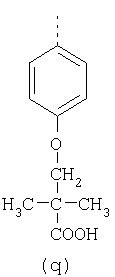
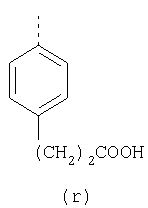
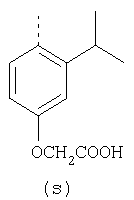
пунктирная линия означает связь, соединяющую группу R с группой SO2, R1 и R2 независимо означают Н, прямой или разветвленный насыщенный углеводород, содержащий не более 12 атомов углерода, включающую одну или две группы R5, и необязательно дополнительно включающую от одной до трех групп X, где Х независимо выбран из группы, состоящей из -О-, -NHCO-, -CONH-, или группа R5 без участия групп R1 или R2 напрямую присоединена к ароматическому кольцу; R3 и R4 независимо представляют собой Н, алкил, содержащий от одного до трех атомов углерода, или O-алкил, содержащий от одного до трех атомов углерода, R5 независимо означает Н или СООН; при условии, что по меньшей мере, одна из групп R1 и R2 не является Н и по меньшей мере, один заместитель R5 не является Н и не более двух групп R5 представляют собой СООН, так что соединение содержит, по меньшей мере, одну, но не более двух групп СООН; и к фармацевтически приемлемым солям указанных соединений. Предпочтительными примерами соединений Формул 1-4 или изомеров соединений Формул 2 и 3, в которых группы ОСН3 и HF2CO, соответственно, находятся в 6 положении бензимидазольного кольца, являются соединения, включающие, по меньшей мере, одну группу X, где, по меньшей мере, один из заместителей X означает О или CONH, и соединения, содержащее две группы R5, означающие СООН, или фармацевтически приемлемые соли указанных соединений. Также предпочтительными примерами соединений по изобретению являются соединения Формул 1-4 или указанные выше изомеры соединений Формул 2 и 3 содержащие, по меньшей мере, одну, но не более двух групп СООН, как описано выше, или их фармацевтически приемлимые соли, а также соединение по изобретению, в котором заместители R2, R3 и R4 представляют собой водород, a R1 представляет собой группу OCH2COOH, присоединенную к 4-положению фенильного кольца относительно сульфонильной группы, или его фармацевтически приемлемая соль. Особенно предпочтительными примерами соединений Формулы 1, Формулы 2, Формулы 3 или Формулы 4, а также изомеров соединений Формул 2 и 3, в которых группы ОСН3 и HF2CO, соответственно, находятся в 6 положении бензимидазольного кольца, являются соединения, где R означает группы, выбранные из Формул (а)-(s), пунктирная линия представляет собой связь, соединяющую группу R с группой SO2,
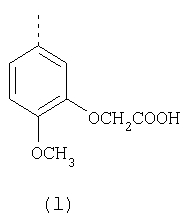
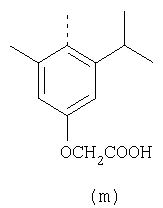
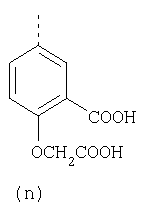
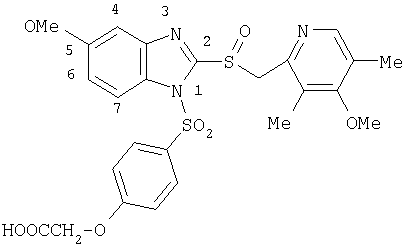
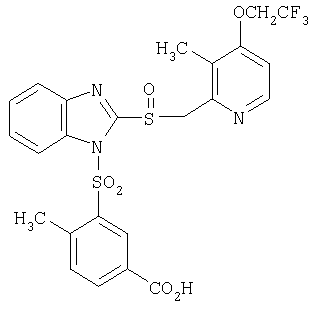
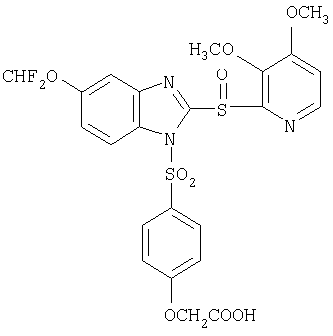
или фармацевтически приемлемые соли указанных соединений. Наиболее предпочтительными примерами соединений по изобретению являются – Соединение, имеющее формулу
или фармацевтически приемлемая соль указанного соединения. – Соединение, имеющее формулу
или его фармацевтически приемлемая соль. – Соединение, имеющее формулу
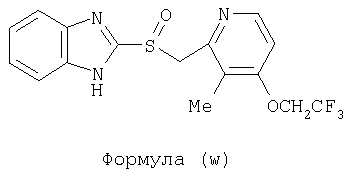
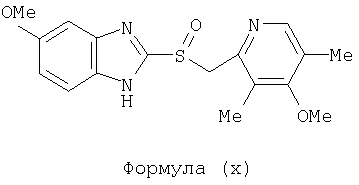
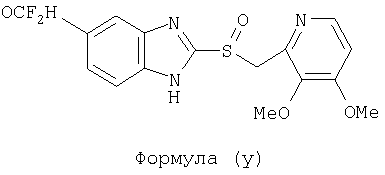
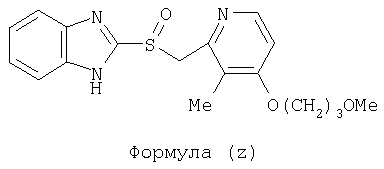
или фармацевтически приемлемая соль указанного соединения. Также изобретение относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении секреции желудочной кислоты и включающей фармацевтически приемлемый наполнитель и любое из соединений по изобретению. Также изобретение относится к указанной фармацевтической композиции, дополнительно включающей лекарственное средство – ингибитор протонного насоса, выбранный из группы, состоящей из формул (w), (x), (y) и (z)
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Определения Термин «алкил» означает и включает в себя все группы, известные как нормальный алкил, разветвленный алкил, циклоалкил, а также циклоалкилалкил. Фармацевтически приемлемая соль может быть приготовлена для любого соединения данного изобретения, содержащего функциональную группу, способную к образованию соли, как, например, карбоксильная группа, или основная (например, аминная) функциональные группы соединений настоящего изобретения. Фармацевтически приемлемой солью является любая соль, сохраняющая активность исходного соединения и не оказывающая никакого пагубного или неблагоприятного воздействия на субъекта, которому она введена, и в тех условиях, в которых она введена. Фармацевтически приемлемые соли могут быть производными органических или неорганических оснований. Соль может представлять собой моно- или поливалентный ион. Особый интерес представляют соли с неорганическими ионами, такими как ионы лития, натрия, калия, кальция и магния. Органические соли могут быть образованы с аминами, в частности, аммониевые соли, такие как моно-, ди- и триалкиламины или этаноламины. Соли также могут быть образованы с кофеином, трометамином и аналогичными молекулами. Соляная кислота или некоторые другие фармацевтически приемлемые кислоты могут образовывать соль с соединением изобретения, если оно содержит основную группу, такую как аминогруппа или пиридиновое кольцо. Некоторые из соединений настоящего изобретения могут содержать один или более хиральных центров и, следовательно, могут существовать в энантиомерных или диастереомерных формах. Объем настоящего изобретения охватывает все изомеры, смеси диастереомеров и рацемические смеси энантиомеров (оптических изомеров). Основные осуществления Химическая структура соединений изобретения представлена и описана общими терминами в разделе «Сущность изобретения» в связи с Формулами 1-4. Как можно видеть из этих формул, соединения изобретения представляют собой пиридилметилсульфинилбензимидазолы, замещенные по пиридиновому и бензимидазольному фрагментам аналогично хорошо известным лекарственным препаратам – ингибиторам протонного насоса – лансопразолу (Патент США 4628098), омепразолу (Патент США 4255431 и 5693818), пантопразолу (Патент США 4758579) и рабепразолу (Патент США 5045552). Описания Патентов США 4628098, 4255431, 5693818, 4758579 и 5045552 специально приведены здесь в качестве ссылки. Кроме того, как видно из Формул 1-4, в соединениях изобретения один из атомов азота бензимидазольного фрагмента замещен группой (обозначенной как R в Формулах 1-4), которая постепенно отщепляется в физиологических условиях и тем самым образует пиридилметилсульфинилбензимидазольное соединение, содержащее свободную функцию NH в бензимидазольном фрагменте. Соединения, образующиеся при таком отщеплении группы R, подвергаются далее перегруппировке при кислотном катализе и превращаются в тиофильные соединения, которые ингибируют фермент Н,К-АТФазу, участвующий в секреции желудочной кислоты. Таким образом, новые соединения настоящего изобретения, содержащие группу R, являются пролекарствами соединений – ингибиторов протонного насоса, которые также могут быть представлены Формулами 1-4, в которых, помимо всего, группа R может быть водородом. Для дополнительного описания пролекарств пиридилметилсульфинилбензимидазольного типа препаратов – ингибиторов протонного насоса, содержащих замещенный арилсульфонил или замещенный арилсульфонильный фрагмент, присоединенный к одному из бензимидазольных атомов азота, делается ссылка на Патент США 6093734, 6599167 и публикацию РСТ WO 00109498, описания которых специально приведены в данной заявке посредством ссылки. Как показано на примере растворимости и аналогичных данных, приведенных ниже, соединения настоящего изобретения представляют собой значительное усовершенствование по сравнению с известными соединениями, поскольку обладают большей растворимостью в физиологической среде и, следовательно, как можно предположить, будут более биодоступны. Что касается группы, обозначенной как R, в настоящее время наиболее предпочтительными являются соединения, в которых R представляет собой замещенный фенил (формула (i)). Обратимся теперь к переменным, обозначенным как R3 и R4 в формуле (i); в настоящее время предпочтительными будут соединения изобретения, в которых эти переменные независимо выбраны из Н, метила, этила, изопропила, метокси, этокси, CF3, СН3О и OCF3. Предпочтительно, чтобы, по меньшей мере, один из этих заместителей не был водородом, и предпочтительно, чтобы эти заместители находились при атоме или атомах углерода, которые расположены смежно с атомом углерода, связанным с сульфонильной группой. Во многих предпочтительных соединениях изобретения, в которых R означает фенил, эти атомы углерода находятся в орто-положении относительно сульфонильной группы. Специалисты в данной области согласятся, что заместители R3 и R4 могут оказывать электронное и стерическое влияние на скорость отрыва или гидролиза сульфонильной группы от бензимидазольного ядра и тем самым влиять на биодоступность пролекарств настоящего изобретения. Если, как в некоторых предпочтительных осуществлениях, заместители R3 и R4 находятся в орто- (или аналогичном) положении относительно сульфонильной группы, то наличие или отсутствие у таких заместителей стерического объема будет особенно сильно влиять на скорость гидролиза сульфонильной группы от бензимидазольного фрагмента. В некоторых из предпочтительных в настоящее время соединений изобретения R означает фенил (формула (i)), а оба заместителя R3 и R4 являются метилами и занимают в фенильном кольце орто- и орто’-положения. Согласно другим примерам предпочтительных соединений изобретения, R означает фенил, одна из групп R3 и R4 является Н, а другая – изопропилом. Орто- и орто’-метильные и орто-изопропильные заместители замедляют скорость гидролиза по сравнению с соединением, обладающим аналогичной во всех отношениях структурой, но в котором отсутствуют такие орто- и орто’-заместители. Что касается переменных R1 и R2, важной особенностью соединений изобретения является то, что одна или обе эта группы содержат карбоксильную функцию. Назначение карбоксильной группы, входящей в состав этих переменных в соединениях изобретения, заключается в увеличении растворимости соединений в водных физиологических средах при физиологическом значении рН по сравнению с известными пролекарствами пиридилметилсульфинилбензимидазольных ингибиторов протонного насоса. Вообще говоря, желательно, чтобы величина рКа фрагмента карбоновой кислоты (или тетразола) соединений изобретения лежала в интервале от 2 до 6, еще более предпочтительно, чтобы величина рКа была в интервале от 2 до 4, и еще более предпочтительно, чтобы рКа равнялась приблизительно 3. Один или два фрагмента карбоновой кислоты, присоединенных к заместителям R1 и/или R2, обеспечивают требуемую величину рКа и, следовательно, требуемую растворимость для соединений изобретения. Специалисты в данной области согласятся, что на кислотность фрагмента карбоновой кислоты влияют электронные эффекты других групп, находящихся по соседству, особенно если карбоксильная группа присоединена непосредственно к ароматическому ядру. Один или два фрагмента карбоновой кислоты могут быть заменены тетразольным кольцом, и, кроме того, физиологически приемлемые соли соединений настоящего изобретения могут иметь такую же или даже лучшую растворимость в физиологических средах, чем соответствующие свободные кислоты. Требование настоящего изобретения, чтобы одна или обе группы R1 и R2 содержали, по меньшей мере, один, но, в общей сложности, не более двух фрагментов карбоновых кислот (или их фармацевтически приемлемых солей), может быть удовлетворено целым рядом групп R1 и R2 или их комбинацией. Например, одна из этих двух групп может представлять собой Н, при этом другая группа будет содержать одну или две карбоксильные группы. Или же каждая из групп R1 и R2 может содержать по одной карбоксильной группе. Карбоксильные группы, обозначенные R5 в формуле (i), могут быть присоединены непосредственно к ароматическим или гетероароматическим ядрам, либо одна или обе группы R1 и R2 могут содержать углеводородный “скелет” или “каркас”, который присоединен непосредственно к ароматическим ядрам. Или же углеводородный “скелет” или “каркас” может быть сам присоединен к ароматическим или гетероароматически ядрам через промежуточную эфирную, тиоэфирную, аминную, сложноэфирную или амидную группу. Эти группы обозначены переменной Х при описании соединений в разделе «Сущность изобретения» данной заявки. Кроме того, эфирная, тиоэфирная, аминная, сложноэфирная или амидная группа или группы могут быть включены в одно или несколько мест углеводородного “скелета” или “каркаса”, при этом карбоксильная (или тетразольная) группа или группы присоединены к R1 и/или R2 или “содержатся” в R1 и/или R2, которые сами содержат эфирные, тиоэфирные, амино, сложноэфирные или амидные звенья. Любая комбинация таких звеньев может быть приемлема для получения соединения в объеме изобретения. Кроме того, “скелет” или “каркас” сам по себе может быть прямой или разветвленной цепью, и ответвление может осуществляться за счет углерод-углеродных связей или связей углерод-группа X. Понимая, что группы R1 и R2 могут быть выбраны независимо и таким образом, чтобы, по меньшей мере, одна, но не более двух, карбоксильных (или тетразольных) групп присутствовала в соединении изобретения, предпочтительными значениями групп R1 и R2 являются следующие: (1) Н, (2)(CH2)mR5. (3)(CH2)mCH(R5)(CH2)n[CH(R5)]q(CH2)oCH3, (4) X(CH2)mR5, (5) X(CH2)mCH(R5)[(CH2)nCH(R5)]q(CH2)oCH3, (6) (CH2)mX(CH2)pCH(R5)[(CH2)nCH(R5)]q(CH2)oCH3, (7) (CH2)mCH(R5)(CH2)nX[(CH2)pCH(R5)]q(CH2)oCH3, (8) (СН2)mХ[(СН2)рСН(R5)]q(СН2)nСН(R5)(СН2)оСН3, (9) (CH2)m[CH(R5)(CH2)n]qХ(СН2)рСН(R5)(СН2)оСН3, (10) Х(СН2)mХСН(R5)[(СН2)nСН(R5)]q(СН2)оСН3, (11) (CH2)m[CH(CH2)s(R5)](CH2)n[CH(CH2)s(R5)]q(CH2)oCH3, (12) X[CH(CH2)s(R5)]mR5, (13) Х(СН2)m[CH(CH2)s(R5)][CH(CH2)s(R5)]q(CH2)oCH3, (14) (CH2)mX(CH2)p[CH(CH2)s(R5)][CH(CH2)s(R5)]q(CH2)oCH3, (15) (CH2)m[CH(CH2)s(R5)](CH2)nX[CH(CH2)s(R5)]q(CH2)oCH3, (16) (CH2)mX[(CH2)p[CH(CH2)s(R5)]]q(CH2)n[CH(CH2)s(R5)](CH2)oCH3, (17) (CH2)m[[CH(CH2)s(R5)](CH2)n]qX(CH2)p[CH(CH2)s(R5)](CH2)oCH3, (18) X(CH2)mX[CH(CH2)s(R5)][(CH2)n[CH(CH2)s(R5)]]q(CH2)oCH3 (19) Х(СН2)mС(СН3)2R5 (20) X(CH2)mX(CH2)nR5, где m означает целое число от 0 до 6; n означает целое число от 0 до 5; q означает целое число – 0 или 1, о означает целое число от 0 до 5, s означает целое число от 0 до 5, сумма чисел m, n, q, о и s не превышает 12 и где остальные переменные имеют значения, определенные выше для Формул 1-4, с учетом изложенного для Формул 1-4, а также при условии, что группа R5 не связана напрямую с О, NHCO или CONH. Используя замещенную фенилсульфонильную группу и СООН (для R5) в качестве предпочтительных фрагментов в соединениях настоящего изобретения, далее представлены структурные формулы, изображающие, не ограничиваясь перечнем, а лишь иллюстрируя в качестве примеров, предпочтительные группы R1 или R2, перечисленные выше (2), (3), (4), (5), (10), (11), (12), (19) и (20).
Наиболее предпочтительные группы R, входящие в соединения изобретения, изображены ниже. Для облегчения ссылки на них эти предпочтительные группы обозначены R с соответствующими индексами, как, например, “R11, R12 и т.д.”
В настоящее время наиболее предпочтительными являются соединения изобретения, в которых одна из групп R11-R34 присоединена к бензимидазольному фрагменту, изображенному в Формулах 1-4. Структуры частных примеров наиболее предпочтительных соединений изобретения представлены ниже в экспериментальной части вместе с описанием получения конкретных примеров. Методики синтеза Вообще говоря, соединения изобретения могут быть получены основным гидролизом замещенных или незамещенных фенилсульфонилэтиловых эфиров моно- или дикарбоновых кислот, которые составляют соединения изобретения. Общий способ предпочтительного в настоящее время способа синтеза представлен на Реакционной схеме 1, где для простоты иллюстрации пиридилметилсульфинилбензимидазольные фрагменты, соответствующие Формулам 1-4, изображены в общем виде Формулой 5, арильные, гетероарильные или алкильные группы Формулы (i) представлены фенильной группой, и показаны только одна группа R1 (как определено выше) и только одна монокарбоновая кислота. Однако специалист в данной области легко поймет, что описанная здесь методика синтеза может быть использована для получения всех соединений в объеме изобретения, но с такими модификациями, которые очевидны для специалиста с точки зрения настоящего раскрытия. Обратимся теперь непосредственно к Формуле 5на Реакционной схеме 1, данная формула изображает пиридилметилсульфинилбензимидазолы, известные как лансопразол, омепразол, пантопразол и рабепразол. Переменная Z означает Н или такой заместитель, который может присутствовать в бензимидазольной части любого из этих соединений, а Z
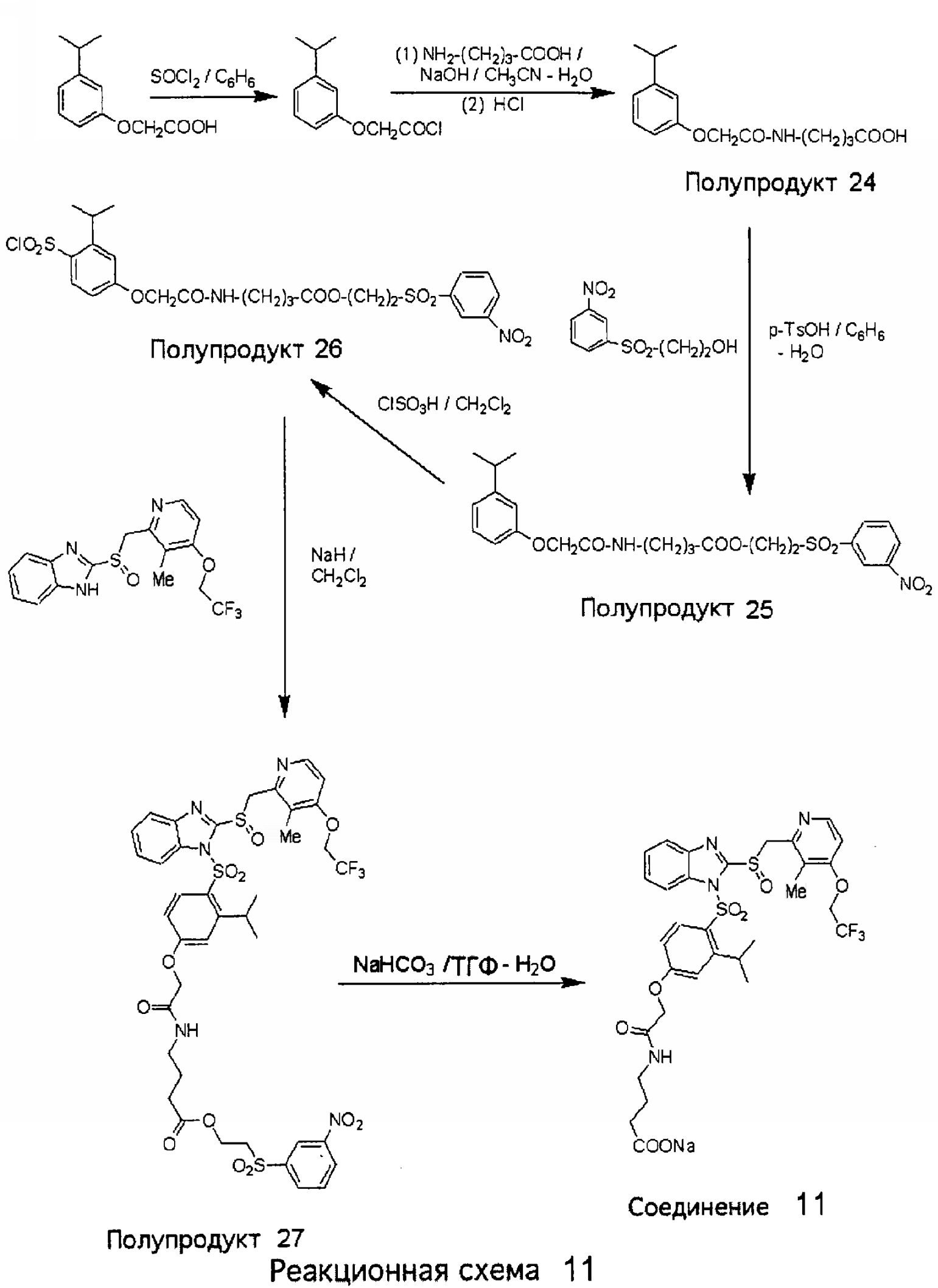
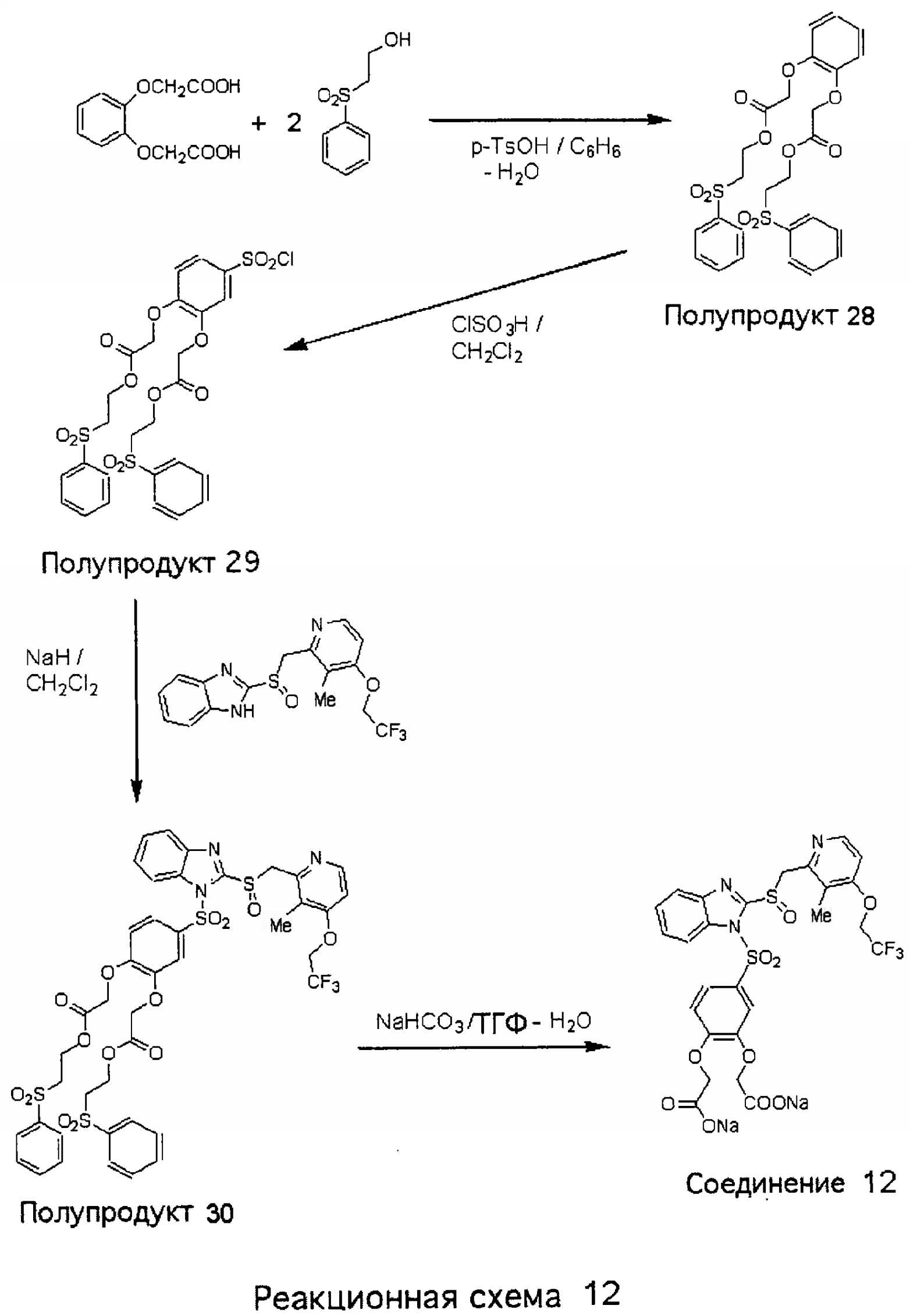
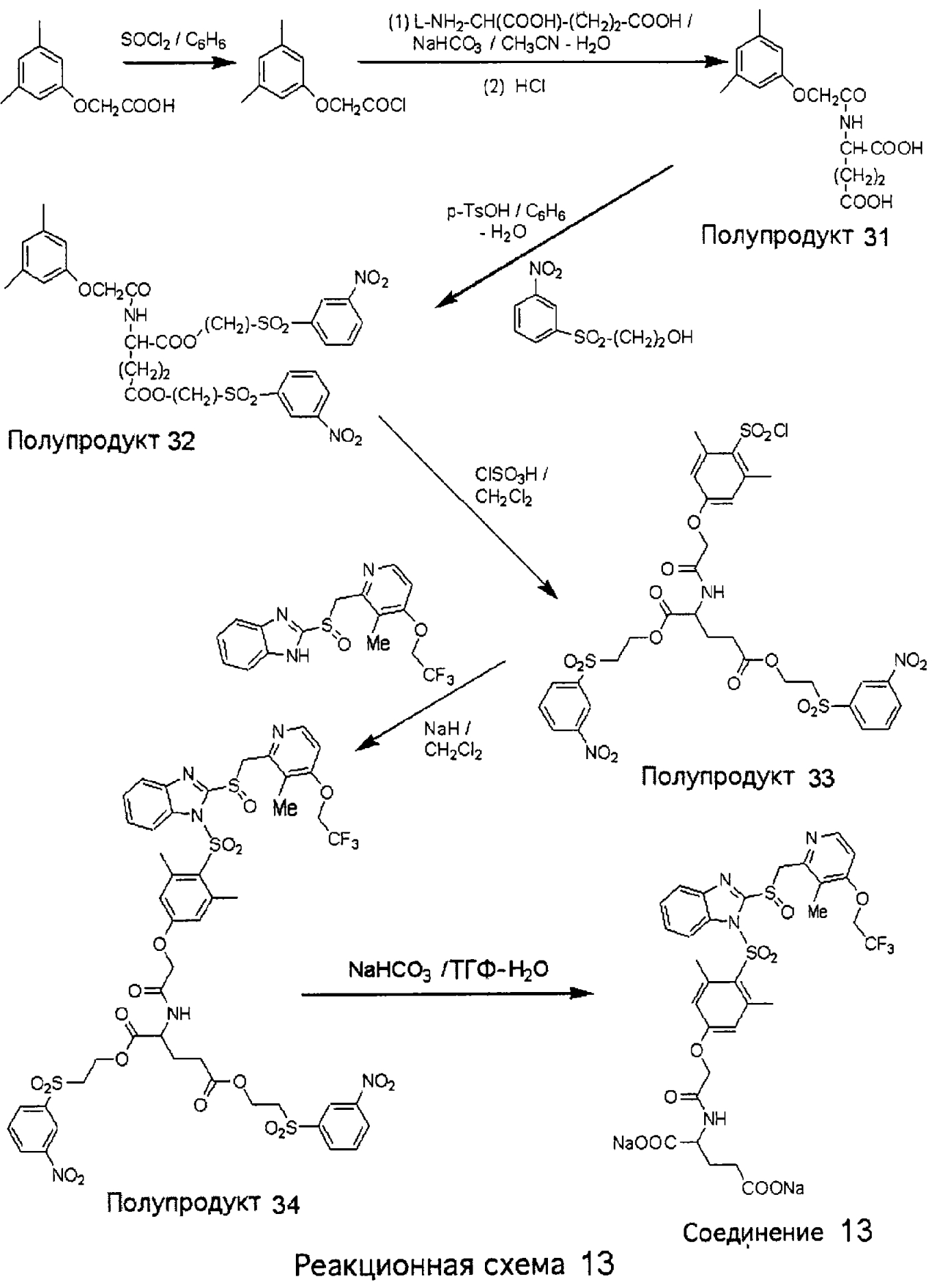
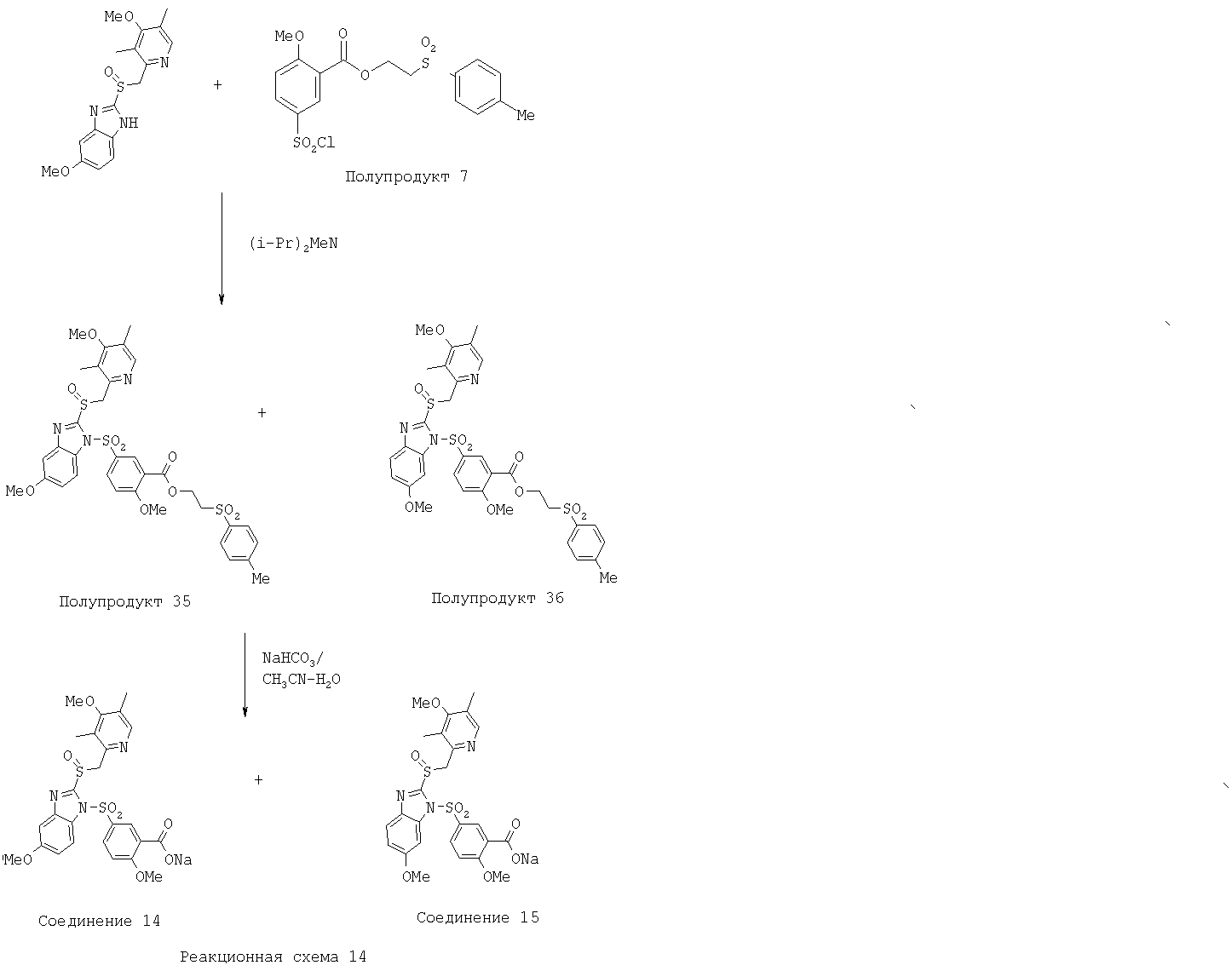
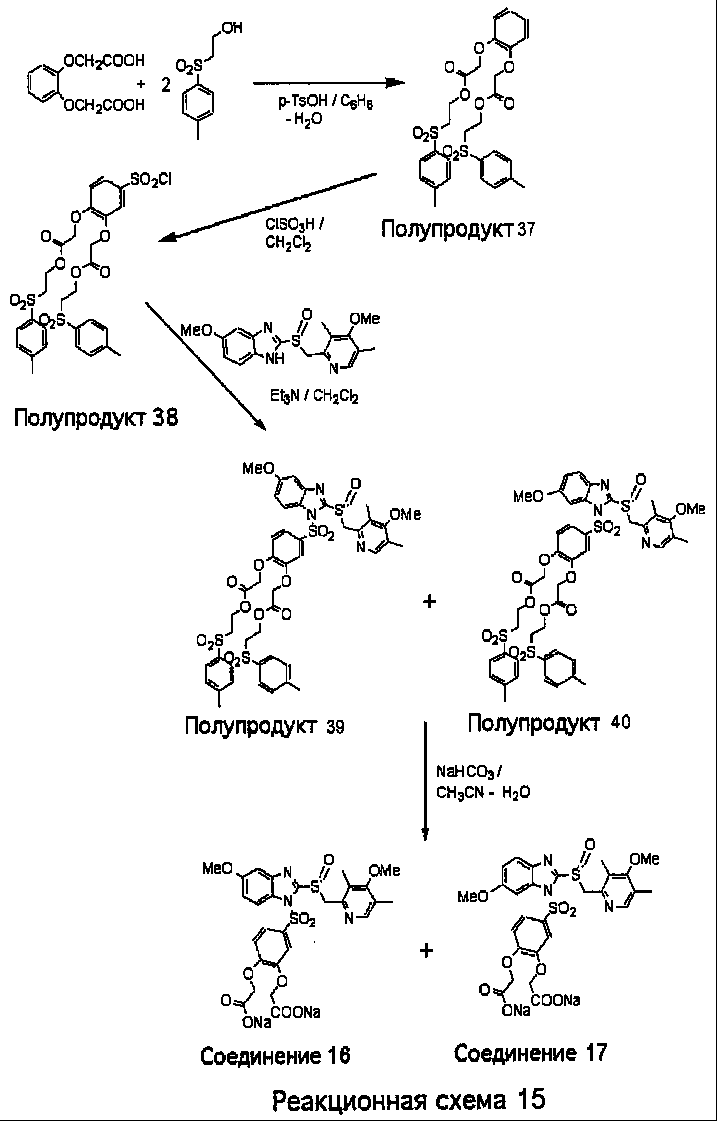
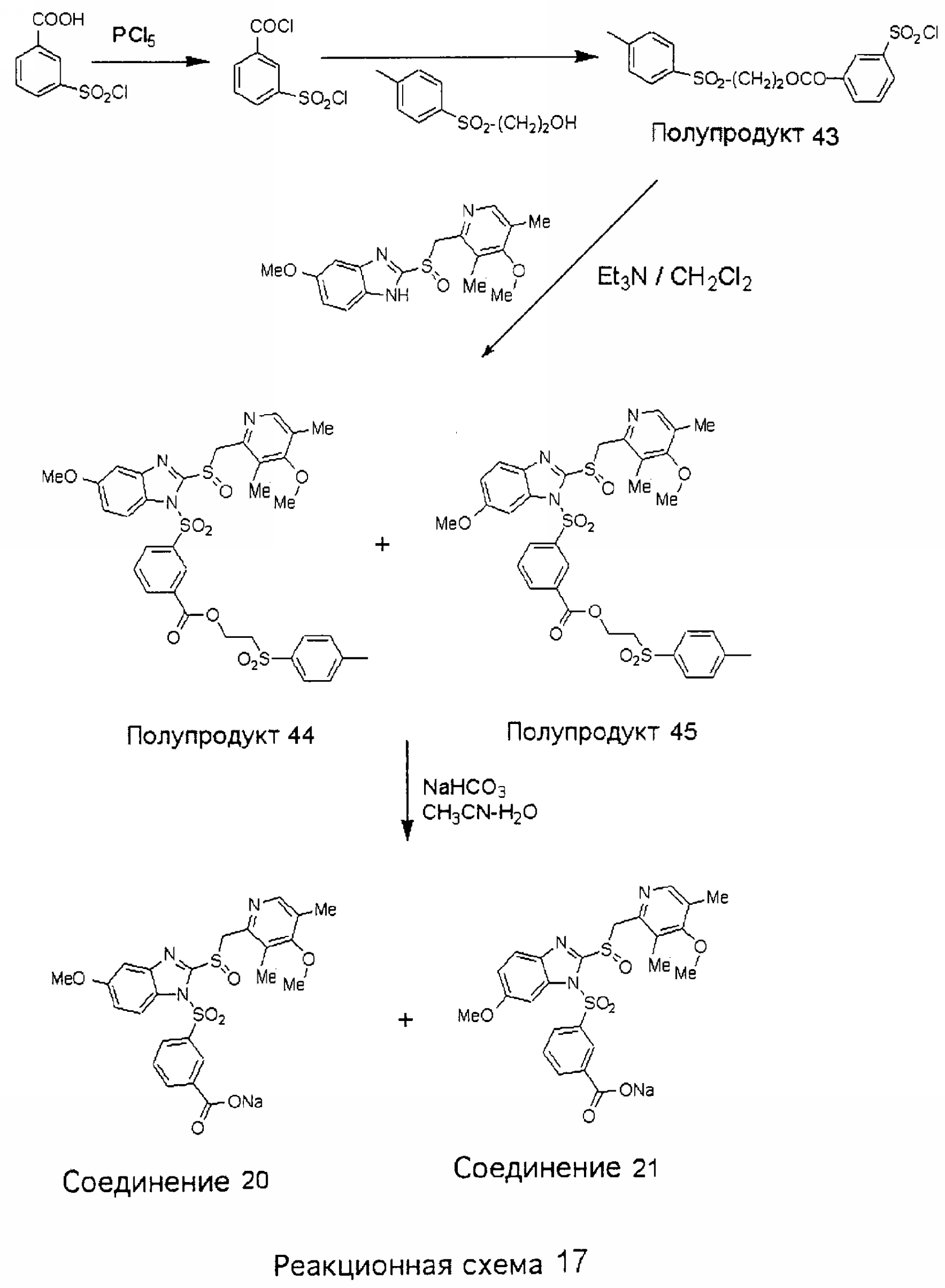
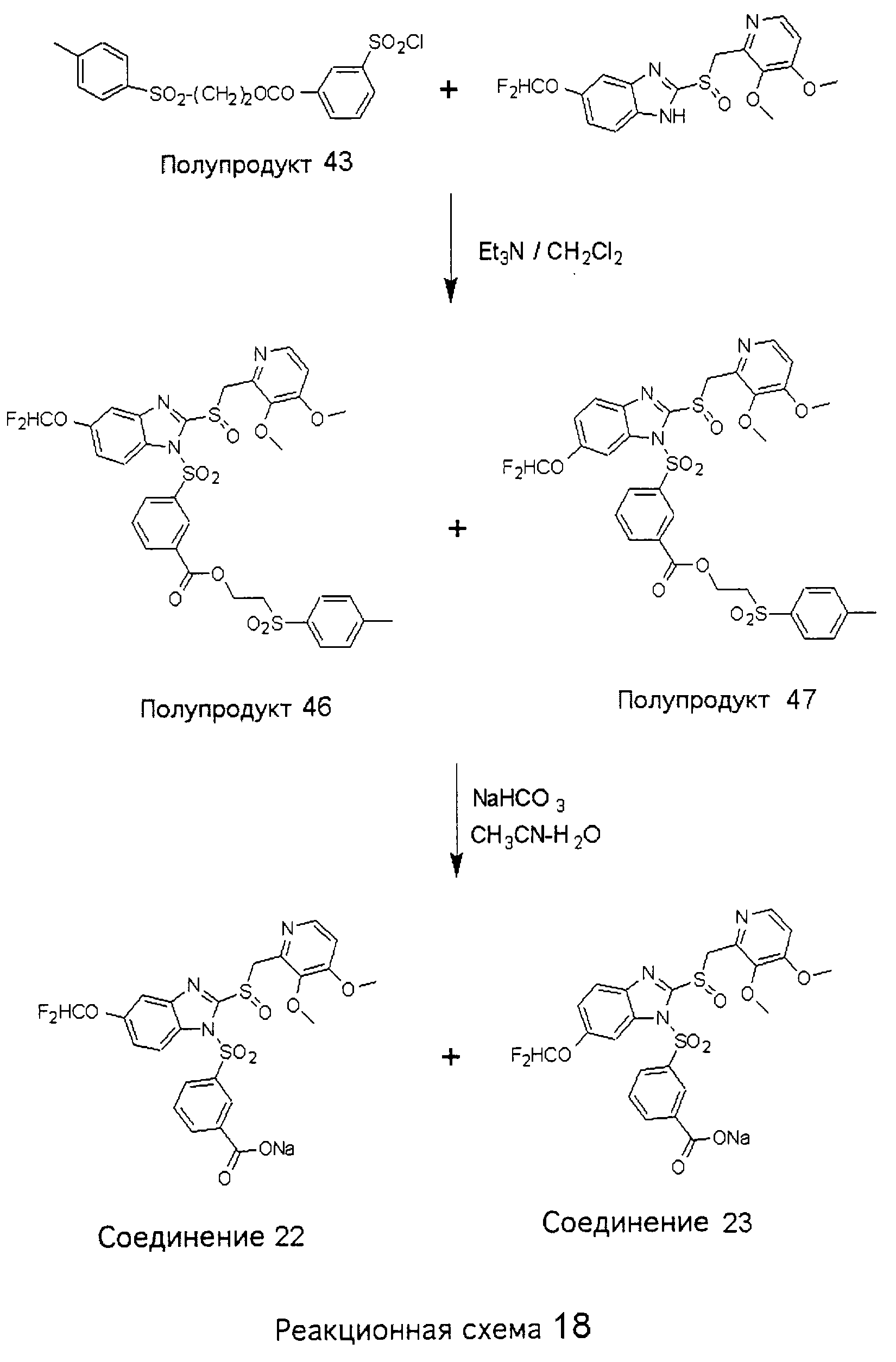
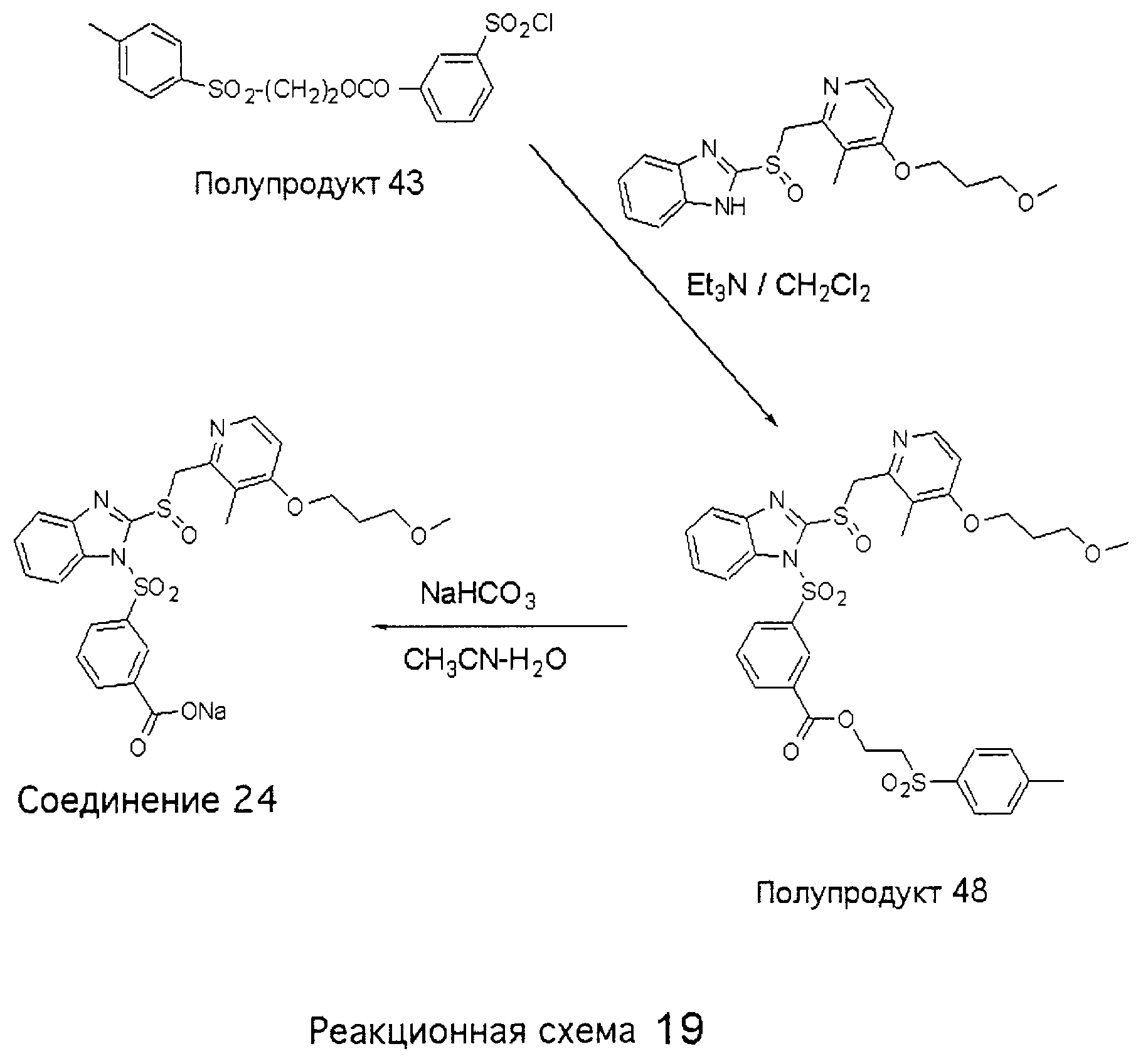
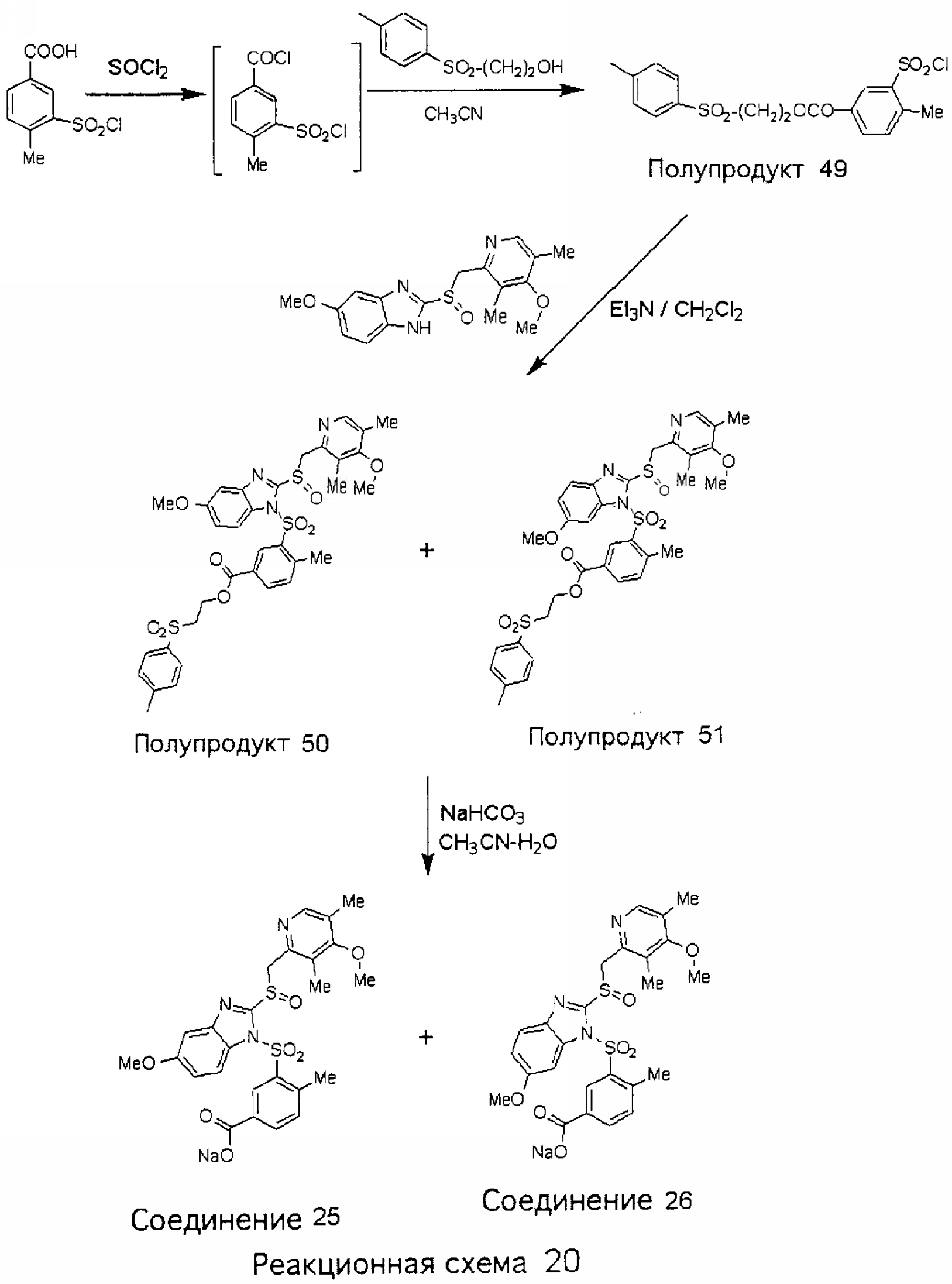
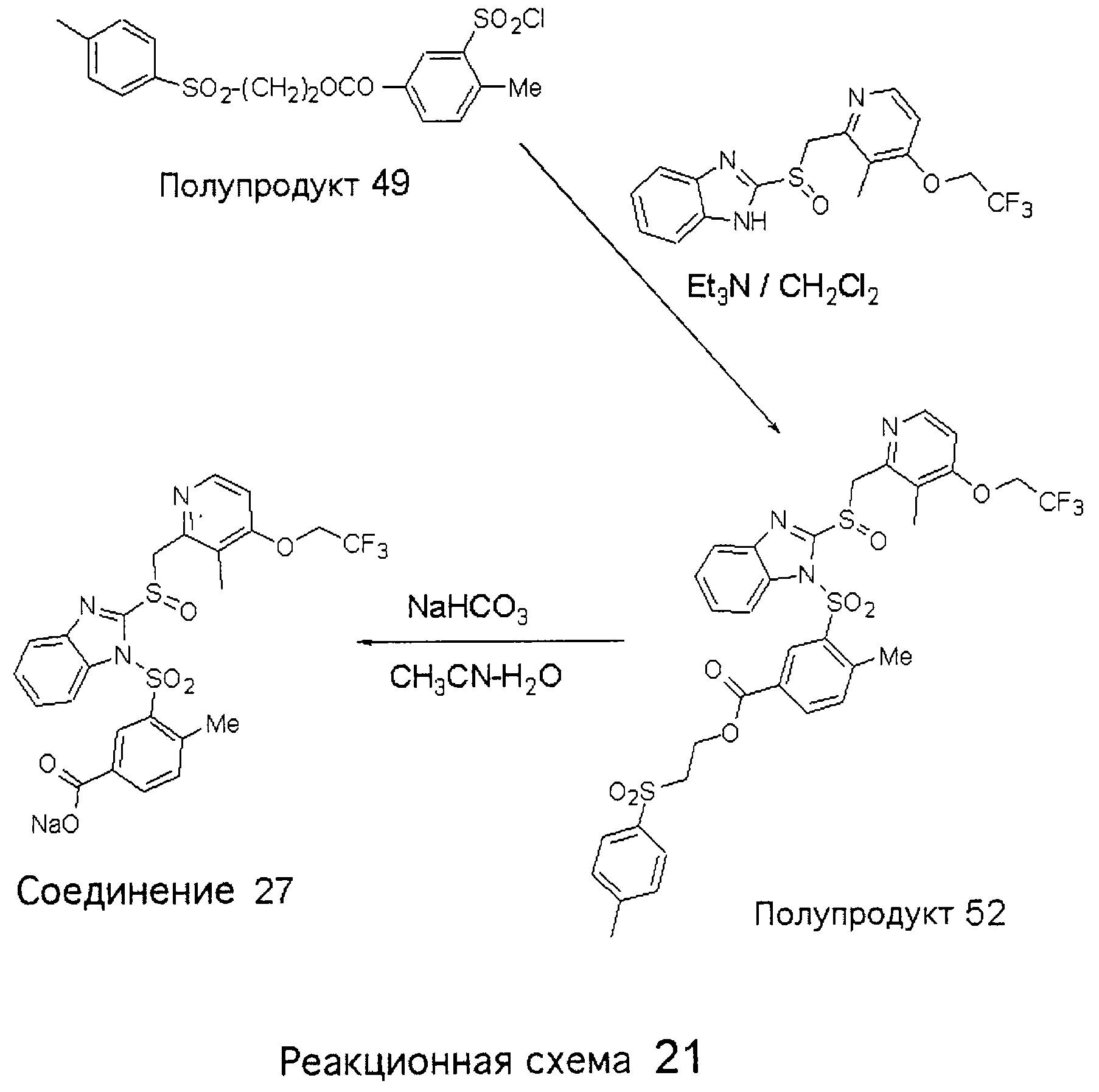
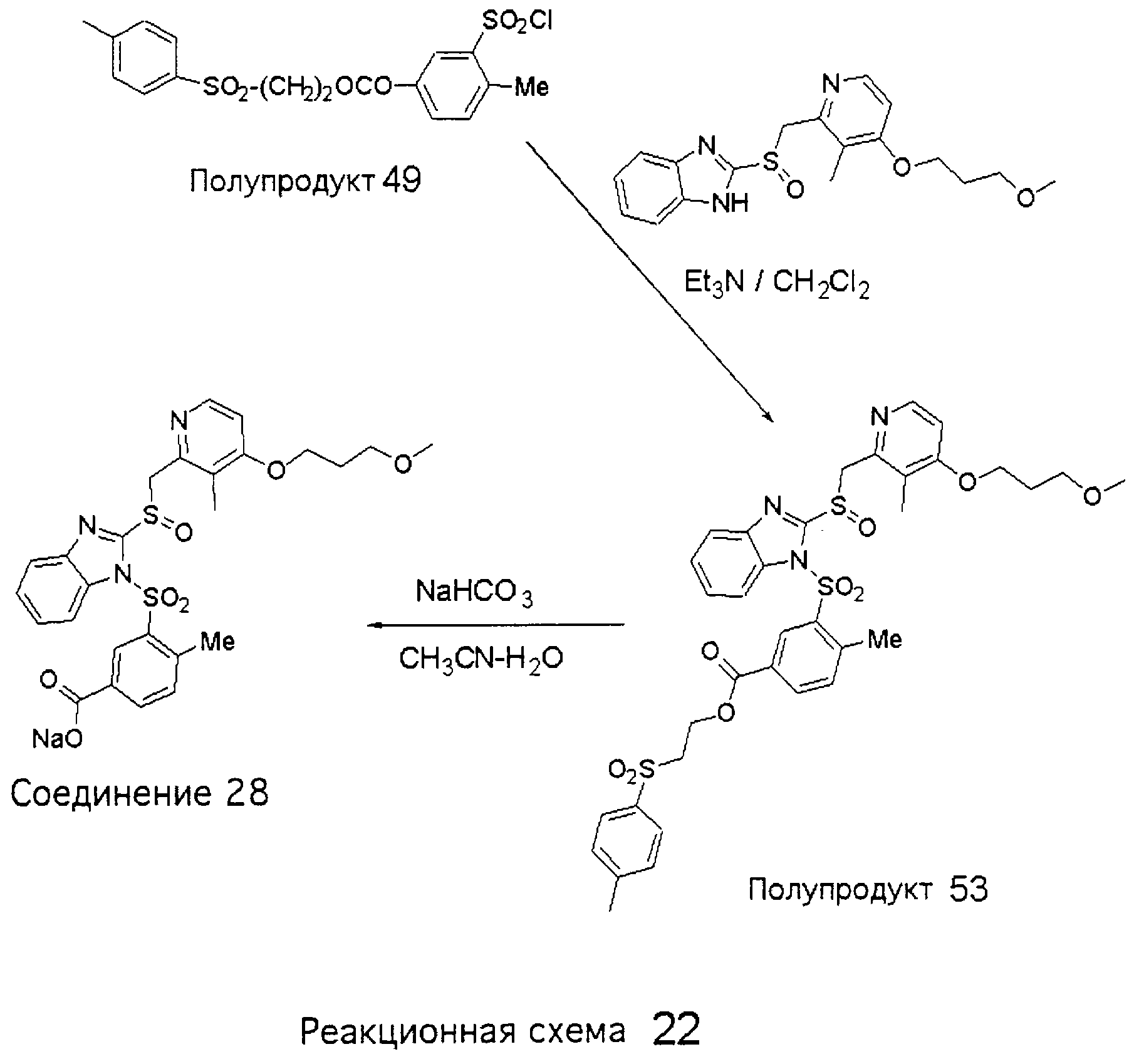
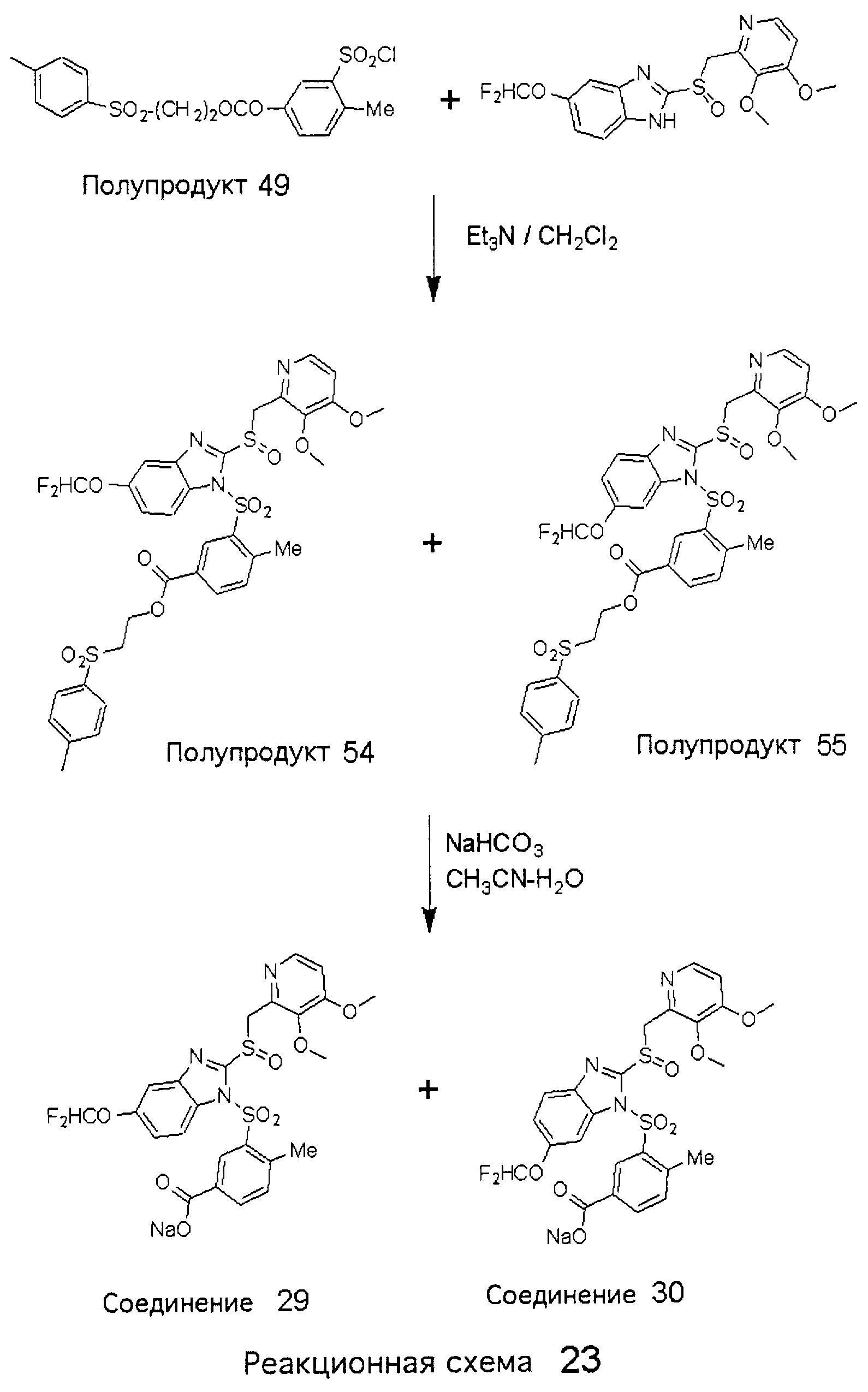
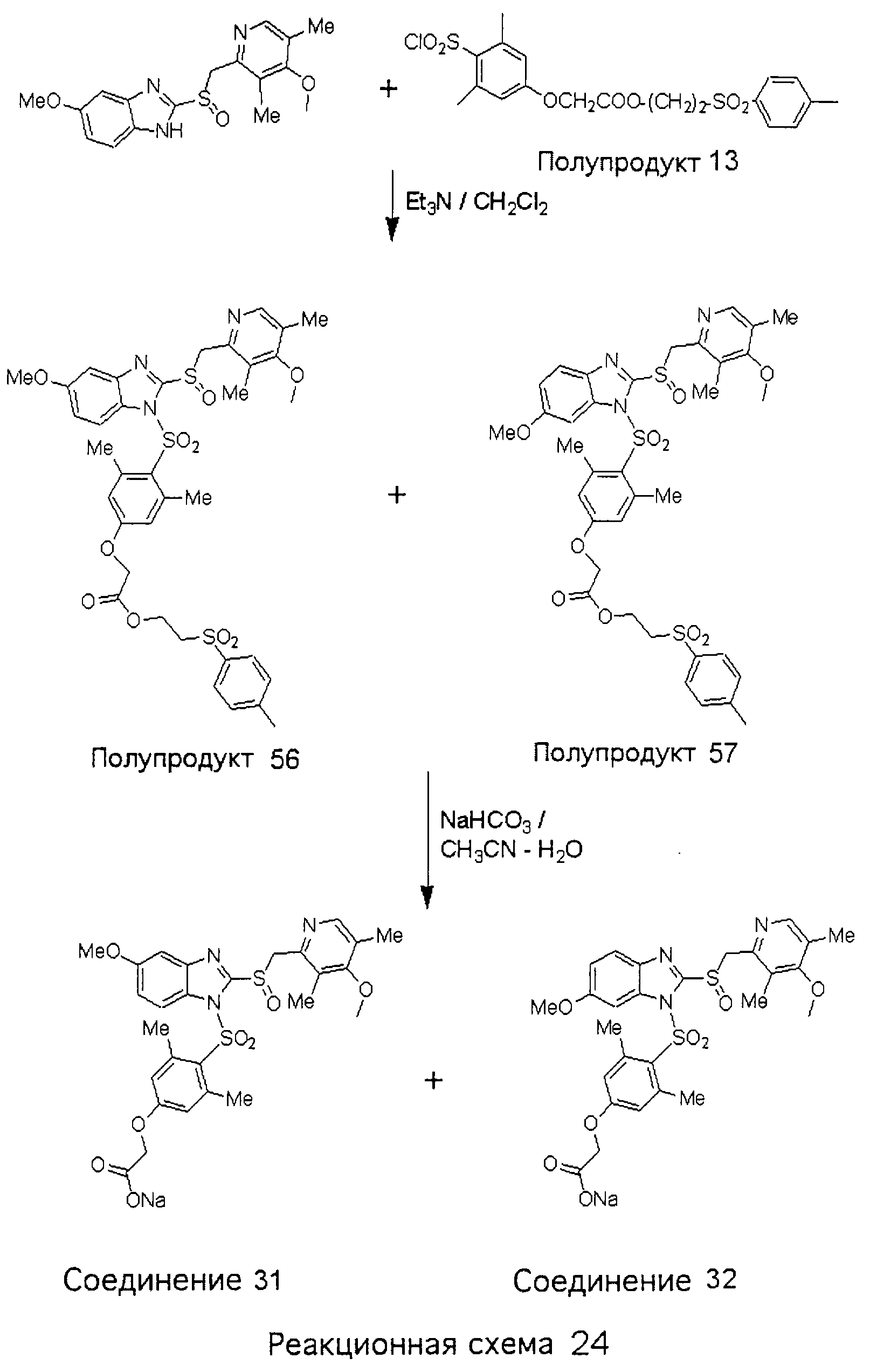
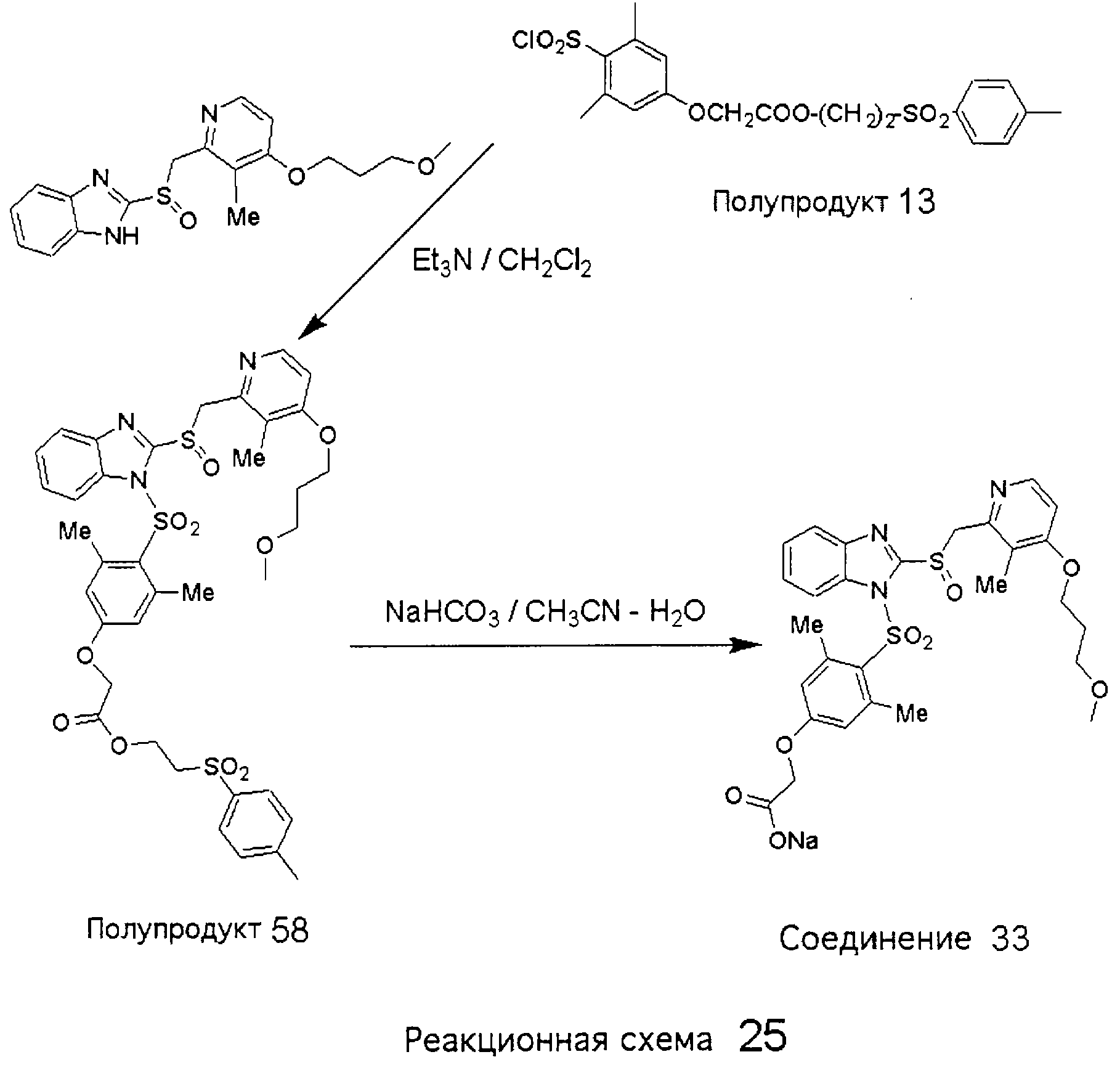
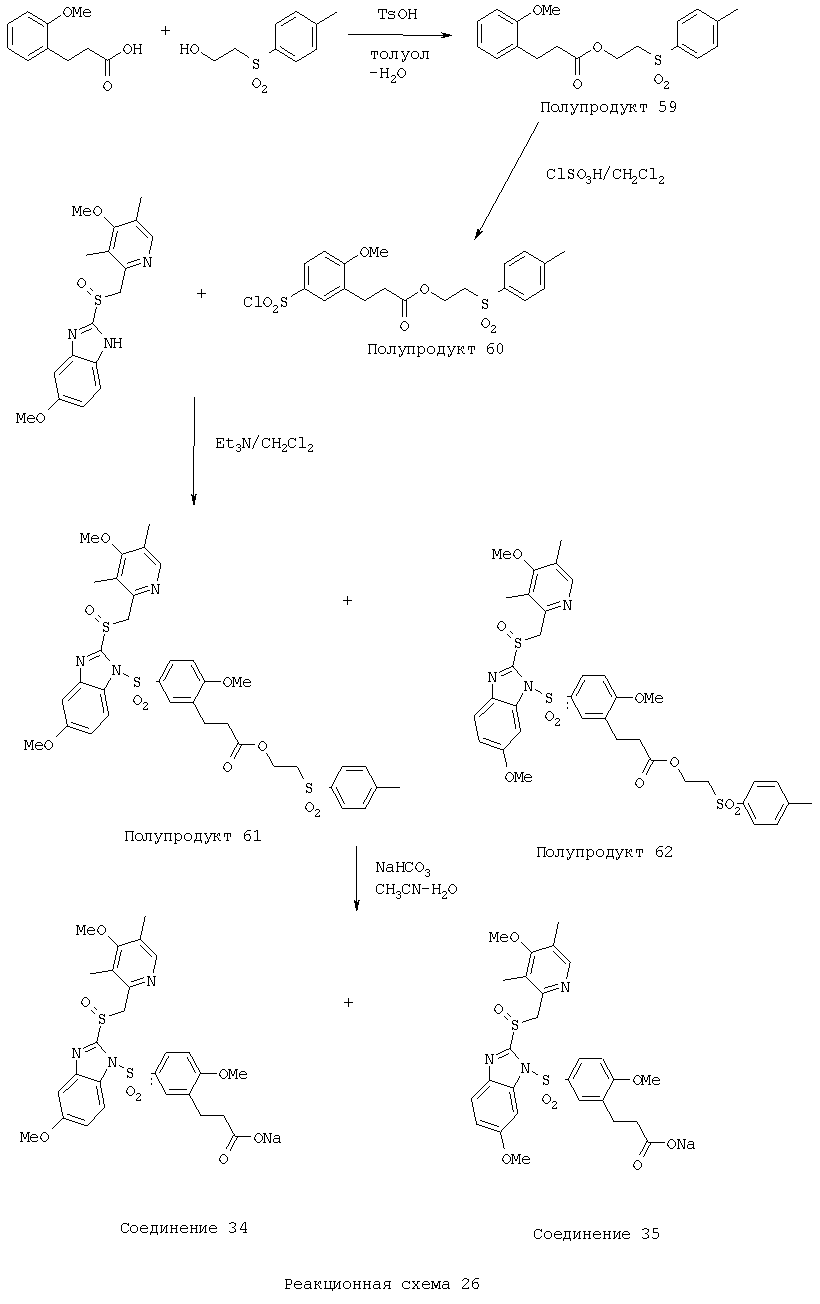
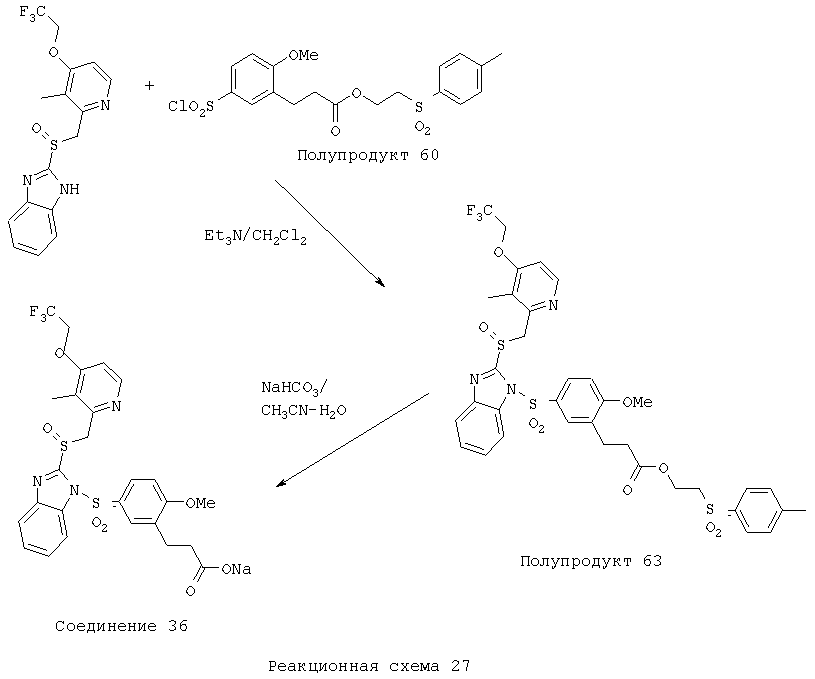
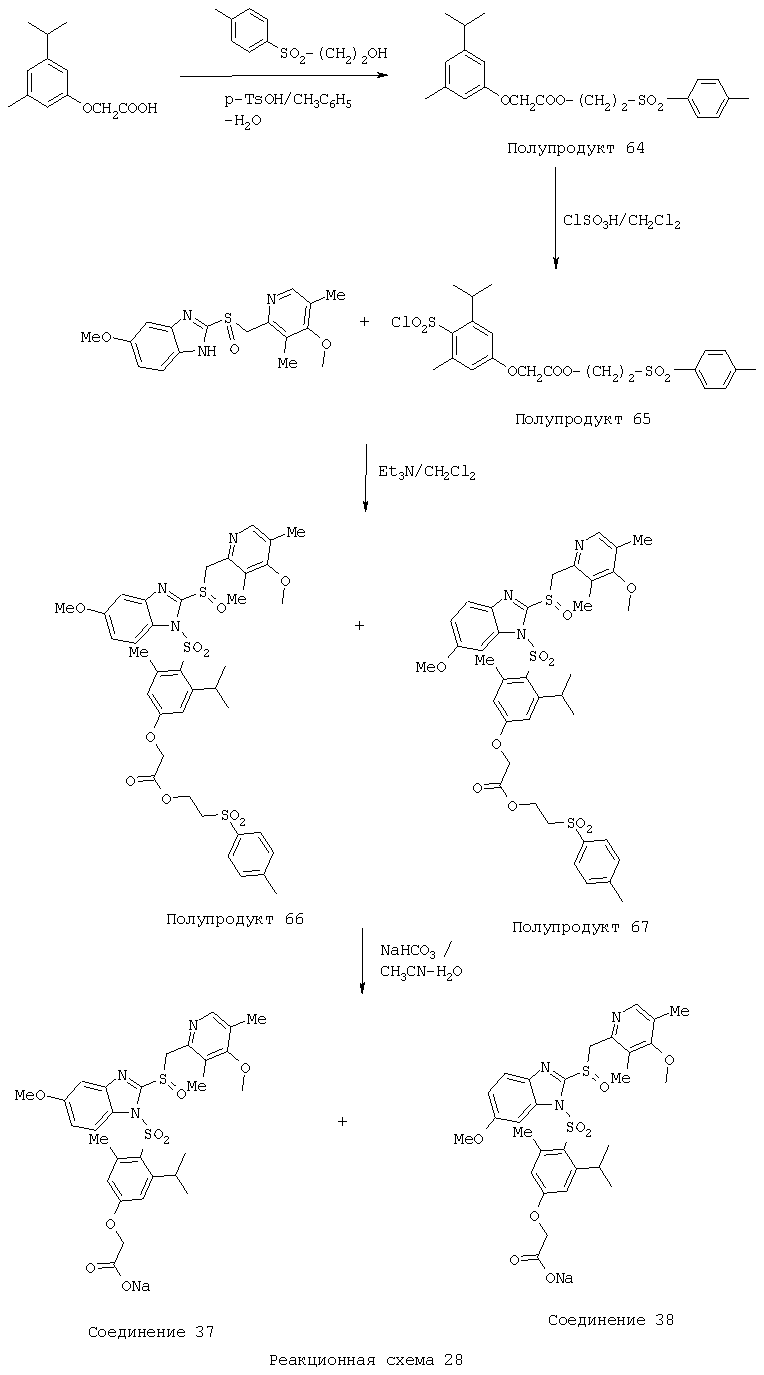
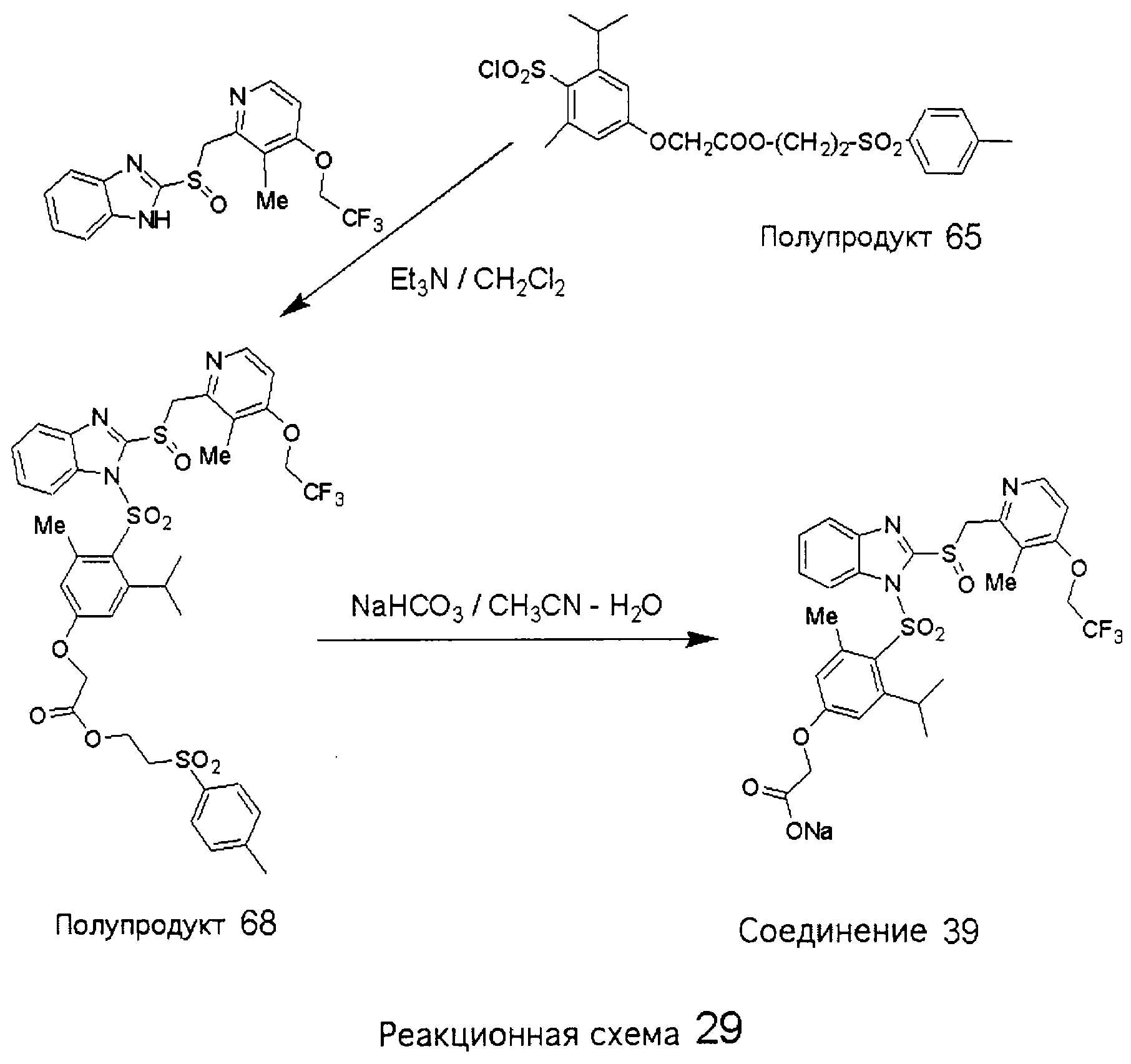
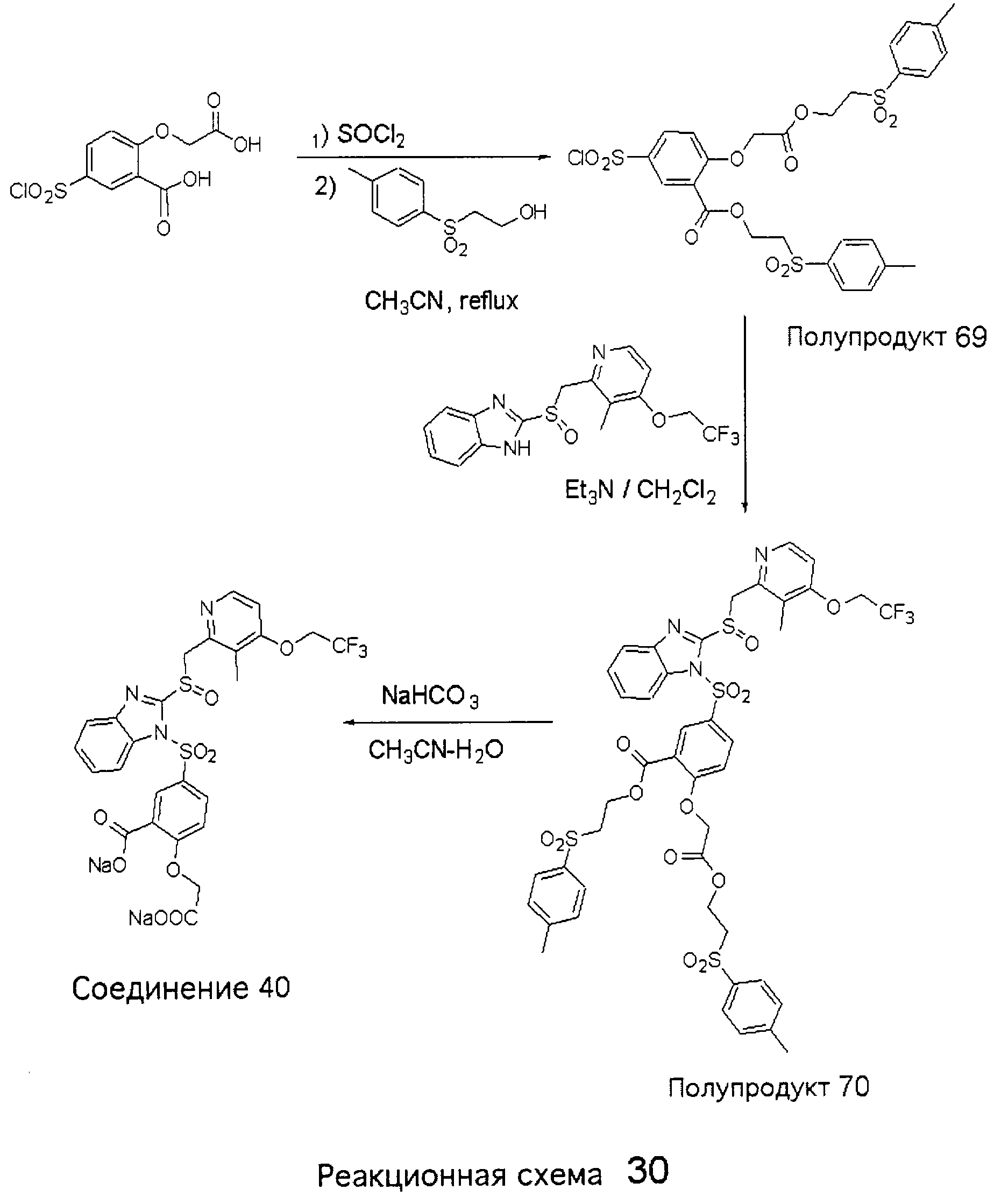
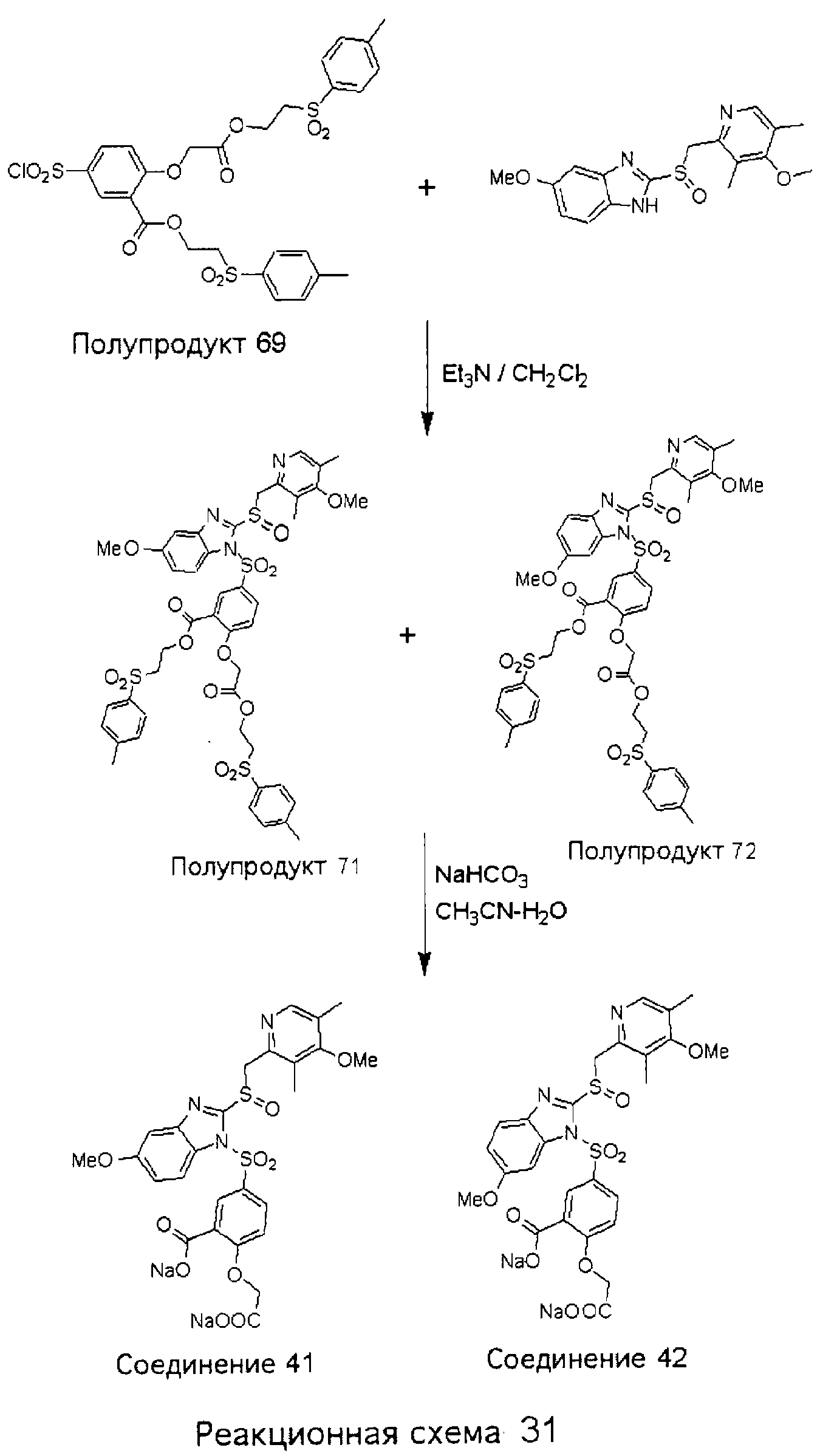
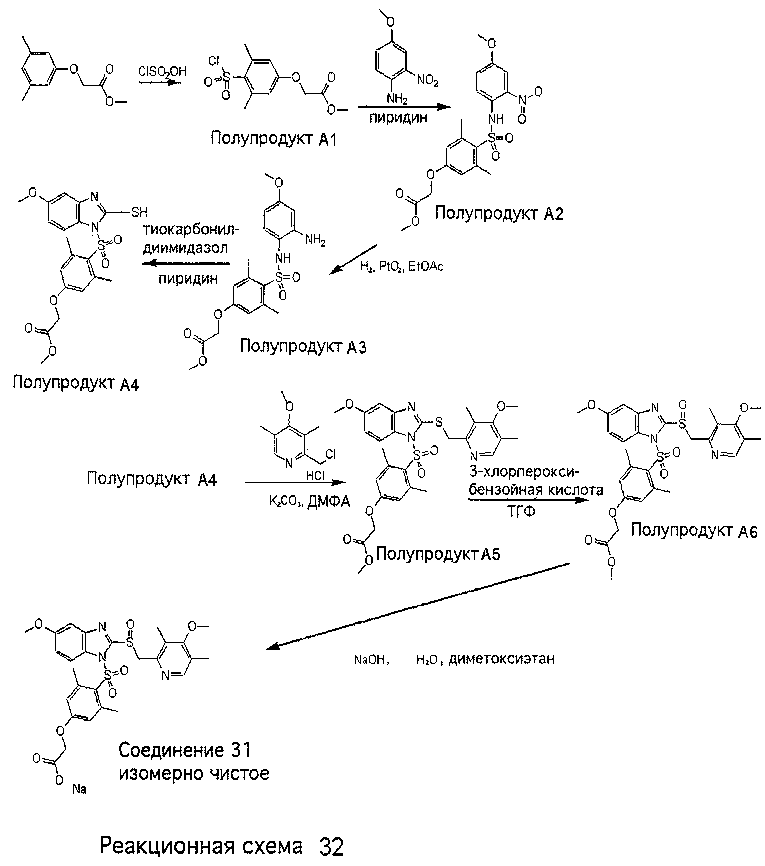
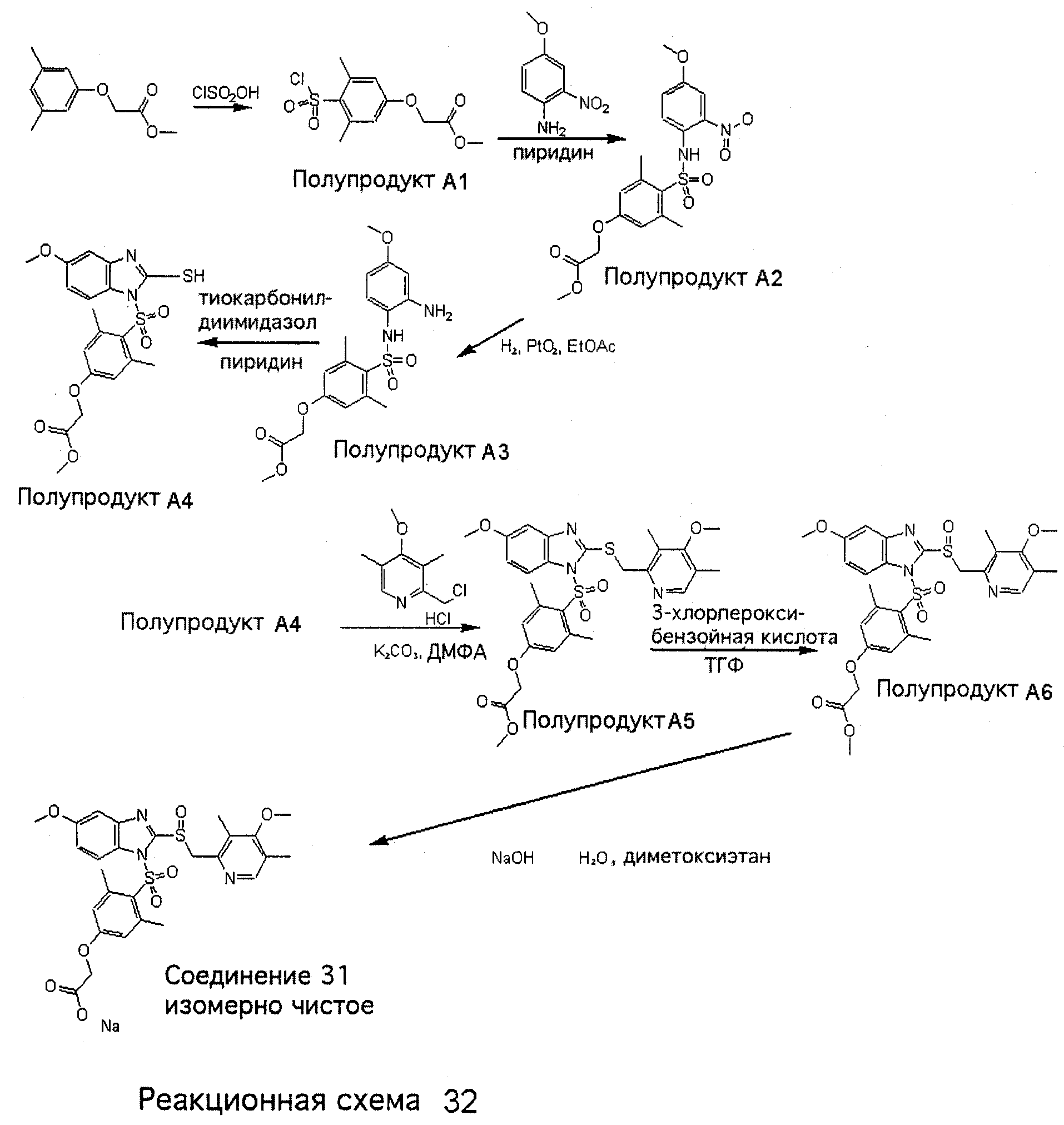
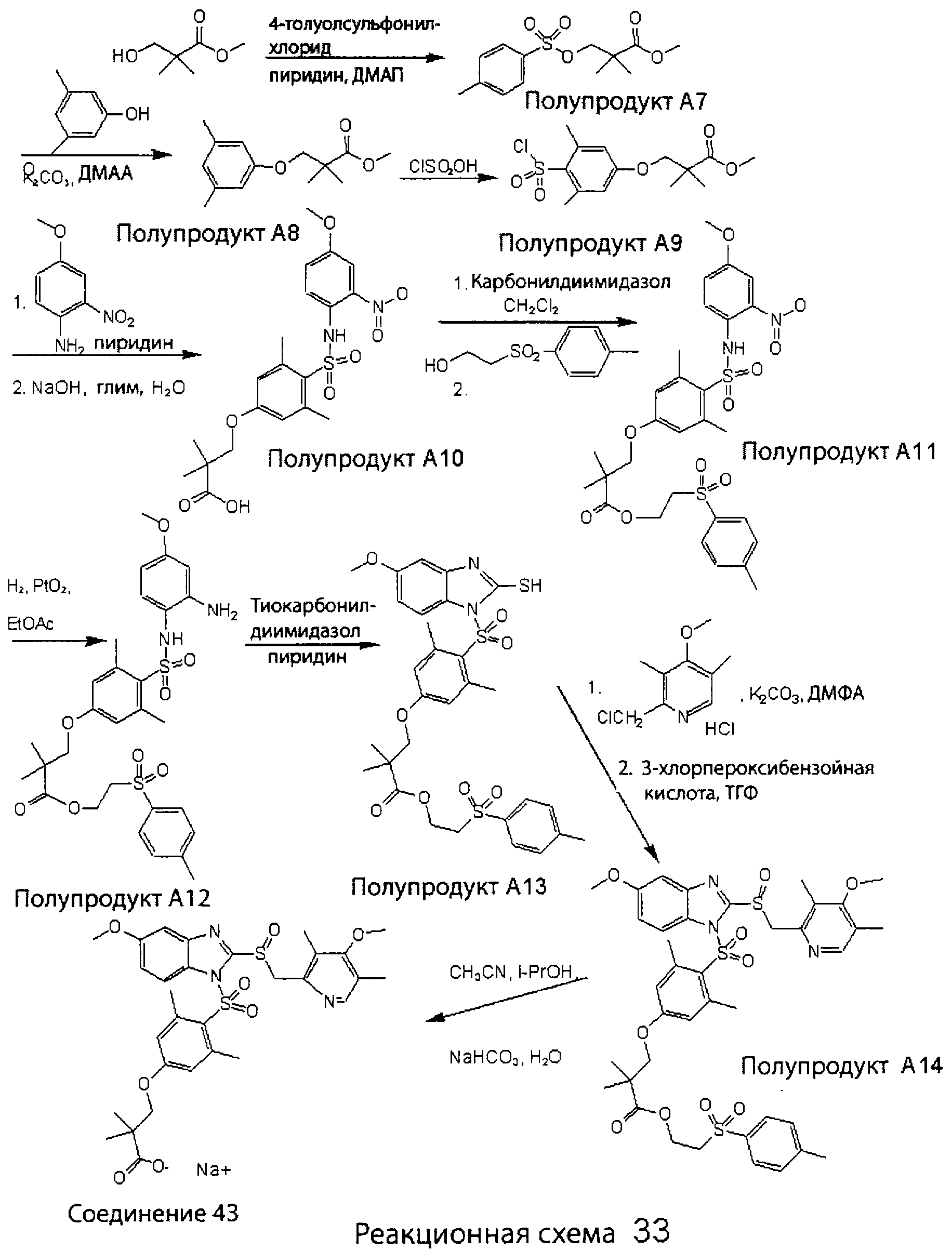
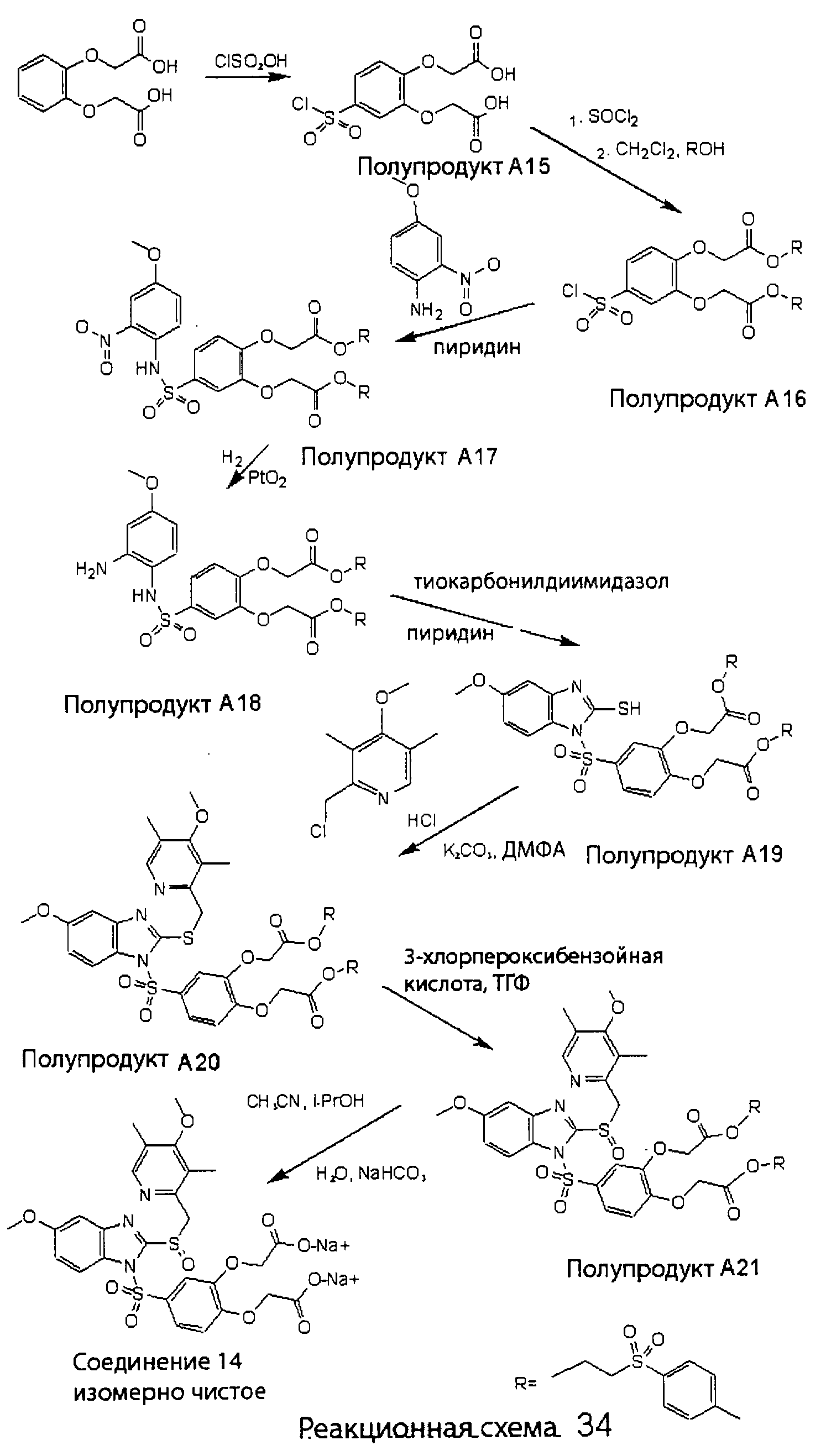
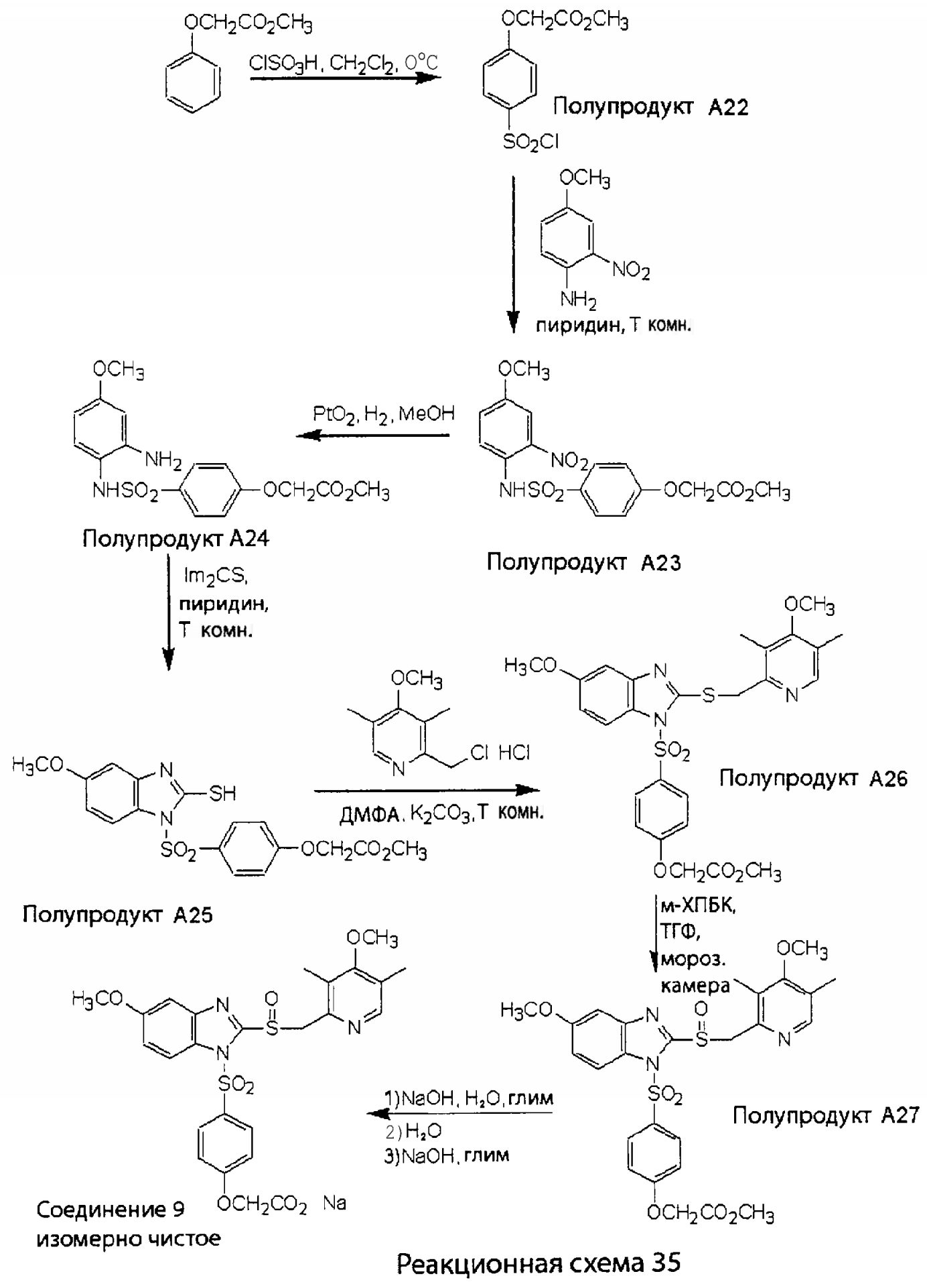
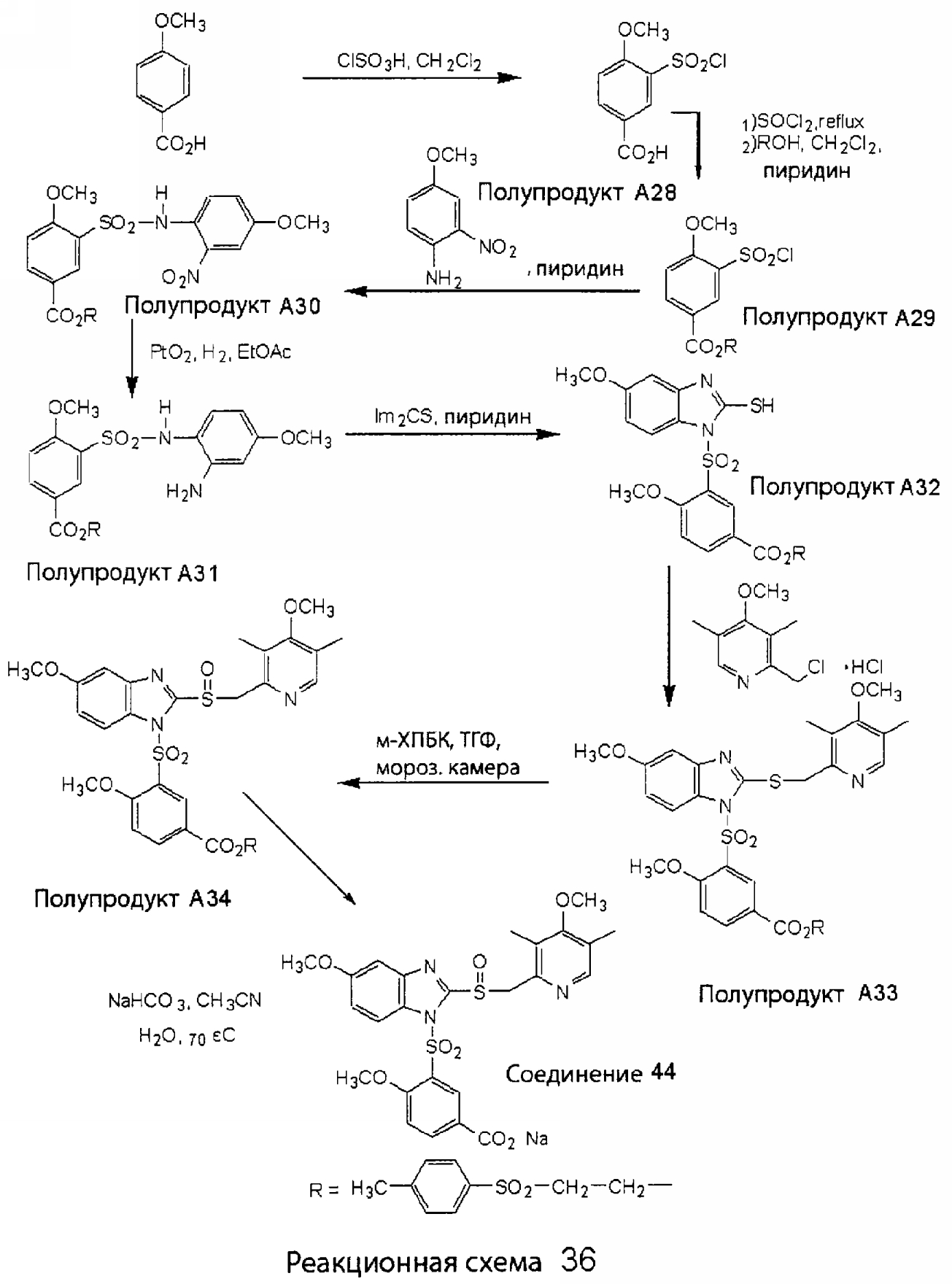
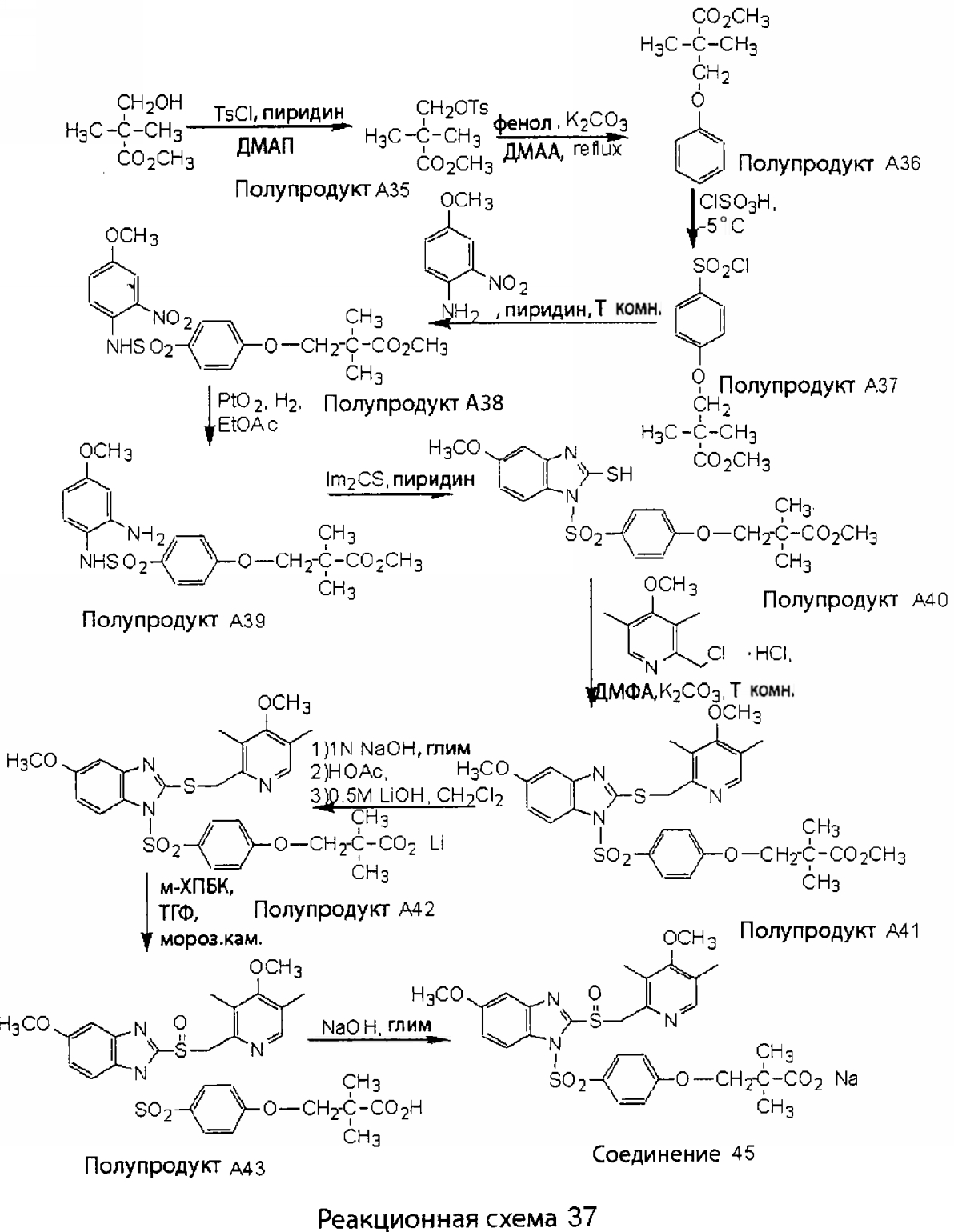
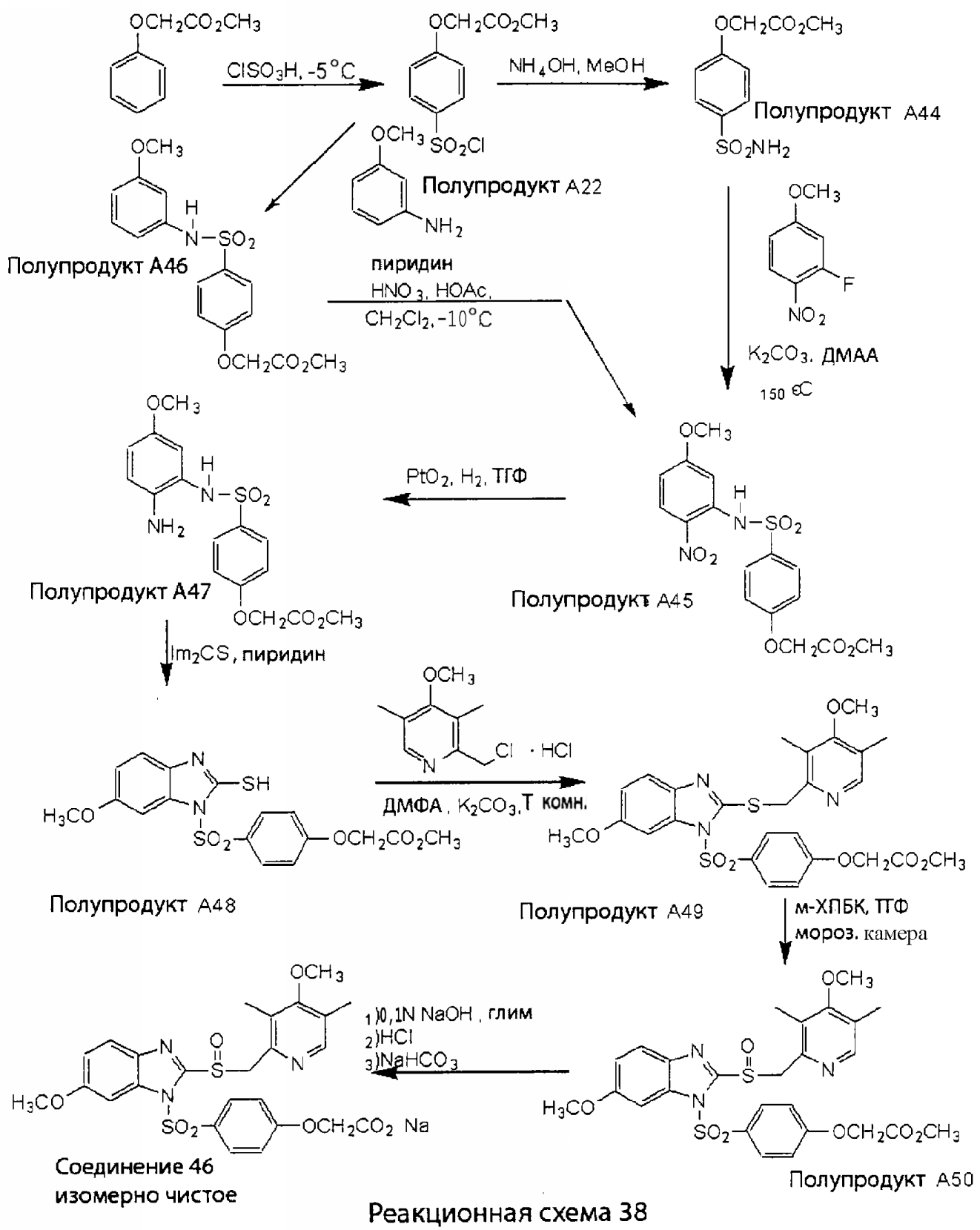
Заместитель в фенилсульфонилэтильной части обозначен символом Z”, который во многих предпочтительных примерах означает метильную группу в пара-положении или нитрогруппу в мета-положении фенильного кольца. Предпочтительные способы синтеза хлорсульфонильных соединений Формулы 6 проиллюстрированы ниже схемами реакций, а также подробно описаны в разделе «Конкретные примеры». Но помимо этого и в общем случае, хлорсульфонильные соединения в объеме Формулы 6 могут быть получены такими модификациями представленных здесь реакций, которые очевидны для специалиста в данной области в свете широко доступной химической патентной и научной литературы. Реакция пиридилметилсульфинилбензимидазолов Формулы 5 с хлорсульфонильными соединениями Формулы 6 приводит к образованию полупродуктов Формулы 7. Полупродукты Формулы 7 гидролизуются в присутствии мягкого основания, такого как NaHCO3, с образованием натриевой соли соединений изобретения. Побочным продуктом этой реакции является замещенный или незамещенный фенилвинилсульфон, изображенный на Реакционной схеме 1. Натриевая соль может быть легко превращена в соединение изобретения в форме свободной кислоты, представленное (в упрощенном виде) Формулой 8. Соединения изобретения, обозначенные Формулами 2 и 3, содержат заместитель в бензимидазольном фрагменте, а именно группу СН3О (Формула 2, производные омепразола) или группу HF2CO (Формула 3, производные пантопразола). По этой причине реакция омепразола и пантопразола (представленных в упрощенном виде Формулой 5) с хлорсульфонильными соединениями Формулы 6 приводит к двум изомерным соединениям, каждое из которых охватывается объемом изобретения. Обычно, но не обязательно, эти два изомера образуются при реакции приблизительно в отношении 1: 1, и для данного изобретения было установлено, что биологическая активность, растворимость и особенно стабильность при этом могут различаться, в некоторых случаях значительно. Хотя при необходимости изомеры могут быть отделены один от другого с помощью современных методов разделения, таких как жидкостная хроматография высокого давления (ЖХВД), разработан также более эффективный синтетический подход к синтезу конкретного изомера таких соединений. Реакционные схемы 1А и 1В раскрывают общие синтетические подходы к получению двух конкретных изомеров производных омепразола данного изобретения. Подробные схемы синтеза предпочтительных конкретных изомеров производных омепразола согласно изобретению показаны и описаны в разделе конкретных примеров.
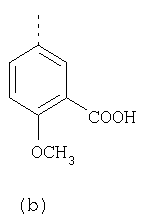
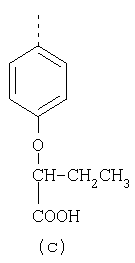
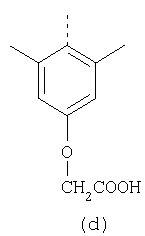
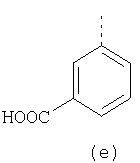
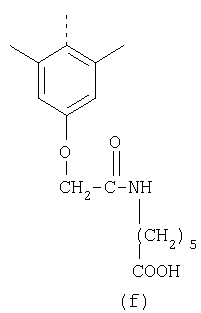
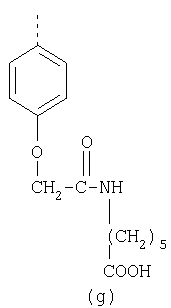
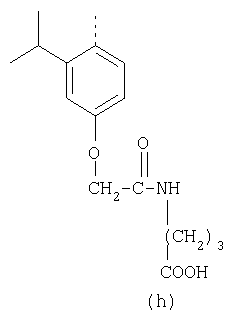
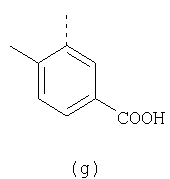
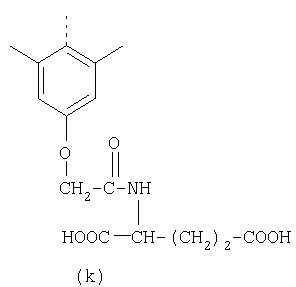
Согласно Реакционной схеме 1А 2-нитро-4-метоксианилин (доступный из Aldrich) взаимодействует с хлорсульфонильным соединением Формулы 9. Как и на Реакционной схеме 1, на Реакционных схемах 1А и 1В для простоты иллюстрации реагент Формулы 9 изображает арильную, гетероарильную или алкильную части Формулы (i) – на примере фенильной группы, и показывает одну группу R1 (как определено выше) и один эфир, приводящий к монокарбоновой кислоте. Однако специалистам в данной области нетрудно понять, что приведенная здесь методика синтеза может быть использована для получения конкретных изомеров производных омепразола настоящего изобретения, содержащих полный набор заместителей R, как определено для Формул 1-4. Реакция 2-нитро-4-метоксианилина с хлорсульфонильным соединением Формулы 9 приводит к производному хлорсульфонил-2-нитро-4-метоксианилина Формулы 10. Нитрогруппу последнего восстанавливают на следующей стадии и получают хлорсульфонил-2-амино-4-метоксианилин Формулы 11. Хлорсульфонил-2-амино-4-метоксианилин Формулы 11 реагирует далее с тиокарбонилдиимидазолом (Im2C=S) (либо с фенилизоцианатом или тиофосгеном) с замыканием кольца и образованием 2-тиобензимидазольного производного Формулы 12, в котором метоксильная группа находится в 5-положении имидазольного кольца, а хлорсульфонильная группа присоединена к атому азота в 1-положении. Соединение Формулы 12 взаимодействует с 2-хлорметил-4-метокси-3,5-диметилпиридином с образованием N-1-сульфонил-5-метокси-2-[(4-метокси-3,5-диметил-(2-пиридил))метилтио]бензимидазола Формулы 13. Реагент 2-хлорметил-4-метокси-3,5-диметилпиридин или его гидрохлорид могут быть получены обработкой 4-метокси-3,5-диметилпиридинметанола тионилхлоридом. Тиоэфирную группу Формулы 13 окисляют до сульфоксидной обработкой 3-хлорпероксибензойной кислотой (мета-хлорперпербензойной кислотой, м-ХПБК) или другим подходящим окислителем и получают N-1-сульфонил-5-метокси-2-[(4-метокси-3,5-диметил-(2-пиридил))метилсульфинил]бензимидазол Формулы 14. При обработке соединений Формулы 14 основанием происходит омыление эфирной группы карбоновой кислоты, присоединенной к арильному кольцу хлорсульфонильной части, и образование соединений изобретения, производных омепразола, Формулы 15, в которых метоксильная группа находится в 5-положении бензимидазольной части (чистые позиционные изомеры). Реакционная схема 1 В иллюстрирует способ синтеза “второго изомера”, в котором метоксильная группа находится в 6-положении бензимидазольной части. Согласно этой схеме 3-метоксианилин (доступный из Aldrich) взаимодействует с реагентом Формулы 9 с образованием хлорсульфонил-3-метоксианилина Формулы 16. Хлорсульфонил-3-метоксианилин Формулы 16 реагирует с азотной кислотой в присутствии серной кислоты с образованием хлорсульфонил-3-метокси-6-нитроанилина Формулы 17. Нитрогруппу соединения Формулы 17 восстанавливают до аминогруппы и полученный хлорсульфонил-3-метокси-6-аминоанилин замыкают обработкой тиокарбонилдиимидазолом с образованием 2-тиобензимидазольного производного Формулы 18, в котором метоксильная группа находится в 6-положении имидазольного кольца, а хлорсульфонильная группа присоединена к атому азота в 1-положении. Соединение Формулы 18 далее подвергают той же последовательности превращений, что и соединение Формулы 12на Реакционной схеме 1А, и получают соединения изобретения, производные омепразола, Формулы 21, в которых метоксильная группа находится в 6-положении бензимидазольного фрагмента (чистые позиционные изомеры). В рамках данного изобретения было установлено, что позиционные изомеры Формулы 21 менее стабильны, чем позиционные изомеры Формулы 15. Реакционные схемы 2-44, приведенные ниже в разделе «Конкретные примеры», раскрывают предпочтительные способы синтеза типичных предпочтительных соединений изобретения. БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ, СПОСОБЫ ВВЕДЕНИЯ Значительным преимуществом соединений настоящего изобретения является их способность самопроизвольно высвобождать активные формы ингибиторов протонного насоса в процессе гидролиза в организме млекопитающих (включая человека). Гидролиз может быть химическим или ферментным. Поскольку соединения настоящего изобретения самопроизвольно высвобождают активную форму лекарственных препаратов – ингибиторов протонного насоса – в процессе протекающего in vivo гидролиза, они могут достигать большей продолжительности эффективной концентрации препарата в организме. Таким образом, соединения настоящего изобретения являются пролекарствами, которые превращаются в активные лекарственные препараты в ходе гидролиза в организме, обеспечивая длительную продолжительность эффективной концентрации. Увеличение длительности ингибирующей активности за счет самопроизвольного гидролиза соединений данного изобретения обеспечивает более эффективное ингибирование секреции желудочной кислоты, что дает возможность лучшей терапии кислотозависимых заболеваний, указанных выше. Соединения данного изобретения для ингибирования секреции желудочной кислоты могут вводиться перорально. Стандартная дневная доза соединений будет зависеть от различных факторов, как, например, индивидуальная потребность каждого пациента. В общем случае пероральная и парентеральная дозы будут составлять от 5 до 300 мг в день. Специалисту в данной области понятно, что для перорального введения соединения изобретения смешивают с фармацевтически приемлемыми наполнителями, хорошо известными per se. В частности, препарат, предназначенный для системного введения, может быть приготовлен в виде порошка, пилюли, таблетки и тому подобного, либо в виде сиропа или эликсира, пригодных для перорального применения. Описание веществ, обычно используемых для приготовления таблеток, порошков, пилюль, сиропов и эликсиров, можно найти в различных известных книгах и монографиях, например в Remington’s Pharmaceutical Science, Edition 17, Mack Publishing Company, Easton, Pennsylvania. Соединения настоящего изобретения могут объединяться с определенным количеством известных ингибиторов протонного насоса, например с лансопразолом, омепразолом, пантопразолом или рабепразолом, для создания комбинации лекарство-пролекарство, которую вводят для ингибирования секреции желудочной кислоты. В соответствии с этим сначала секрецию желудочной кислоты у пациента блокирует ингибитор протонного насоса (лекарство). Указанные выше известные и широко используемые ингибиторы протонного насоса имеют период полувыведения из плазмы 60-90 минут. По мере того, как эффективная концентрация ингибитора протонного насоса (лекарства) снижается в процессе метаболизма, соединение настоящего изобретения (пролекарство) непрерывно подвергается гидролизу и обеспечивает и поддерживает концентрацию нового активного ингибитора в организме млекопитающего, включая организм человека, более длительное время, чем немодифицированный ингибитор протонного насоса. Это приводит к более быстрому и эффективному ингибированию секреции кислоты. Недостаток многих используемых в настоящее время ингибиторов протонного насоса заключается в том, что для терапии путем инъекции в жидкой форме они должны быть восстановлены из лиофилизированного порошка в среде с высоким значением рН, приблизительно 9,5-10,5. Пролекарства настоящего изобретения устраняют этот недостаток – необходимость в восстановительной среде со столь высоким значением рН, поскольку соединения настоящего изобретения могут быть восстановлены до инъецируемой жидкости в среде с величиной рН приблизительно 7-8. Специалисту очевидно, что жидкость, восстанавливающая лекарственный препарат для инъекции его в жидкой форме, должна быть фармацевтически приемлемым водным раствором, хорошо известным per se. Такие фармацевтически приемлемые растворы, применяемые для введения препаратов в инъецируемой форме, описаны, например, в монографии PHARMACEUTICAL DOSAGE FORMS (Parenteral Medications, Volume 1, Edited by K.E.Avis, H.A.Lieberman и L.Lachman (1992). Еще одним полезным качеством лекарственных препаратов соединений изобретения типа пре-ингибиторов протонного насоса (П-ИПН) является их способность обеспечивать более эффективное лечение эрозивного эзофагита и менее тяжелых рефлюкс-заболеваний. Это связано с тем, что для эффективного лечения эрозивного эзофагита (и в меньшей степени малых рефлюкс-заболеваний) требуется предотвращение обратного заброса содержимого желудка при величине рН 3,0 или ниже. Современные лекарственные ИПН-препараты допускают отклонение кислотности до рН<2 несколько раз в день, что приводит к умеренному уменьшению интенсивности симптомов. Однако заживление требует повышения значения рН до >4,0 в течение примерно 16 часов или более. Если, как это происходит при обычном лечении современными препаратами ИПН, в оставшиеся 8 часов кислотность будет периодически падать до рН 3,0 и ниже, пациенты по-прежнему будут испытывать боль. Более эффективное и продолжительное подавление кислотности препаратами настоящего изобретения, по всей видимости, приведет к значительно лучшему лечению такого заболевания, а также к более быстрому излечиванию всех кислотозависимых эрозий или язв. Лекарственные препараты настоящего изобретения типа пре-ингибиторов протонного насоса (П-ИПН) обеспечивают улучшенную двойную терапию уничтожения H.pylori. Это происходит благодаря тому, что препараты ИПН действуют совместно с антибиотиками, влияющими на клеточное деление, такими как амоксициллин (amoxicillin) (биосинтез клеточных стенок) и кларитромицин (clarithromycin) (синтез белка), путем повышения рН желудочной поверхности до величины, позволяющей большей части популяции бактерий в момент нахождения антибиотика в желудочной полости пребывать в фазе деления или роста. Однако воздействие препаратов на внутрижелудочный рН ограничено временем их пребывания в плазме. Лекарственные препараты настоящего изобретения типа пре-ингибиторов протонного насоса (П-ИПН) способны постоянно повышать внутрижелудочную рН до величины, близкой к нейтральной, при обычной терапии один раз в день. Поэтому при двойной терапии пролекарствами изобретения (например, пролекарством омепразола согласно изобретению) вместе с эффективным антибиотиком, таким как амоксициллин, можно ожидать 100% уничтожения бактерий. Даже монотерапия препаратами настоящего изобретения типа пре-ингибиторов протонного насоса (П-ИПН) будет, по всей вероятности, успешной для уничтожения Н.pylori. Это обусловлено тем, что в отсутствие кислоты фермент Н.pylori уреаза повышает величину рН окружающей среды до значения >8,3, которое токсично для микроорганизма. ИПН в современной рецептуре ингибируют рост или присутствие микроорганизма в полости за счет повышения антральной величины рН до значений, близких к нейтральным. Повышение в течение 24 часов величины рН до нейтральной, которое может достигаться с помощью лекарственных препаратов настоящего изобретения, вероятно, приводит к «самоуничтожению» бактерий. Примерно 30% больных желудочно-кишечными расстройствами имеют симптомы без поддающегося количественной оценке основного заболевания (неязвенная диспепсия). Наиболее вероятной причиной этих симптомов является повышенная желудочно-кишечная афферентная нервная чувствительность к “желудочной” кислоте. Только ликвидация кислоты снимает (облегчает) эти симптомы, и этого можно достичь с помощью лекарственных препаратов по данному изобретению. РАСТВОРИМОСТЬ И СТАБИЛЬНОСТЬ Растворимость Еще одним значительным преимуществом пролекарств ингибиторов протонного насоса настоящего изобретения по сравнению с пролекарствами ингибиторов протонного насоса, раскрытыми в Патентах США 6093734, 6559167 и WO 00109498, а также по сравнению со всеми другими известными препаратами, является их повышенная растворимость. В качестве иллюстрации этого водная растворимость каждого из известных соединений с (а) по (f), приведенных ниже, была менее 0,01 мкг/мл (<0,01 мкг/мл), если эти соединения были пролекарствами препарата лансопразол (соединения (а)-(с)), и составляла от 5 до 8 мкг/мл (5-8 мкг/мл), если эти соединения являлись пролекарствами препарата омепразол (соединения (d)-(f)). Напротив, растворимость в дистиллированной воде свободных карбоновых кислот Соединений 2 и 9 изобретения оказалась выше 100 мкг/мл (>100 мкг/мл).
Определяли растворимость натриевых солей некоторых соединений настоящего изобретения в фосфатно-буферном растворе с величиной рН 7,4 (50 мМ фосфата натрия, 10 мМ KCl, 0,1 М NaCl), а также в более кислом буферном растворе Бриттона-Робинсона с величиной рН 3 (40 мМ уксусной кислоты, 40 мМ фосфорной кислоты, 40 мМ борной кислоты, 36 мМ NaOH и 19,6 мг/мл KCl). Результаты, полученные для буферного раствора с величиной рН 7,4, приведены в Таблице 1. Как видно из Таблицы 1, было обнаружено, что изученные соединения хорошо растворимы в фосфатно-буферном растворе с величиной рН 7,4. Изученные соединения оказались значительно менее растворимыми в буферном растворе Бриттона-Робинсона с величиной рН 3, но при этом более растворимыми, чем известные соединения (а)-(f). Это понятно с точки зрения того, что в буферном растворе с величиной рН 7,4 соединения изобретения образуют натриевую соль, тогда как в буферном растворе с величиной рН 3 соединения существуют в форме менее растворимых свободных карбоновых кислот.Таким образом, раствор натриевой или другой фармацевтически приемлемой соли соединений изобретения или приготовленных другим способом соединений изобретения при рН 7 или выше является хорошо растворимым и, следовательно, пригоден для введения путем внутривенной инъекции.
Стабильность в буферных растворах Стабильность Соединений 1,2, 3, 4 и 5 изобретения в водном растворе (0,1 мг/мл) изучали на буферных растворах Бриттона-Робинсона с величинами рН 3, рН 7 и рН 9 соответственно. Для каждого соединения в каждом буфере готовили раствор с концентрацией 0,1 мг/мл, приготовленные растворы выдерживали при температуре 37°С в течение 1 часа и затем с помощью ВЭЖХ определяли концентрации тестируемых соединений. Было установлено, что тестируемые соединения стабильны в водных растворах при нейтральных условиях. Полупериод гидролиза при величине рН 7 для пролекарства с образованием соответствующего ингибитора протонного насоса оказался свыше 50 часов. Это показывает, что пролекарства сами по себе являются достаточно стабильными для внутривенной инъекции. Стабильность в плазме Типичные тестируемые соединения изобретения (пролекарства) инкубировали в плазме при температуре 37°С, после чего концентрацию этих соединений, а также соответствующих соединений – ингибиторов протонного насоса (ИПН) – определяли методом градиентной ВЭЖХ-УФ. Результаты представлены в Таблице 2. Хотя период полувыведения из плазмы тестируемых соединений варьировался в зависимости от их структуры, тем не менее очевидно, что в плазме соединения изобретения превращаются в соответствующие ингибиторы протонного насоса со скоростью, большей, чем скорость гидролиза соединений при нейтральной или близкой к нейтральной величине рН. Это свидетельствует о том, что пролекарства изобретения могут использоваться в качестве пролекарств при внутривенной инъекции и что in vivo они высвобождают активный ингибитор протонного насоса.
ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ СОЕДИНЕНИЙ ИЗОБРЕТЕНИЯ НА СЕКРЕЦИЮ ЖЕЛУДОЧНОЙ КИСЛОТЫ У НАХОДЯЩИХСЯ В СОЗНАНИИ КРЫС-САМЦОВ ПРИ ОПРЕДЕЛЕННЫХ ВРЕМЕННЫХ ИНТЕРВАЛАХ Пероральное введение В этом эксперименте использовали крыс-самцов (линии Sprague-Dawley). Натриевую соль омепразола или натриевую соль лансопразола (20 мг) суспендировали в 10 мл 0,1N раствора NaHCO3. По 40 мг натриевых солей каждого из соединений: Соединения 1, Соединения 2, Соединения 3, Соединения 4, Соединения 6 Соединения 9, Соединения 12, смеси (1:1) Соединений 16 и 17, смеси (1:1) Соединений 18 и 19, смеси (1:1) Соединений 20 и 21, смеси (1:1) Соединений 22 и 23, Соединения 27, смеси (1:1) Соединений 34 и 35 и Соединения 36 растворяли в 10 мл 50 мМ фосфатно-натриевого буферного раствора, рН 7,4. Крысам вводили в следующих дозах (мкмоль/кг веса тела крысы): омепразол (10 мкмоль/кг), лансопразол (10 мкмоль/кг). Соединение 1 (20 мкмоль/кг), Соединение 2 (40 мкмоль/кг), Соединение 3 (20 мкмоль/кг), Соединение 4 (20 мкмоль/кг), Соединение 6 (20 мкмоль/кг), Соединение 9 (40 мкмоль/кг), Соединение 12 (40 мкмоль/кг), смесь (1:1) Соединений 16 и 17 (40 мкмоль/кг), смесь (1:1) Соединений 18 и 19 (40 мкмоль/кг), смесь (1:1) Соединений 20 и 21 (40 мкмоль/кг), смесь (1:1) Соединений 22 и 23 (40 мкмоль/кг), Соединение 27 (40 мкмоль/кг), смесь (1:1) Соединений 34 и 35 (40 мкмоль/кг) и Соединение 36 (40 мкмоль/кг). Данные растворы лекарственных препаратов вводили перорально. Через 3 часа после введения брюшную полость крыс рассекали и при легкой анестезии эфиром лигировали привратник. Для стимулирования кислоты подкожно инъецировали гистамин (30 мг/кг) и карбахол (30 мкг/кг). Брюшную полость сразу же закрывали. Двумя часами позже перевязывали пищевод и удаляли желудок. Собирали желудочный сок и количественно оценивали выделение кислоты титрованием 0,1N раствором NaOH. В качестве контрольного опыта перорально вводили 1 мл 50 мМ фосфатного буферного раствора (рН 7,4) без добавления какого-либо пролекарства или лекарства – ингибитора протонного насоса. Количество выделившейся кислоты оценивали тем же способом, что описан выше, показывая максимум стимулированной гистамином и карбахолом секреции желудочной кислоты. Процент ингибирования рассчитывали исходя из частных реакций, вызванных тестируемым соединением и полученных в контрольном опыте.
Внутривенное введение Также было изучено ингибирование секреции желудочной кислоты после внутривенного введения соединений изобретения. Натриевую соль лансопразола (20 мг) растворяли в 40% гидроксипропил-бета-циклодекстрине. Соединения изобретения, использованные в данном эксперименте, растворяли в фосфатно-буферном растворе с величиной рН 7,4. Каждое соединение инъецировали внутривенно в дозе 5 мкмоль/кг или 10 мкмоль/кг (мкмоль/кг веса тела крысы), как показано в Таблице 4. Между 2 и 4 часами после инъекции собирали желудочный сок и определяли процентное ингибирование, как описано выше.
КОНКРЕТНЫЕ ПРИМЕРЫ
2-(3-Нитробензолсульфонил)этиловый эфир 3-хлорсульфонилбензойной кислоты (Полупродукт 1) К твердой 3-хлорсульфонилбензойной кислоте (3,2 г, 14,4 ммоль) при комнатной температуре прибавляли при перемешивании твердый PCl5 (3,0 г, 14,4 ммоль). Смесь нагревали до температуры 70°С до образования жидкости коричневого цвета, которую грели в течение еще 2 часов. Побочный POCl3 отгоняли в вакууме, а маслянистый остаток коричневого цвета растворяли в 15 мл CH3CN, после чего прибавляли 2-(3-нитробензолсульфонил)этанол (2,8 г, 12,0 ммоль). Смесь нагревали при кипении в течение 36 часов. После этого добавляли воду и смесь экстрагировали СН2Cl2. Полученное масло очищали хроматографией на короткой колонке (элюент: от СН2Cl2 до 1% МеОН в СН2Cl2) и получали полутвердое вещество коричневого цвета, которое при обработке смесью эфир-EtOAc превращалось в кристаллы белого цвета (выход 2,75 г (53%)). 1Н ЯМР (ДМСО-d6, 400 МГц) 2-(3-Нитробензолсульфонил)этиловый эфир 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукт 2) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (600 мг, 1,62 ммоль) в CH2Cl2 (10 мл) при комнатной температуре прибавляли NaH (45 мг, 1,95 ммоль), получая прозрачный раствор. К полученной прозрачной смеси при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 1, 845 мг, 1,95 ммоль, 1,2 экв.) в СН2Cl2, после чего массу перемешивали в течение 2 часов. Затем добавляли воду и смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали. Маслянистый остаток очищали колоночной хроматографией (3% МеОН в СН2Cl2) и получали 1,0 г (80%) пены желтого цвета. 1H ЯМР (CDCl3, 400 МГц) 3-{2-[3-Метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойная кислота (Соединение 1) и ее натриевая соль Раствор 2-(3-нитробензолсульфонил)этилового эфира 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукта 2, 900 мг, 1,17 ммоль) и NaHCO3 (118 мг, 1,41 ммоль, 1,2 экв.) в смеси ТГФ – H2O (6 мл – 3 мл) нагревали при температуре 70°С в течение 20 минут, за это время гетерогенная смесь становилась прозрачной. После этого все летучие компоненты удаляли в вакууме, оставшуюся смолу растворяли в СН2Cl2, смесь фильтровали для удаления твердых частиц. Фильтрат упаривали в вакууме досуха и оставшуюся пену желтого цвета обрабатывали смесью эфир – EtOAc (5:1) до появления осадка. Осадок отфильтровывали и получали 630 мг (94%) натриевой соли 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (натриевая соль Соединения 1). 1H ЯМР (ДМСО-d6, 400 МГц) 3-{2-[3-Метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойная кислота (Соединение 1) Натриевую соль 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (100 мг) растворяли в 10 мл воды и подкисляли до рН 3 1N раствором HCl, после чего экстрагировали дихлорметаном. Дихлорметановый слой сушили над безводным сульфатом магния, упаривали досуха и получали свободную кислоту (Соединение 1, 76 мг).
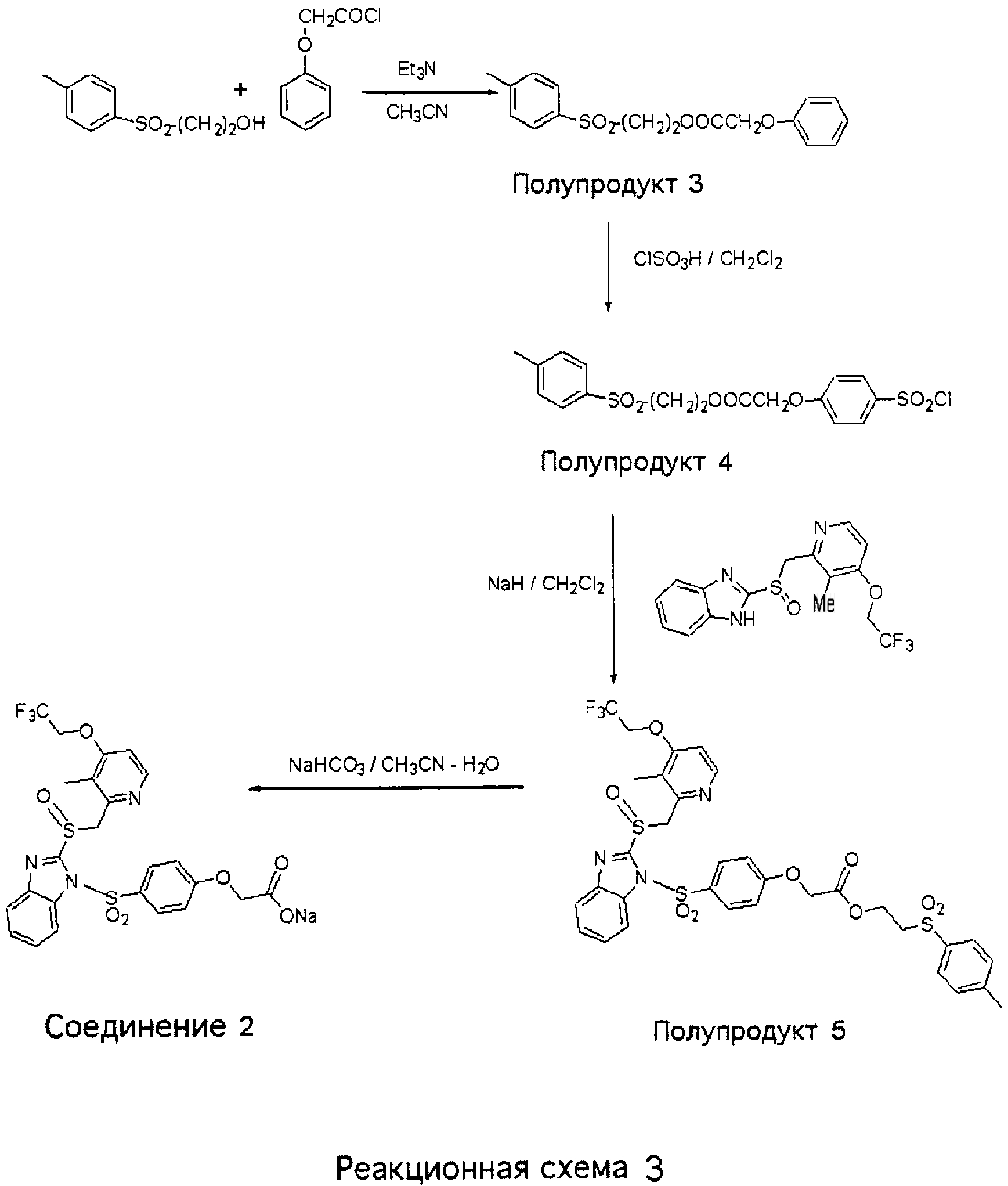
2-(Толуол-4-сульфонил)этиловый эфир феноксиуксусной кислоты (Полупродукт 3) К раствору феноксиацетилхлорида (доступного из Aldrich, 5,0 г) и триэтиламина (Et3N) (3 г) в 50 мл СН3CN прибавляли раствор 2-(п-толилсульфонил)этанола (5,0 г) при 0°С. После этого добавляли воду и реакционную массу экстрагировали CH2Cl2. Объединенные органические слои промывали 1N раствором HCl и насыщенным раствором NaHCO3, сушили над безводным MgSO4, концентрировали и получали 8,0 г (97%) эфира (Полупродукта 3) в виде кристаллов светло-желтого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир (4-хлорсульфонилфенокси)уксусной кислоты (Полупродукт 4) К охлажденной на ледяной бане смеси 2-(толуол-4-сульфонил)этилового эфира феноксиуксусной кислоты (Полупродукта 3, 3,0 г) и 1 мл CH2Cl2 по каплям прибавляли 3,0 мл хлорсульфоновой кислоты (5,2 г, 5,0 экв.). По окончании прибавления хлорсульфоновой кислоты ледяную баню убирали. Смесь продолжали перемешивать при комнатной температуре в течение 3 часов. После этого густую сиропообразную массу выливали при интенсивном перемешивании на колотый лед. Кристаллический осадок белого цвета отфильтровывали, промывали метанолом и холодным бензолом, сушили в течение ночи в глубоком вакууме и получали 3,4 г (87%) Полупродукта 4 в виде порошка белого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир (4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 5) К гетерогенному раствору 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (900 мг, 2,44 ммоль) в 30 мл CH2Cl2 при комнатной температуре прибавляли 70 мг NaH (2,92 ммоль), за это время масса гомогенизировалась. К полученной прозрачной реакционной массе прибавляли 2-(толуол-4-сульфонил)этиловый эфир 4-(хлорсульфонилфенокси)уксусной кислоты (Полупродукт 4, 1,26 г, 2,92 ммоль, 1,2 экв.). После полного растворения хлорсульфоната в реакционную массу добавляли около 1 г твердого NaHCO3. (Без добавления NaHCO3 смесь к моменту завершения реакции чернеет.) После этого отгоняли растворитель, а маслянистый остаток очищали колоночной хроматографией (силикагель, от CH2Cl2 до 4% МеОН в СН2Cl2) и получали 1,75 г (94%) требуемого продукта (Полупродукт 5) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль (4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Соединение 2) 2-(Толуол-4-сульфонил)этиловый эфир (4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 5, 400 мг, 0,54 ммоль) растворяли в 4 мл CH3CN и 2 мл изопропанола, после чего прибавляли раствор NaHCO3 (48 мг, 0,57 ммоль, 1,1 экв.) в 3 мл Н2О. Смесь нагревали при температуре 70°С в течение 2 часов. После этого в вакууме отгоняли все летучие вещества, а маслянистый остаток перерастворяли в EtOAc и полученную смесь фильтровали от нерастворимых твердых частиц. Фильтрат концентрировали, сушили в вакууме и получали пену белого цвета с желтоватым оттенком. Пену промывали этиловым эфиром для удаления побочного продукта (винилтолуолсульфона) и получали 300 мг Соединения 2 (натриевой соли) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (ДМСО-d6, 400 МГц)
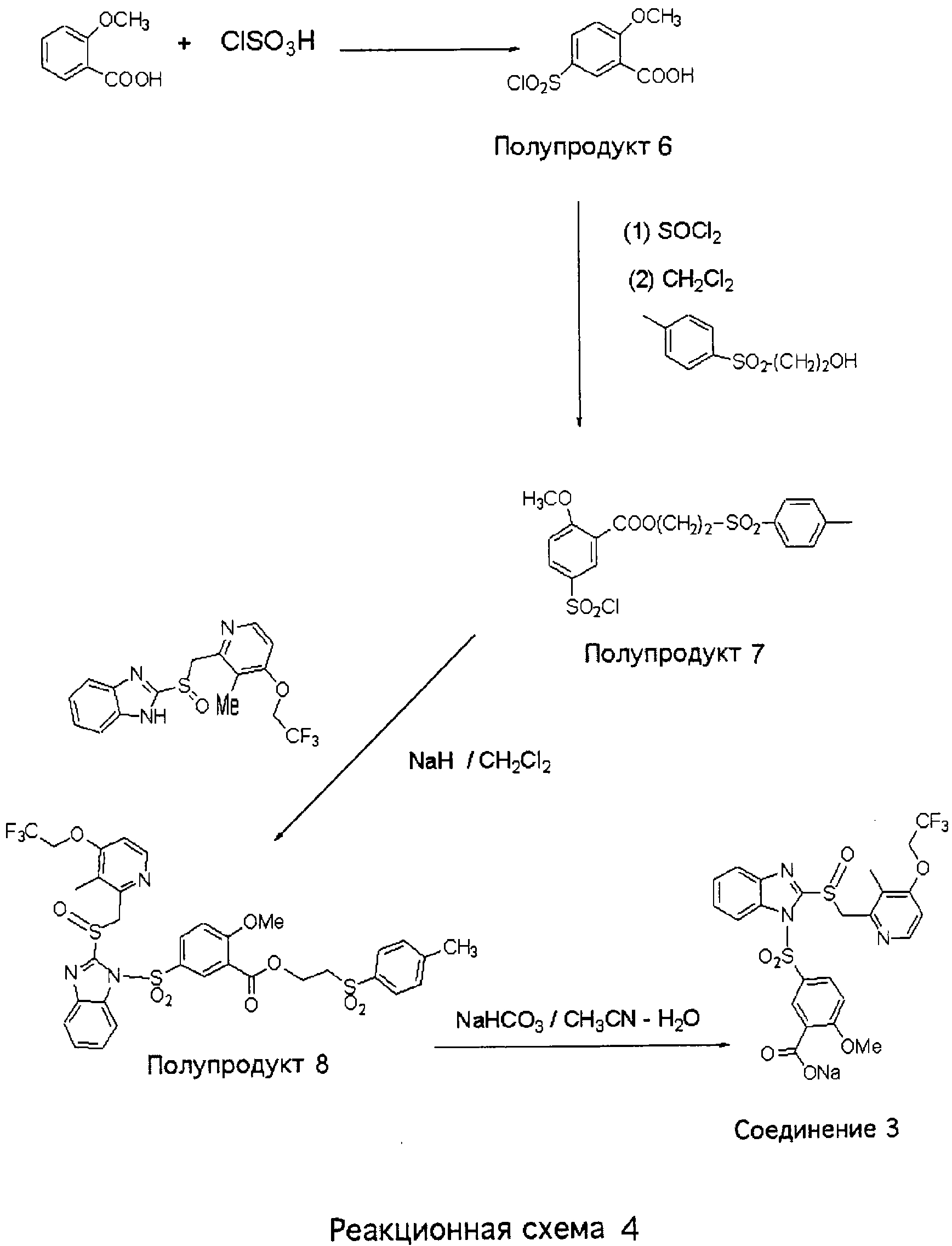
5-Хлорсульфонил-2-метоксибензойная кислота (Полупродукт 6) 2-Метоксибензойную кислоту (доступную из Aldrich, 5,0 г, 32,9 ммоль) нагревали с хлорсульфоновой кислотой (164 ммоль, 5,0 экв., 19,1 г, 11,0 ммоль) при температуре 50°С в течение 2 часов. Полученную густую жидкость коричневого цвета выливали при интенсивном перемешивании на колотый лед. Образовавшийся осадок белого цвета отфильтровывали, промывали Н2О, сушили в течение ночи в глубоком вакууме и получали Полупродукт 6 (4,8 г, 58%) в виде порошка белого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц) 2-(n-Толуолсульфонил)этил-5-хлорсульфонил-2-метоксибензоат (Полупродукт 7) Смесь 5-хлорсульфонил-2-метоксибензойной кислоты (Полупродукта 6, 3,0 г, 12,0 ммоль) и SOCl2 (6 мл) нагревали при температуре кипения в течение 2 часов. Затем отгоняли избыток SOCl2. Полученный маслянистый остаток растворяли в 20 мл СН2Cl2 и при комнатной температуре прибавляли 2-(п-толуолсульфонил)этанол (доступный из Aldrich 2,0 г, 10 ммоль) в 10 мл СН2Cl2. Полученную смесь нагревали при температуре кипения в течение 3 часов. После этого при пониженном давлении отгоняли летучие вещества, маслянистый остаток очищали колоночной хроматографией (силикагель, от гексан – СН2Cl2, 1:1, до СН2Cl2) и получали 3,5 г (81%) 2-(n-толуолсульфонил)этил-5-хлорсульфонил-2-метоксибензоата (Полупродукта 7) в виде прозрачного вязкого масла. 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир 2-метокси-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукт 8) К прозрачному раствору 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (740 мг, 2,0 ммоль) и NaH (60 мг, 2,5 ммоль) в 10 мл CH2Cl2 прибавляли 2-(п-толуолсульфонил)этил-5-хлорсульфонил-2-метоксибензоат (Полупродукт 7, 1,0 г, 2,3 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. После этого добавляли воду и смесь экстрагировали CH2Cl2. Дихлорметановый слой сушили над безводным сульфатом магния, упаривали при пониженном давлении и получали вязкое масло. Масло очищали колоночной хроматографией (силикагель, от СН2Cl2 до 2% МеОН в CH2Cl2) и получали 1,0 г (65%) 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукт 8) 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 2-метокси-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Соединение 3) Раствор 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукта 8, 400 мг, 0,52 ммоль) и NaHCO3 (52 мг, 0,62 ммоль, 1,2 экв.) в смеси СН3СН (3 мл) – Н2O (2 мл) – i-PrOH (1 мл) нагревали при температуре 70°С в течение 1,5 часов. Затем летучие вещества отгоняли, маслянистый остаток растворяли в смеси СН2Cl2-МеОН, массу фильтровали от нерастворимых твердых частиц. Фильтрат концентрировали при упаривании. Оставшийся сироп растворяли в Н2O и смесь экстрагировали СН2Cl2 (2 раза) для извлечения исходного вещества, лансопразола и толилвинилсульфона. Водный раствор сушили вымораживанием и получали 200 мг (65%) натриевой соли Соединения 3 в виде кристаллов светло-желтого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц)
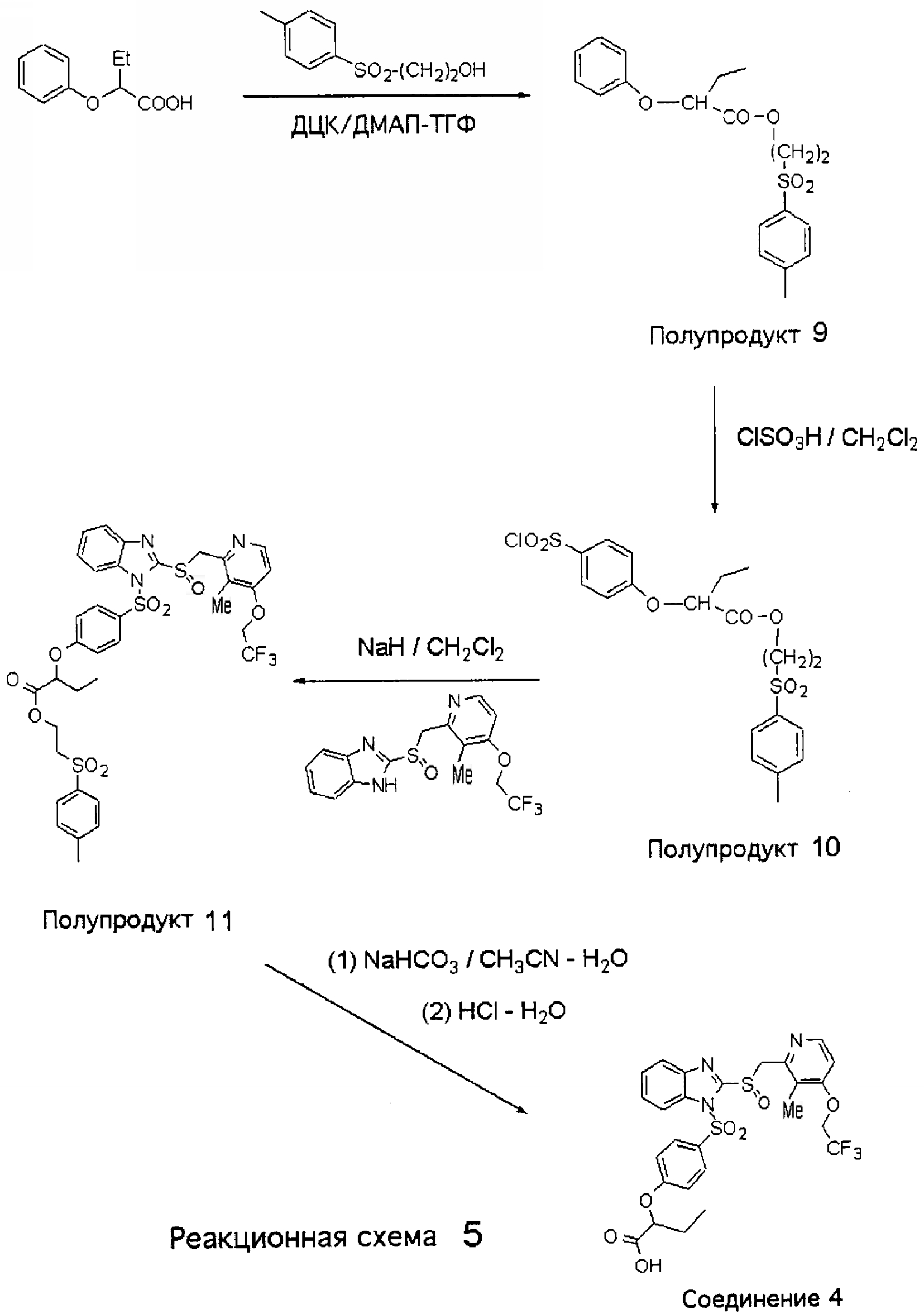
2-(n-Толуолсульфонил)этиловый эфир фенокси-2-масляной кислоты (Полупродукт 9) К смеси фенокси-2-масляной кислоты (доступной из Aldrich, 2,51 г, 13,8 ммоль), 2-(п-толуолсульфонил)этанола (2,8 г, 13,8 ммоль) и 0,5 г п-диметиламинопиридина (ДМАП) в 50 мл тетрагидрофурана (ТГФ) прибавляли N,N’-дициклогексилкарбодиимид (ДЦК) (3,14 г, 15,2 ммоль, 1,1 экв.) в 15 мл ТГФ при 0°С. Полученную массу перемешивали в течение ночи при комнатной температуре. Затем из реакционной массы отфильтровывали осадок белого цвета, а фильтрат концентрировали при упаривании. Полученное масло очищали колоночной хроматографией (от гексан-СН2Cl2 до СН2Cl2) и получали Полупродукт 9 (4,1 г, 82%) в виде прозрачного масла. 1Н ЯМР (CDCl3, 400 МГц) 2-(n-Толуолсульфонил)этиловый эфир 4-хлорсульфонилфенокси-2-масляной кислоты (Полупродукт 10) К охлажденной на ледяной бане смеси 2-(n-толуолсульфонил)этилового эфира фенокси-2-масляной кислоты (Полупродукта 9, 4,0 г, 11,0 ммоль) и 1 мл CH2Cl2 медленно прибавляли хлорсульфоновую кислоту (3,7 мл, 6,4 г, 5,0 экв.). По окончании прибавления хлорсульфоновой кислоты ледяную баню убирали. Полученную смесь перемешивали при комнатной температуре в течение 3 часов. После этого вязкую сиропообразную массу при интенсивном перемешивании выливали на измельченный лед и получали смолистый осадок. Смесь осадка, льда и воды экстрагировали СН2Cl2, органические фракции сушили над MgSO4, концентрировали упариванием и получали 4,1 г (81%) 1-(n-толуолсульфонил)этилового эфира 4-хлорсульфонилфенокси-2-масляной кислоты (Полупродукт 10). 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир 2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)масляной кислоты (Полупродукт 11) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (500 мг, 1,36 ммоль) в CH2Cl2 (10 мл) при комнатной температуре прибавляли NaH (35 мг, 1,45 ммоль) и получали прозрачный раствор. К образовавшейся прозрачной смеси при комнатной температуре прибавляли 2-(n-толуолсульфонил)этиловый эфир 4-хлорсульфонилфенокси-2-масляной кислоты (Полупродукт 10, 700 мг, 1,52 ммоль, 1,12 экв.) в СН2Cl2, и полученную массу перемешивали в течение 2 часов. После этого добавляли воду и смесь экстрагировали CH2Cl2. Органические слои сушили и концентрировали при упаривании. Маслянистый остаток очищали колоночной хроматографией (3% МеОН в СН2Cl2) и получали Полупродукт 11 (1,0 г, 93%) в виде пены белого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(4-{2-[3-Метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)масляная кислота (Соединение 4) Раствор 2-(толуол-4-сульфонил)этилового эфира 2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)масляной кислоты (Полупродукта 11, 400 мг, 0,50 ммоль) и NaHCO3 (47 мг, 0,55 ммоль, 1,1 экв.) в смеси CH3CN-H2O (7 мл – 3 мл) нагревали при температуре 70°С в течение 3 часов. После этого летучие вещества отгоняли, а оставшееся смолистое масло растворяли в CH3CN. Раствор в СН3СN фильтровали от нерастворимых твердых частиц. Фильтрат сушили и оставшуюся пену желтого цвета с целью кристаллизации обрабатывали смесью эфир – EtOAc (5:1). Осадок повторно обрабатывали СН3СN раствор в СН3СN фильтровали и получали 180 мг (58%) натриевой соли 2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)масляной кислоты (Соединение 4, натриевая соль). Натриевую соль растворяли в воде, подкисляли до рН 3 1N раствором HCl и экстрагировали дихлорметаном. Дихлорметановые фракции упаривали и получали Соединение 4 в виде свободной кислоты, которую дополнительно очищали на короткой колонке с силикагелем и получали 125 мг 2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)масляной кислоты (Соединение 4). 1Н ЯМР (ДМСО-d6, 400 МГц)
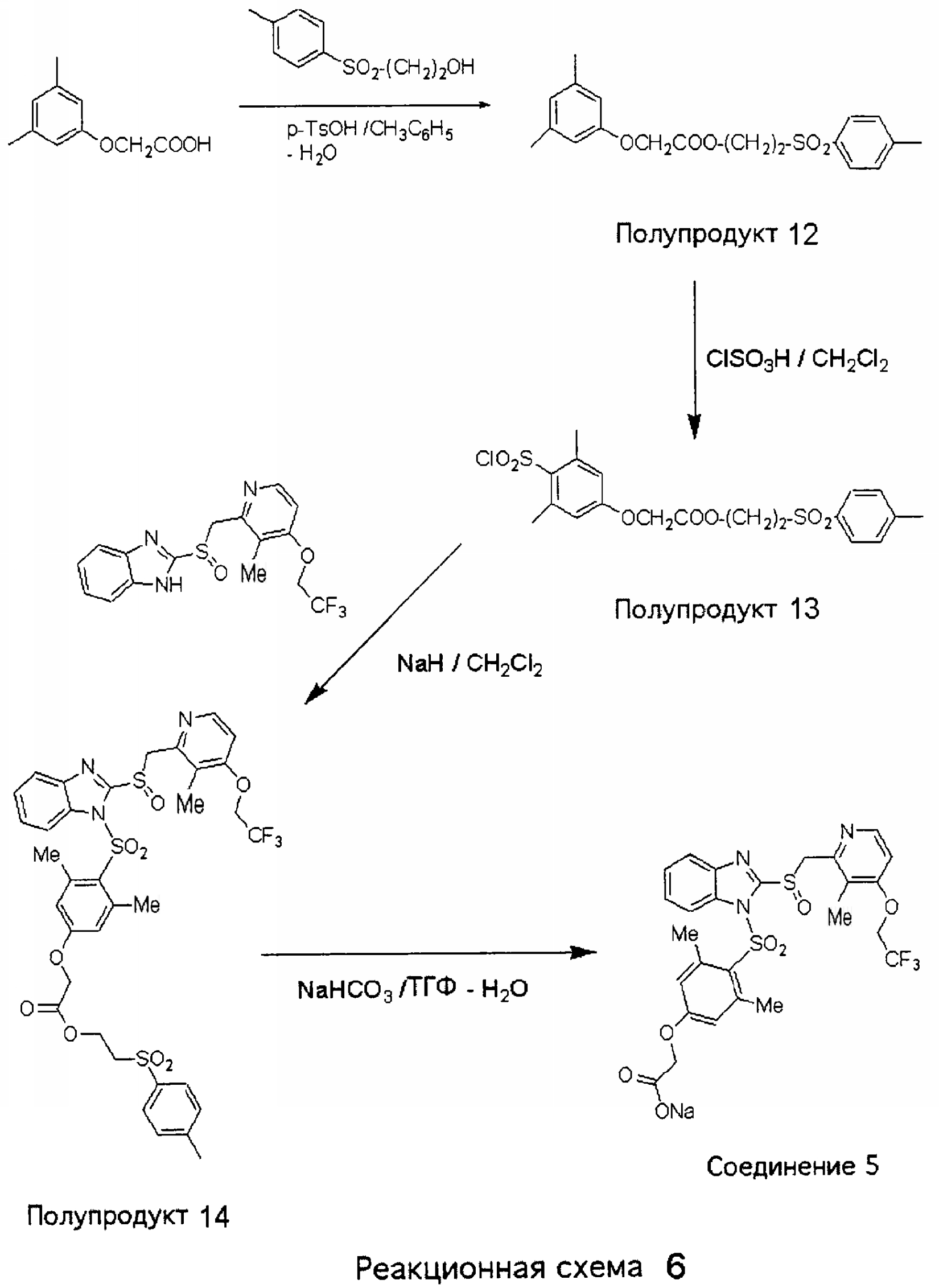
2-(n-Толуолсульфонил)этиловый эфир 3,5-диметилфеноксиуксусной кислоты (Полупродукт 12) 3,5-Диметилфеноксиуксусную кислоту получали по известной методике следующим образом. 3,5-Диметилфенол (12,2 г, 0,1 моль) и хлоруксусную кислоту (18,9 г, 0,2 моль) растворяли в 50% водном изопропаноле (300 мл) и медленно прибавляли 3-эквимоль NaOH (12 г, 0,3 моль). Реакционную массу кипятили с обратным холодильником в течение ночи, после чего концентрировали дистилляцией. Остаток разбавляли водой до 300 мл и подкисляли до рН 1. Смесь экстрагировали дихлорметаном (500 мл). Дихлорметановую фракцию концентрировали при пониженном давлении и получали продукт с небольшой примесью непрореагировавшего фенола. Осадки кристаллизовали из смеси бензол – гексан и получали чистый продукт, 15,6 г (86,6%). Смесь 3,5-диметилфеноксиуксусной кислоты (полученной, как описано выше, 2,3 г, 12,6 ммоль), 2-(п-толуолсульфонил)этанола (2,3 г, 11,5 ммоль) и гидрата п-толуолсульфоновой кислоты (p-TsOH·H2O) (0,5 г) в 100 мл толуола кипятили с насадкой Дина-Старка в течение 4 часов. Затем добавляли воду и смесь экстрагировали CH2Cl2. Объединенные органические слои промывали насыщенным раствором NaHCO3 (2х), сушили над MgSO4, концентрировали и получали Полупродукт 12 (4,4 г, 97%) в виде вязкого желтоватого масла. 1Н ЯМР (CDCl3, 400 МГц) 2-(n-Толуолсульфонил)этиловый эфир 4-хлорсульфонил-3,5-диметилфеноксиуксусной кислоты (Полупродукт 13) К смеси 2-(n-толуолсульфонил)этилового эфира 3,5-диметилфеноксиуксусной кислоты (Полупродукт 12,4,0 г, 11,0 ммоль) и CH2Cl2 (1,5 мл) при охлаждении по каплям прибавляли ClSO3Н, после чего смесь интенсивно перемешивали в течение 2 часов при 0°С. Образовавшееся вязкое масло при интенсивном перемешивании выливали на колотый лед. Смесь экстрагировали СН2Cl2. Органический слой сушили над MgSO4, концентрировали при упаривании и получали вязкое масло. После обработки диэтиловым эфиром (2 мл) и гексаном (2 мл) масло затвердевало. Кристаллы сушили в вакууме и получали 2-(n-толуолсульфонил)этиловый эфир 4-хлорсульфонил-3,5-диметилфеноксиуксусной кислоты (Полупродукт 13, 4,7 г, 94%) в виде кристаллов белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир (3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 14) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (500 мг, 1,36 ммоль) в СН2Cl2 (10 мл) при комнатной температуре прибавляли NaH (40 мг, 1,65 ммоль) и получали прозрачный раствор. К полученному прозрачному раствору при комнатной температуре прибавляли 2-(n-толуолсульфонил)этиловый эфир 4-хлорсульфонил-3,5-диметилфеноксиуксусной кислоты (Полупродукт 13, 760 мг, 1,65 ммоль, 1,2 экв.) в СН2Cl2 (5 мл) и смесь перемешивали в течение 4 часов. После этого добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали при упаривании. Маслянистый остаток очищали колоночной хроматографией (3% МеОН в СН2Cl2) и получали 2-(толуол-4-сульфонил)этиловый эфир (3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 14, 700 мг, 65%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль (3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил] бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Соединение 5) Раствор 2-(толуол-4-сульфонил)этилового эфира (3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукта 14, 400 мг, 0,50 ммоль) и NaHCO3 (51 мг, 0,60 ммоль, 1,2 экв.) в смеси ТТФ-Н2О (6 мл -3 мл) нагревали при температуре 70°С в течение 3 часов. Затем летучие вещества удаляли, оставшееся смолистое масло растворяли в ТТФ и смесь фильтровали от нерастворившихся твердых частиц. Фильтрат сушили, отгоняли растворитель и получали пену желтого цвета, которую для кристаллизации обрабатывали смесью эфир – EtOAc (5:1). Осадок обрабатывали СН3СН, отфильтровывали и получали Соединение 5 (230 мг, 72%) в виде кристаллов светло-желтого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц)
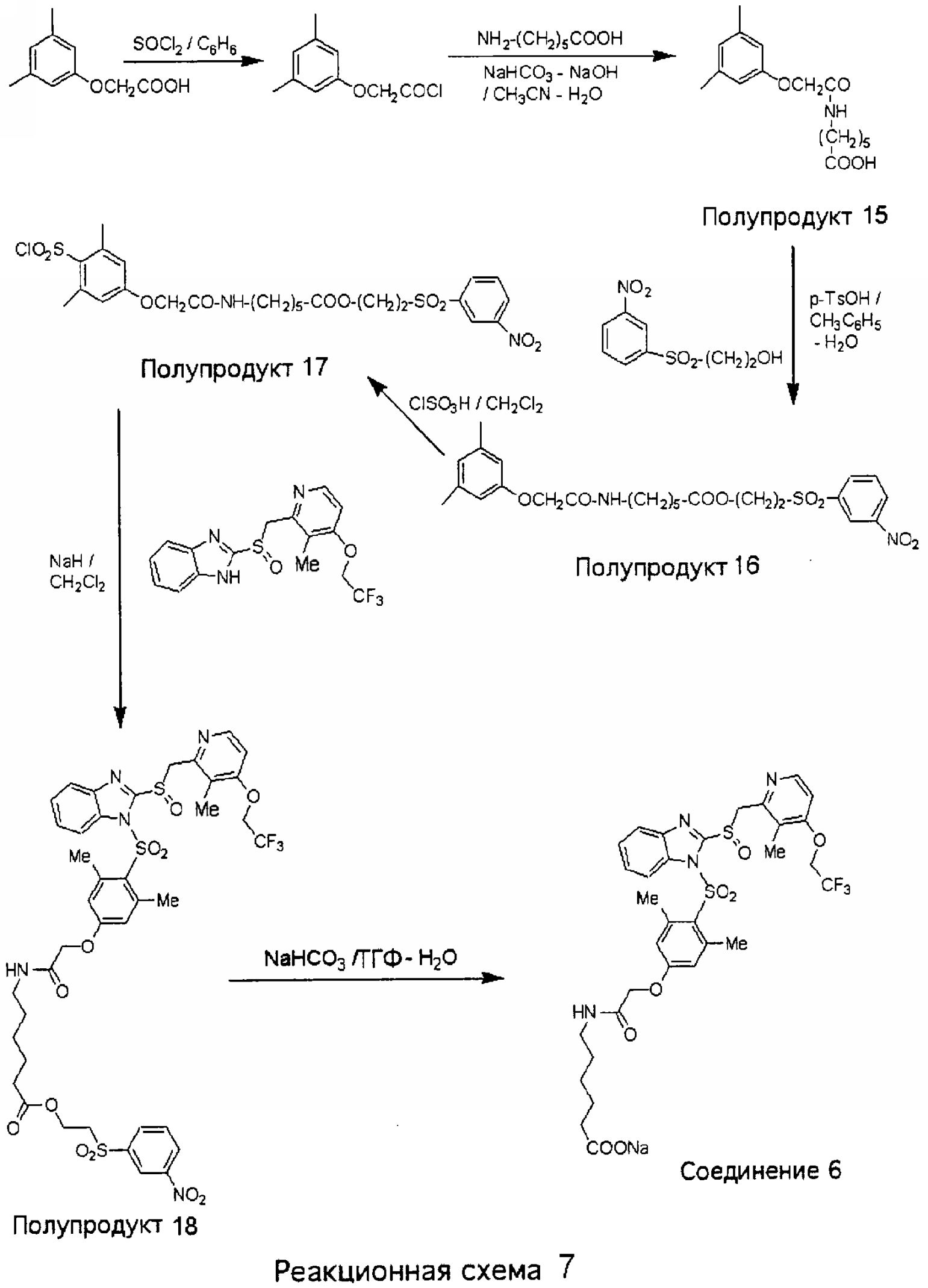
6-[2-(3,5-Диметилфенокси)ацетиламино]гексеновая кислота (Полупродукт 15) Смесь 3,5-диметилфеноксиуксусной кислоты (полученной, как описано выше, 9,3 г, 51,6 ммоль) и SOCl2 (11,3 мл, 18,5 г, 156 ммоль, 3,0 экв.) в 10 мл бензола кипятили с обратным холодильником в течение 2 часов. Затем в вакууме отгоняли летучие вещества и получали хлорангидрид 3,5-диметилфеноксиуксусной кислоты в виде масла светло-коричневого цвета. К охлажденному на ледяной бане раствору 6-амино-н-капроновой кислоты (13,5 г, 103 ммоль, 2,0 экв.) и NaOH (4,2 г, 105 ммоль) в 100 мл Н2O и 130 мл CH3CN по каплям прибавляли раствор хлорангидрида кислоты (полученного на предыдущей стадии) в 100 мл СН3CN и раствор NaHCO3 (6,5 г, 77,0 ммоль, 1,5 экв.) в 80 мл Н2O. Реакционную массу интенсивно перемешивали в течение ночи. Затем при пониженном давлении отгоняли большую часть CH3CN и смесь подкисляли при комнатной температуре до рН 2 концентрированной HCl. Выпавший осадок белого цвета отфильтровывали, промывали сначала Н2O, затем гексаном и после этого сушили в глубоком вакууме и получали Полупродукт 15 (14,5 г, 95%) в виде кристаллического вещества белого цвета. 1H ЯМР (CDCl3, 400 МГц) 2-(3-Нитробензолсульфонил)этиловый эфир 6-[2-(3,5-диметилфенокси)ацетиламино]гексеновой кислоты (Полупродукт 16) Смесь 6-[2-(3,5-диметилфенокси)ацетиламино]гексеновой кислоты (Полупродукта 15, 3,0 г, 10,2 ммоль), 2-(м-нитробензолсульфонил)этанола (доступного из Aldrich, 2,3 г, 10,0 ммоль) и гидрата п-толуолсульфоновой кислоты (0,5 г) в 100 мл толуола кипятили в течение ночи с насадкой Дина-Старка. Затем добавляли воду и смесь экстрагировали CH2Cl2. Объединенные органические слои дважды промывали насыщенным раствором NaHCO3, сушили над безводным MgSO4 и концентрировали при пониженном давлении. Полученное масло очищали колоночной хроматографией (1% МеОН в СН2Cl2) и получали Полупродукт 16 (4,8 г, 89%) в виде вязкого желтоватого масла. 1Н ЯМР (CDCl3, 400 МГц) 2-(3-Нитробензолсульфонил)этиловый эфир 6-[2-(4-хлорсульфонил-3,5-диметилфенокси)ацетиламино] гексеновой кислоты (Полупродукт 17) К охлажденному раствору 2-(3-нитробензолсульфонил)этилового эфира 6-[2-(3,5-диметилфенокси)ацетиламино]гексеновой кислоты (Полупродукта 16, 4,6 г, 9,1 ммоль) в 3 мл СН2Cl2 по каплям при 0°С прибавляли ClSO3Н (3 мл, 5 экв., 45,5 ммоль). В ходе реакции отбирали небольшие аликвотные пробы реакционной массы в качестве образцов, обрабатывали льдом, экстрагировали этилацетатом и этилацетатные фракции анализировали методом тонкослойной хроматографии (ТСХ). Анализ методом ТСХ показал, что реакция завершилась через 30 минут. Затем полученную вязкую реакционную массу при интенсивном перемешивании выливали на колотый лед и получали смолистое вещество желтого цвета, сосредоточенное в основном на дне колбы. Эту смесь экстрагировали СН2Cl2, органические слои сушили над безводным MgSO4, концентрировали при пониженном давлении и получали Полупродукт 17 (2,2 г, 40%) в виде пены белого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(3-Нитробензолсульфонил)этиловый эфир 6-[2-(3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновой кислоты (Полупродукт 18) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (500 мг, 1,35 ммоль) в CH2Cl2 при комнатной температуре прибавляли NaH (40 мг, 1,65 ммоль) и получали прозрачный раствор. К полученному прозрачному раствору при комнатной температуре прибавляли 2-(3-нитробензолсульфонил)этиловый эфир 6-[2-(4-хлорсульфонил-3,5-диметилфенокси)ацетиламино]гексеновой кислоты (Полупродукт 17, 1,0 г, 1,65 ммоль, 1,2 экв.) в CH2Cl2 и смесь перемешивали в течение 4 часов. После этого добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали при пониженном давлении. Полученное масло очищали колоночной хроматографией (3% МеОН в CH2Cl2) и получали Полупродукт 18 (860 мг, 68%) в виде пены белого цвета. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 6-[2-(3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновой кислоты (Соединение 6) Раствор 2-(3-нитробензолсульфонил)этилового эфира 6-[2-(3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновой кислоты (Полупродукта 18, 420 мг, 0,45 ммоль) и NaHCO3 (42 мг, 0,49 ммоль) в смеси ТГФ-Н2О (6 мл – 3 мл) нагревали при температуре 70°С в течение 1 часа. Затем удаляли летучие вещества, оставшееся смолистое масло растворяли в СН2Cl2 и полученный раствор фильтровали от нерастворимых твердых частиц. Фильтрат концентрировали досуха, полутвердый остаток обрабатывали диэтиловым эфиром и получали кристаллы, которые кратковременно обрабатывали CH3CN. Осадок из суспензии в CH3CN отфильтровывали и получали Соединение 6 (170 мг, 50%) в виде кристаллического вещества белого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц)
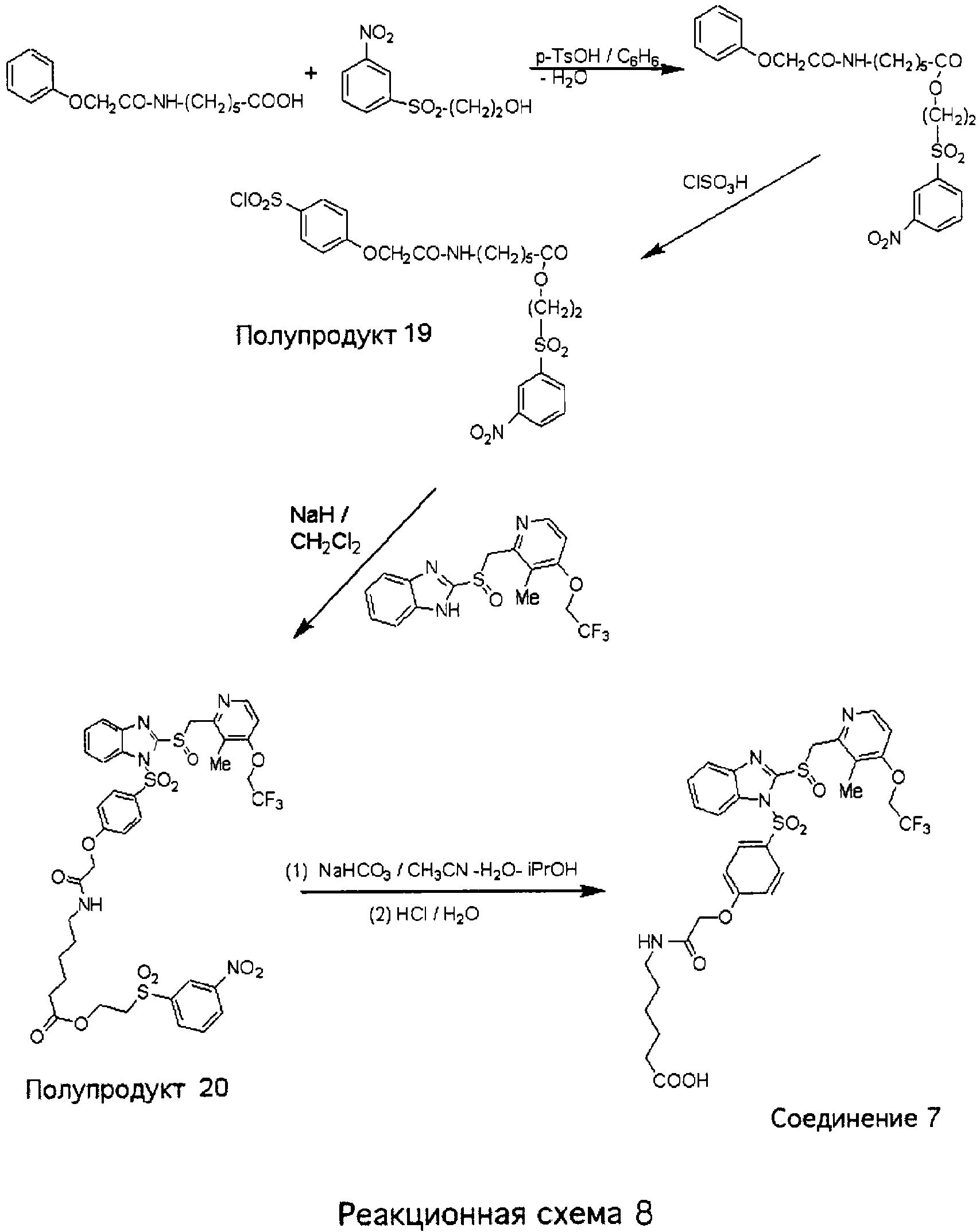
2-(3-Нитробензолсульфонил)этиловый эфир 6-[2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновой кислоты (Полупродукт 20) Смесь 6-(2-феноксиацетиламино)-н-гексеновой кислоты (синтезированной аналогично Полупродукту 15, 3,15 г, 11,9 ммоль, 1,1 экв.), 2-(м-нитробензолсульфонил)этанола (2,5 г, 10,8 ммоль) и гидрата п-толуолсульфоновой кислоты (0,5 г) в 100 мл бензола кипятили в течение ночи с насадкой Дина-Старка. После этого добавляли воду, и смесь экстрагировали СН3Cl2. Объединенные органические слои промывали насыщенным раствором NaHCO3, сушили над MgSO4 и концентрировали при пониженном давлении. Маслянистый остаток очищали колоночной хроматографией (1% МеОН в CH2Cl2) и получали 4,6 г (89%) 2-(3-нитробензолсульфонил)этилового эфира 6-(2-феноксиацетиламино)-н-гексеновой кислоты. К охлажденному раствору 2-(3-нитробензолсульфонил)этилового эфира 6-(2-феноксиацетиламино)-н-гексеновой кислоты (4,5 г, 9,4 ммоль) в 10 мл CH2Cl2 при 0°С по каплям прибавляли ClSO3Н (3,3 мл, 5,5 г, 5 экв., 47,0 ммоль). Реакция хлорсульфонилирования протекала за 30 минут. Вязкую реакционную массу при интенсивном перемешивании выливали на измельченный лед, в результате чего в основном на дне колбы осаждалось смолистое вещество желтого цвета. Смесь экстрагировали CH2Cl2, органические слои сушили над безводным сульфатом магния, концентрировали при пониженном давлении и получали сиропообразное вещество, которое очищали колоночной хроматографией (от CH2Cl2 до 1% МеОН в CH2Cl2) и получали 2-(3-нитробензолсульфонил)этиловый эфир 6-[2-(4-хлорсульфонилфенокси)ацетиламино]-н-гексеновой кислоты (Полупродукт 19, 3,3 г, 60%) в виде пены белого цвета. 2-(3-Нитробензолсульфонил)этиловый эфир 6-[2-(4-хлорсульфонилфенокси)ацетиламино]-н-гексеновой кислоты (Полупродукт 19, 1,0 г, 1,73 ммоль, 1,27 экв.) в CH2Cl2 (5 мл) при комнатной температуре прибавляли к гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (500 мг, 1,36 ммоль) в СН2Cl2 (10 мл) и NaH (40 мг, 1,65 ммоль) и массу перемешивали в течение 2 часов. После этого добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали при пониженном давлении. Маслянистый остаток очищали колоночной хроматографией (от СН2Cl2 до 3% МеОН в СН2Cl2) и получали 2-(3-нитробензолсульфонил)этиловый эфир 6-[2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновой кислоты (Полупродукт 20, 1,15 г, 94%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) 6-[2-(4-{2-[3-Метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновая кислота (Соединение 7) Раствор 2-(3-нитробензолсульфонил)этилового эфира 6-[2-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]гексеновой кислоты (Полупродукт 20, 450 мг, 0,50 ммоль) и NaHCO3 (50 мг, 0,60 ммоль, 1,2 экв.) в смеси CH3CN-H2O-i-PrOH (2 мл : 1 мл : 1 мл) нагревали при температуре 70°С в течение 1,5 часов. После этого отгоняли летучие вещества, оставшееся смолистое масло растворяли в СН2Cl2 и смесь фильтровали от нерастворимых твердых частиц. Фильтрат сушили и концентрировали при пониженном давлении. Маслянистый остаток очищали на колонке с силикагелем и получали 150 мг Соединения 7 в виде пены светло-коричневого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц)
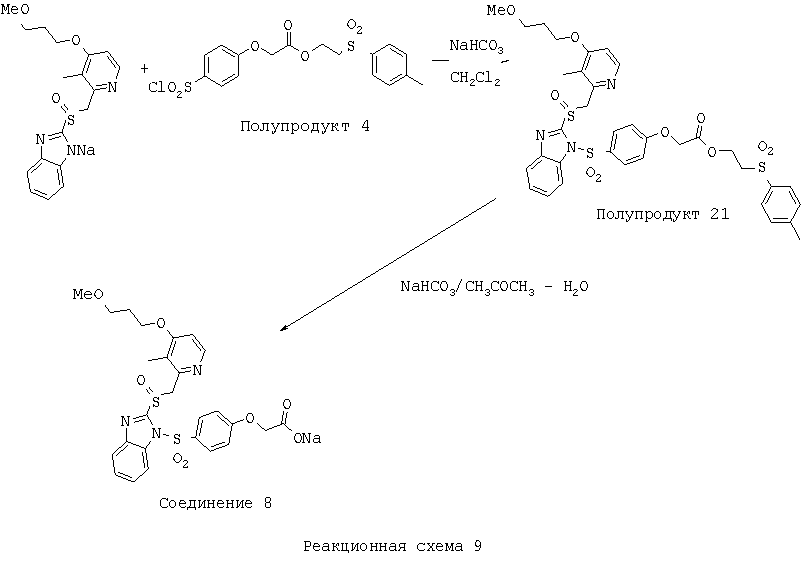
2-(Толуол-4-сульфонил)этиловый эфир (4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 21) К раствору натриевой соли 2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]-1Н-бензимидазола (760 мг, 2,0 ммоль) в 10 мл CH2Cl2 прибавляли 2-(толуол-4-сульфонил)этиловый эфир (4-хлорсульфонилфенокси)уксусной кислоты (Полупродукт 4, 1,04 г, 2,4 ммоль, 1,2 экв.). В реакционную массу также добавляли твердый NaHCO3 (1 г). Реакционную массу перемешивали в течение 8 часов при комнатной температуре, после чего отфильтровывали твердый NaHCO3. Дихлорметановые фракции упаривали при пониженном давлении и получали маслянистый остаток, который очищали колоночной хроматографией (силикагель, от CH2Cl2 до 3% МеОН в СН2Cl2) и получали Полупродукт 21 (1,4 г, 92%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль (4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Соединение 8) 2-(Толуол-4-сульфонил)этиловый эфир (4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 21, 400 мг, 0,53 ммоль) растворяли в 6 мл ацетона и прибавляли раствор NaHCO3 (50 мг, 0,597 ммоль, 1,1 экв.) в 4 мл Н2О. Смесь нагревали при температуре 70°С в течение 2 часов. После этого в вакууме отгоняли летучие вещества, маслянистый остаток растворяли в EtOAc-i-PrOH (5:1) и смесь фильтровали от нерастворимых примесей. Фильтрат концентрировали, остаток сушили в вакууме и получали пену белого цвета с желтоватым оттенком. Пену отмывали этилацетатом от побочного продукта (винилтолуолсульфона) и получали натриевую соль (4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Соединение 8, 300 мг) в виде кристаллов белого цвета с желтоватым оттенком. 1Н ЯМР (ДМСО-d6, 400 МГц)
Смесь 2-(толуол-4-сульфонил)этилового эфира {4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Полупродукта 22) и 2-(толуол-4-сульфонил)этилового эфира {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Полупродукт 23) К гетерогенному раствору 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (840 мг, 2,44 ммоль) в 20 мл СН2Cl2 при комнатной температуре прибавляли 90 мг NaH (3,75 ммоль, 1,5 экв.) и получали гомогенную смесь. К полученной прозрачной реакционной массе прибавляли измельченный 2-(толуол-4-сульфонил)этиловый эфир (4-хлорсульфонилфенокси)уксусной кислоты (Полупродукт 4, 1,26 г, 2,92 ммоль, 1,2 экв.). В реакционную смесь также добавляли твердый NaHCO3 (1 г). Реакционную массу перемешивали в течение 8 часов при комнатной температуре, после чего твердый NaHCO3 отфильтровывали. Дихлорметановые фракции упаривали при пониженном давлении до маслянистого остатка, который очищали колоночной хроматографией (силикагель, от CH2Cl2 до 4% МеОН в СН2Cl2) и получали смесь Полупродукта 22 и Полупродукта 23 (1,6 г, 88%; отношение 5- и 6-изомеров 1:1) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли {4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Соединение 9) и натриевой соли {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Соединение 10) Смесь 2-(толуол-4-сульфонил)этилового эфира {4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Полупродукта 22) и 2-(толуол-4-сульфонил)этилового эфира {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Полупродукта 23), 2,2 г (2,97 ммоль), растворяли в 20 мл ацетонитрила и прибавляли раствор NaHCO3 (250 мг, 2,97 ммоль) в 10 мл Н2О. Полученную смесь нагревали при температуре 60°С в течение 3 часов. После этого реакционную массу концентрировали приблизительно до 10 мл при пониженном давлении, после чего концентрат отмывали этилацетатом от побочного продукта (винилтолуолсульфона). При необходимости для более четкого деления двух слоев добавляли 1 мл концентрированного раствора NaCl. Водный слой сушили в глубоком вакууме и получали пену белого цвета с желтоватым оттенком. Твердую пену экстрагировали хлороформом. Хлороформенные экстракты концентрировали при пониженном давлении, обрабатывали этилацетатом, выдерживали в течение 2 часов при 0°С и получали осадок. Осадок отфильтровывали, сушили и получали 1,37 г изомерной смеси Соединения 9 (натриевая соль) и Соединения 10 (натриевая соль) (отношение 5- и 6-изомеров 1:1) в виде кристаллического вещества белого цвета. 1Н ЯМР (D2O, 400 МГц)
4-(3-Изопропилфеноксиацетамидо)масляная кислота (Полупродукт 24) Смесь 3-изопропилфеноксиуксусной кислоты (полученной реакцией 3-изопропилфенола с хлоруксусной кислотой в присутствии гидроксида натрия, 5,0 г, 25,8 ммоль) и SOCl2 (5,0 мл, 8,2 г, 68,8 ммоль) в 10 мл бензола кипятили с обратным холодильником в течение 2 часов. После этого в вакууме отгоняли летучие вещества и получали хлорангидрид 3-изопропилфеноксиуксусной кислоты в виде масла светло-коричневого цвета. К охлажденному на ледяной бане раствору 4-аминомасляной кислоты (5,3 г, 51,6 ммоль, 2,0 экв.) и NaOH (2,0 г, 51,6 ммоль) в 60 мл Н2О и 80 мл СН3CN по каплям прибавляли хлорангидрид кислоты в 40 мл CH3CN и NaHCO3 (3,3 г, 38,7 ммоль, 1,5 экв.) в 50 мл Н2O. Полученную смесь интенсивно перемешивали в течение 5 часов. Затем при пониженном давлении отгоняли основную часть СН3CN, смесь подкисляли при комнатной температуре концентрированной HCl до рН 2. Выпавший осадок белого цвета отфильтровывали, промывали сначала Н2O, затем гексаном, сушили в глубоком вакууме и получали Полупродукт 24 (6,6 г, 92%) в виде кристаллического вещества белого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(3-Нитробензолсульфонил)этиловый эфир 4-(3-изопропилфеноксиацетамидо)масляной кислоты (Полупродукт 25) Смесь 4-(3-изопропилфеноксиацетамидо)масляной кислоты (Полупродукта 24, 3,1 г, 11,1 ммоль, 1,1 экв.), 2-(3-нитробензолсульфонил)этанола (2,3 г, 10,0 ммоль) и TsOH·H2O (0,5 г) в 100 мл бензола нагревали в течение ночи при температуре кипения с насадкой Дина-Старка. После этого смесь концентрировали упариванием, полученное масло очищали колоночной хроматографией (от CH2Cl2 до 1% МеОН в СН2Cl2) и получали Полупродукт 25 (4,8 г, 97%) в виде масла светло-желтого цвета, кристаллизующегося при хранении. 1Н ЯМР (CDCl3, 400 МГц) 2-(3-Нитробензолсульфонил)этиловый эфир 4-(4-хлорсульфонил-3-изопропилфеноксиацетамидо)масляной кислоты (Полупродукт 26) К смеси 2-(3-нитробензолсульфонил)этилового эфира 4-(3-изопропилфеноксиацетамидо)масляной кислоты (Полупродукт 25, 4,8 г, 9,75 ммоль) и 2 мл СН2Cl2 при 0°С по каплям при интенсивном перемешивании прибавляли ClSO3Н (3,3 мл, 5,8 г, 50 ммоль). Массу перемешивали при 0°С в течение 40 минут и затем при интенсивном перемешивании выливали на колотый лед, при этом образовывалась вязкая масса. Образовавшуюся массу экстрагировали СН2Cl2. Органические слои сушили и концентрировали при пониженном давлении. Маслянистый остаток пропускали через короткую колонку с силикагелем для извлечения вещества, которое не движется на ТСХ. Элюент концентрировали и получали Полупродукт 26 (3,3 г, 57%) в виде пены светло-коричневого цвета. 2-(3-Нитробензолсульфонил)этиловый эфир 4-[2-(3-изопропил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]масляной кислоты (Полупродукт 27) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (500 мг, 1,36 ммоль) в CH2Cl2 (10 мл) при комнатной температуре прибавляли NaH (40 мг, 1,65 ммоль) и получали прозрачный раствор. К полученному прозрачному раствору при комнатной температуре прибавляли 2-(3-нитробензолсульфонил)этиловый эфир 4-(4-хлорсульфонил-3-изопропилфеноксиацетамидо)масляной кислоты (Полупродукт 26, 1,0 г, 1,70 ммоль, 1,25 экв.) в СН2Cl2 (5 мл) и затем массу перемешивали в течение 2 часов. После этого добавляли воду, смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали при пониженном давлении. Маслянистый остаток очищали колоночной хроматографией (от CH2Cl2 до 3% МеОН в CH2Cl2) и получали Полупродукт 27 (1,0 г, 80%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 4-[2-(3-изопропил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]масляной кислоты (Соединение 11) Раствор 2-(3-нитробензолсульфонил)этилового эфира 4-[2-(3-изопропил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]масляной кислоты (Полупродукта 27, 1,0 г, 1,08 ммоль) и NaHCO3 (109 мг, 1,30 ммоль, 1,2 экв.) в смеси ТГФ-Н2O (6 мл – 3 мл) нагревали при температуре 70°С в течение 0,5 часа. После этого отгоняли летучие вещества, маслянистый остаток растворяли в CH2Cl2 и смесь фильтровали от нерастворившихся твердых частиц. Фильтрат упаривали при пониженном давлении и полутвердый остаток обрабатывали смесью эфир-EtOAc (10:1) с образованием кристаллов, которые в свою очередь кратковременно обрабатывали СН3CN. Осадок из суспензии в CH3CN отфильтровывали и получали Соединение 11 (550 мг, 69%) в виде кристаллов белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц)
2-Бензолсульфонилэтиловый эфир [2-(2-бензолсульфонилэтоксикарбонил-метокси)фенокси]уксусной кислоты (Полупродукт 28) Смесь 1,2-фенилендиоксидиуксусной кислоты (доступной из Aldrich, 3,0 г, 13,3 ммоль), 2-(фенилсульфонил)этанола (5,0 г, 26,5 ммоль) и n-TsOH·H2O (0,5 г) в 100 мл бензола кипятили в течение ночи с насадкой Дина-Старка. После этого смесь концентрировали упариванием при пониженном давлении, добавляли воду и экстрагировали CH2Cl2. Объединенные органические фракции промывали насыщенным раствором NaHCO3 и водой, сушили и концентрировали при пониженном давлении. Полученный маслянистый остаток очищали на короткой хроматографической колонке (силикагель, CH2Cl2) и получали Полупродукт 28 (7,4 г, 99%) в виде масла светло-коричневого цвета. 1H ЯМР (CDCl3, 400 МГц) 2-Бензолсульфонилэтиловый эфир [2-(2-бензолсульфонилэтоксикарбонилметокси)-4-хлорсульфонилфенокси]уксусной кислоты (Полупродукт 29) К раствору 2-бензолсульфонилэтилового эфира [2-(2-бензолсульфонилэтоксикарбонилметокси)фенокси]уксусной кислоты (Полупродукта 28, 7,4 г, 13,2 ммоль) в 10 мл CH2Cl2 при 0°С по каплям при интенсивном перемешивании прибавляли ClSO3Н (5,0 мл, 8,8 г, 75,8 ммоль, 5,7 экв.). Массу перемешивали при 0°С в течение 1,5 часов и затем при интенсивном перемешивании выливали на колотый лед, при этом образовывалась вязкая масса. Образовавшуюся массу экстрагировали CH2Cl2. Органические слои сушили и концентрировали при пониженном давлении. Маслянистый остаток сушили в вакууме в течение ночи и получали Полупродукт 29 (8,0 г, 92%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) 2-Бензолсульфонилэтиловый эфир (2-(2-бензолсульфонилэтоксикарбонилметокси)-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукт 30) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазола (лансопразола) (500 мг, 1,35 ммоль) в CH2Cl2 (10 мл) при комнатной температуре прибавляли NaH (40 мг, 1,63 ммоль) и получали прозрачный раствор. К полученной прозрачной смеси при комнатной температуре прибавляли 2-бензолсульфонилэтиловый эфир [2-(2-бензолсульфонилэтоксикарбонилметокси)-4-хлорсульфонилфенокси]уксусной кислоты (Полупродукт 29, 1,0 г, 1,63 ммоль, 1,2 экв.) в CH2Cl2 (5 мл) и массу перемешивали в течение 3 часов. После этого добавляли воду, смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали при пониженном давлении. Маслянистый остаток очищали колоночной хроматографией (от CH2Cl2 до 2% МеОН в CH2Cl2) и получали Полупродукт 30 (1,05 г, 78%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Динатриевая соль (2-карбоксиметокси-4- {2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Соединение 12) Раствор 2-бензолсульфонилэтилового эфира (2-(2-бензолсульфонилэтоксикарбонилметокси)-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Полупродукта 30, 500 мг, 0,50 ммоль) и NaHCO3 (90 мг, 1,10 ммоль, 2,2 экв.) в смеси ТГФ-Н2О (6 мл -3 мл) нагревали при температуре 70°С в течение 2 часов. После этого при пониженном давлении отгоняли летучие вещества и полутвердый остаток кратковременно обрабатывали смесью MeOH-CH2Cl2 (1:1). Полученные кристаллы отфильтровывали и получали Соединение 12 (300 мг, 74%) в виде кристаллов белого цвета с желтоватым оттенком. 1H ЯМР (ДМСО-d6, 400 МГц)
N-(3,5-Диметилфеноксиацетил)-L-глутаминовая кислота (Полупродукт 31) К раствору 3,5-диметилфеноксиуксусной кислоты (9,0 г, 50 ммоль) в бензоле (100 мл) прибавляли хлористый тионил (10,89 мл). Полученную смесь кипятили с обратным холодильником в течение 2 часов, после чего бензол отгоняли. Остаток сушили в вакууме и получали 3,5-диметилфеноксиацетилхлорид, который растворяли в 20 мл ацетонитрила и использовали in situ. L-Глутаминовую кислоту (8,8 г, 60 ммоль) растворяли в растворе карбоната натрия (12,72 г, 150 мл, 120 ммоль) и медленно прибавляли раствор 3,5-диметилфеноксиацетилхлорида в ацетонитриле, поддерживая рН 8 периодическим прибавлением раствора карбоната натрия. Реакционную массу перемешивали при комнатной температуре в течение 2 часов и после подкисления до рН 2 получали кристаллический осадок. Осадок отфильтровывали, обрабатывали горячим бензолом, снова фильтровали, промывали бензолом и получали Полупродукт 31 (9,2 г). 1Н ЯМР (ДМСО-d6, 400 МГц) бис-[2-(3-Нитрофенилсульфонил)этиловый]эфир 3,5-диметилфеноксиацетилглутаминовой кислоты (Полупродукт 32) Смесь N-(3,5-диметилфеноксиацетил)-L-глутаминовой кислоты (Полупродукта 31, 3,0 г, 10,0 ммоль), 2-(3-нитрофенилсульфонил)этанола (4,6 г, 20,0 ммоль) и п-TsOH·H2O (0,5 г) в 100 мл бензола кипятили в течение ночи с насадкой Дина-Старка. После этого смесь концентрировали при упаривании, добавляли воду и экстрагировали СН2Cl2. Объединенные органические фракции промывали насыщенным раствором NaHCO3 и водой, сушили и концентрировали упариванием при пониженном давлении. Маслянистый остаток очищали хроматографией на короткой колонке (силикагель, СН2Cl2) и получали Полупродукт 32 (4,6 г, 62%) в виде масла светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) бис-[2-(3-Нитробензолсульфонил)этиловый] эфир 2-[2-(4-хлорсульфонил-3,5-диметилфенокси)ацетиламино]глутаровой кислоты (Полупродукт 33) К смеси бис-[2-(3-нитрофенилсульфонил)этилового] эфира 3,5-диметилфеноксиацетилглутаминовой кислоты (Полупродукта 32, 4,4 г, 5,99 ммоль) и 10 мл СН2Cl2 при 0°С по каплям при интенсивном перемешивании прибавляли ClSO3Н (2,0 мл, 3,5 г, 29,9 ммоль, 5,0 экв.). Массу перемешивали при 0°С в течение 1 часа, после чего выливали при интенсивном перемешивании на колотый лед и получали вязкую массу. Полученную массу экстрагировали CH2Cl2. Органические фракции сушили и концентрировали при пониженном давлении. Маслянистый остаток сушили в вакууме в течение ночи и получали Полупродукт 33 (2,5 г, 44%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) бис-[2-(3-Нитробензолсульфонил)этиловый] эфир 2-[2-(3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]глутаровой кислоты (Полупродукт 34) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазола (лансопразола) (550 мг, 1,49 ммоль) в СН2Cl2 (10 мл) при комнатной температуре прибавляли NaH (43 мг, 1,80 ммоль) и получали прозрачный раствор. К полученному прозрачному раствору при комнатной температуре прибавляли бис-[2-(3-нитробензолсульфонил)этиловый] эфир 2-[2-(4-хлорсульфонил-3,5-диметилфенокси)ацетиламино]глутаровой кислоты (Полупродукт 33, 1,5 г, 1,80 ммоль, 1,2 экв.) в CH2Cl2 (5 мл), и образовавшуюся реакционную массу перемешивали в течение 4 часов. После этого добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали упариванием при пониженном давлении. Масло очищали колоночной хроматографией (от CH2Cl2 до 2% МеОН в СН2Cl2) и получали Полупродукт 34 (900 мг, 52%) в виде пены светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 2-[2-(3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]глутаровой кислоты (Соединение 13) Раствор бис-[2-(3-нитробензолсульфонил)этилового] эфира 2-[2-(3,5-диметил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)ацетиламино]глутаровой кислоты (Полупродукт 34, 800 мг, 0,69 ммоль) и NaHCO3 (120 мг, 1,41 ммоль, 2,05 экв.) в смеси ТГФ-Н2O (6 мл – 3 мл) нагревали при температуре 70°С в течение 3 часов. После этого при пониженном давлении отгоняли летучие вещества, полутвердый остаток обрабатывали EtOAc и смесь фильтровали от нерастворимых примесей. Фильтрат концентрировали до общего объема приблизительно 3 мл и затем к смеси прибавляли диэтиловый эфир. Выпавший осадок отфильтровывали и получали Соединение 13 (400 мг, 73%) в виде кристаллов белого цвета с желтоватым оттенком. 1Н ЯМР (ДМСО-d6, 400 МГц)
Смесь 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 35) и 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукт 36) 2-Метокси-5-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол (1,5 г, 4,35 ммоль) и 2-(n-толуолсульфонил)этил-5-хлорсульфонил-2-метоксибензоат (Полупродукт 7, 2,2 г, 5,09 ммоль) растворяли в ди(изопропил)метиламине (4 мл) и дихлорметане (20 мл). Реакционную массу перемешивали при комнатной температуре в течение 6 часов. Прибавляли дихлорметан (100 мл) и реакционную массу промывали водой. Дихлорметановый слой сушили над безводным сульфатом магния, концентрировали при пониженном давлении и получали сироп, который очищали хроматографией на колонке с силикагелем (элюент: 2% МеОН в дихлорметане). Изомерную смесь 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 35) и 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 36), 2,96 г (92%), получали в виде пены белого цвета. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 2-метокси-5-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединение 14) и натриевая соль 2-метокси-5-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединение 15) Смесь 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 35) и 2-(толуол-4-сульфонил)этилового эфира 2-метокси-5-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 36) (2,8 г, 3,78 ммоль) растворяли в ацетонитриле (28 мл) и прибавляли водный раствор бикарбоната натрия (350 мг, 4,15 ммоль, 1,1 экв.) в воде (14 мл). Реакционную массу перемешивали при температуре 60°С в течение 2 часов, затем концентрировали при пониженном давлении приблизительно до 14 мл. Концентрат экстрагировали этилацетатом (25 мл) и водный слой лиофилизировали в вакууме. Остаток растворяли в дихлорметане и фильтровали. Фильтрат концентрировали приблизительно до 5 мл, затем добавляли этилацетат (50 мл). Смесь выдерживали при температуре – 20°С в течение 2 часов, после чего выпавший за это время осадок отфильтровывали. Получали смесь (1:1) натриевой соли 2-метокси-5-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединения 14) и натриевой соли 2-метокси-5-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил] бензойной кислоты (Соединения 15), 1,6 г (72%). Вторую порцию продукта, 0,15 г, выделяли из фильтрата после его концентрации и кристаллизации. 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир {2-[2-(толуол-4-сульфонил)этокси-карбонилметокси]фенокси}уксусной кислоты (Полупродукт 37) 1,2-Фенилендиоксидиуксусную кислоту (доступную из Aldrich, 5 г, 22,1 ммоль) и 2-(п-толилсульфонил)этанол (доступный из Aldrich, 8,8 г, 44,2 ммоль) добавляли к толуолу (100 мл). Прибавляли каталитическое количество гидрата п-толуолсульфоновой кислоты (0,5 г) и реакционную массу кипятили с обратным холодильником с отгонкой воды, используя насадку Дина-Старка. После 6 часов кипячения отгоняли толуол. Остаток растворяли в дихлорметане (250 мл), промывали водой (200 мл) и 6N раствором бикарбоната натрия (150 мл). Дихлорметановый слой сушили над безводным сульфатом магния, концентрировали при пониженном давлении и получали 13 г (99%) 2-(толуол-4-сульфонил)этилового эфира {2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 37). 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир {4-хлорсульфонил-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 38) 2-(Толуол-4-сульфонил)этиловый эфир {2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 37) (13 г, 22 ммоль) прибавляли к дихлорметану (30 мл) и охлаждали на ледяной бане. Медленно прибавляли хлорсульфоновую кислоту (10 мл) и реакционную массу перемешивали в течение 2 часов при 0°С, затем в течение 1 часа при комнатной температуре. После этого реакционную массу при перемешивании выливали на колотый лед (200 г). Осадки экстрагировали дихлорметаном. Дихлорметановый слой сушили над безводным сульфатом магния и концентрировали, остаток сушили в вакууме и получали 17,3 г 2-(толуол-4-сульфонил)этилового эфира {4-хлорсульфонил-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 38). 1Н ЯМР (CDCl3, 400 МГц) Смесь 2-(толуол-4-сульфонил)этилового эфира {4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 39) и 2-(толуол-4-сульфонил)этилового эфира {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 40) 2-(Толуол-4-сульфонил)этиловый эфир {4-хлорсульфонил-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукт 38) (7,6 г, 11 ммоль) и 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол (3,5 г, 10 ммоль) прибавляли к раствору, состоящему из дихлорметана (50 мл) и триэтиламина (6 мл). Реакционную массу перемешивали при комнатной температуре в течение 6 часов. Добавляли дихлорметан (200 мл) и дихлорметановый слой промывали водой (200 мл). Дихлорметановый слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Концентрат очищали хроматографией на колонке с силикагелем и получали 6,7 г смеси 2-(толуол-4-сульфонил)этилового эфира {4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукта 39) и 2-(толуол-4-сульфонил)этилового эфира {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукта 40). 1Н ЯМР (CDCl3, 400 МГц) Динатриевая соль {2-карбоксиметокси-4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Соединение 16) и динатриевая соль {2-карбоксиметокси-4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Соединение 17) Смесь 2-(толуол-4-сульфонил)этилового эфира {4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукта 39) и 2-(толуол-4-сульфонил)этилового эфира {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]фенокси}уксусной кислоты (Полупродукта 40) (6,5 г, 6,5 ммоль) растворяли в ацетонитриле (50 мл) и прибавляли раствор бикарбоната натрия (1,15 г, 13,7 ммоль) в воде (30 мл). Реакционную массу перемешивали при температуре 60°С в течение 5 часов. Затем реакционную массу концентрировали при пониженном давлении приблизительно до 30 мл и промывали этилацетатом. Водный слой лиофилизировали и остаток экстрагировали хлороформом (200 мл). Хлороформенные экстракты фильтровали и концентрировали приблизительно до 7 мл. К концентрату прибавляли этилацетат до появления осадка белого цвета. Суспензию выдерживали в течение 3 часов при 0°С, после чего осадок отфильтровывали. Получали смесь (1:1) динатриевой соли {2-карбоксиметокси-4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Соединения 16) и динатриевой соли {2-карбоксиметокси-4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Соединения 17), 3,1 г. 1Н ЯМР (D2O, 400 МГц)
Смесь 2-(толуол-4-сульфонил)этилового эфира 4-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (Полупродукта 41) и 2-(толуол-4-сульфонил)этилового эфира 4-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (Полупродукта 42) К гетерогенному раствору натриевой соли 5-дифторметокси-2-[(3,4-диметоксипиридин-2-ил)метансульфинил]-1Н-бензимидазола (4,3 г, 10,0 ммоль) и 6 мл Et3N в 50 мл CH2Cl2 прибавляли хлорсульфониловый эфир (Полупродукт 4, 5,0 г, 11,5 ммоль, 1,15 экв.). После полного растворения хлорсульфонилового эфира в реакционную массу добавляли около 2 г твердого NaHCO3. (Без добавления NaHCO3 смесь после завершения реакции почернеет). После этого отгоняли растворитель, а маслянистый остаток очищали колоночной хроматографией (силикагель, от CH2Cl2 до 2% МеОН в CH2Cl2) и получали 7,8 г (99%) смеси 2-(толуол-4-сульфонил)этилового эфира 4-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты и 2-(толуол-4-сульфонил)этилового эфира 4-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (Полупродукты 41 и 42; отношение 1:1) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 4-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (Соединение 18) и натриевой соли 4-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (Соединение 19) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 4-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты и 2-(толуол-4-сульфонил)этилового эфира 4-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (Полупродукты 41 и 42, 7,7 г, 9,9 ммоль) в 70 мл СН3CN прибавляли раствор NaHCO3 (0,94 г, 11,1 ммоль, 1,2 экв.) в 35 мл Н2О. Смесь нагревали при температуре 65°С в течение 5 часов. После этого в вакууме отгоняли все летучие вещества, смесь промывали EtOAc и затем водный слой лиофилизировали в течение ночи. Осадок растворяли в СН2Cl2, после чего смесь фильтровали от нерастворимых примесей. Фильтрат концентрировали и полученное масло растворяли в 20 мл EtOAc. К смеси прибавляли диэтиловый эфир до выпадения осадка белого цвета. Осадок отфильтровывали, промывали эфиром, сушили в вакууме и получали 4,5 г (73%) смеси натриевой соли 4-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты и натриевой соли 4-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]феноксиуксусной кислоты (отношение 1:1) в виде кристаллов белого цвета с желтоватым оттенком. 1Н ЯМР (ДМСО-d6, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3-хлорсульфонилбензойной кислоты (Полупродукт 43) Гетерогенную смесь 3-хлорсульфонилбензойной кислоты (11,0 г, 50,0 ммоль) в 18 мл SOCl2 кипятили с обратным холодильником в течение 3 часов. После этого отгоняли избыток SOCl2, маслянистый остаток коричневого цвета растворяли в 60 мл CH3CN и прибавляли 2-(n-толилсульфонил)этанол (9,4 г, 47,0 ммоль, 0,95 экв.). Смесь нагревали при температуре кипения в течение 20 часов. После этого удаляли основную часть CH3CN, образовавшееся масло очищали хроматографией на короткой колонке (силикагель, СН2Cl2) и получали масло светло-коричневого цвета, которое дополнительно сушили в вакууме и получали 19,1 г (95%) кристаллов светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) Смесь 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 44) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 45) К гетерогенной смеси 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (омепразол, 1,0 г, 2,90 ммоль), 5 мл Et3N и приблизительно 1 г NaHCO3 в CH2Cl2 (20 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 43, 1,4 г, 3,48 ммоль, 1,2 экв.) в СН2Cl2, после чего массу перемешивали в течение 2 часов. После этого добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали. Масло очищали колоночной хроматографией (от СН2Cl2 до 1% МеОН в CH2Cl2) и получали 1,67 г (81%) смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 44) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 45) в виде пены белого цвета с желтоватым оттенком (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединения 20) и натриевой соли 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединения 21) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 44) и 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 45) (1,6 г, 2,25 ммоль) в 15 мл CH3CN при комнатной температуре прибавляли раствор NaHCO3 (225 мг, 2,7 ммоль, 1,2 экв.) в 8 мл Н2О и затем смесь нагревали при температуре 65°С в течение 2 часов. После этого удаляли основную часть CH3CN, смесь экстрагировали EtOAc и водный слой далее лиофилизировали в течение ночи. Полученные кристаллы растворяли в CH2Cl2 и смесь фильтровали от нерастворимых твердых частиц. Фильтрат концентрировали почти досуха. Полученный маслянистый остаток растворяли приблизительно в 2 мл CH2Cl2 и к смеси прибавляли EtOAc до появления осадка белого цвета. Выпавший осадок белого цвета отфильтровывали, промывали смесью EtOAc-эфир (3:1), сушили в вакууме и получали 900 мг (72%) смеси натриевой соли 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединения 20) и натриевой соли 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Соединения 21) в виде кристаллического вещества белого цвета (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (D2O, 400 МГц)
Смесь 2-(толуол-4-сульфонил)этилового эфира 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 46) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 47) К гетерогенной смеси натриевой соли 5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}-1Н-бензимидазола (пантапразола, натриевой соли, 1,0 г, 2,3 ммоль), 5 мл Et3N и приблизительно 1 г NaHCO3 в СН2Cl2 (15 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 43, 1,12 г, 2,77 ммоль, 1,2 экв.) в CH2Cl2, после чего массу перемешивали в течение 1,5 часов. Далее добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали. Полученное масло очищали колоночной хроматографией (от СН2Cl2 до 1% МеОН в СН2Cl2) и получали 1,0 г (58%) смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 46) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 47) в виде пены белого цвета с желтоватым оттенком (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Соединения 22) и натриевой соли 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Соединения 23) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 46) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 47) (1,0 г, 1,33 ммоль) в 10 мл CH3CN при комнатной температуре прибавляли раствор NaHCO3 (125 мг, 1,47 ммоль, 1,2 экв.) в 5 мл Н2О, затем смесь нагревали при температуре 65°С в течение 2,5 часов. После этого удаляли основную часть CH3CN, смесь экстрагировали EtOAc и водный слой лиофилизировали в течение ночи. Осадок растворяли в СН2Cl2 и смесь фильтровали от нерастворимых твердых частиц. Фильтрат концентрировали почти досуха и получали масло. Масло растворяли приблизительно в 2 мл СН2Cl2 и к раствору прибавляли смесь EtOAc-эфир (2:1) до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали смесью EtOAc-эфир (2:1), сушили в вакууме и получали 560 мг (71%) смеси натриевой соли 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Соединения 22) и натриевой соли 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]бензойной кислоты (Соединения 23) в виде кристаллического вещества белого цвета (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукт 48) К раствору натриевой соли 2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]-1Н-бензимидазола (рабепразола, натриевой соли, 1,0 г, 2,62 ммоль), 5 мл Et3N и приблизительно 1 г NaHCO3 в CH2Cl2 (15 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 43, 1,27 г, 3,15 ммоль, 1,2 экв.) в CH2Cl2, после чего массу перемешивали в течение 1,5 часов. Затем добавляли воду, смесь экстрагировали СН2Cl2, органические слои сушили и концентрировали. Полученное масло очищали колоночной хроматографией (от СН2Cl2 до 1% МеОН в CH2Cl2) и получали 1,5 г (76%) 2-(толуол-4-сульфонил)этилового эфира 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукта 48) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 3-[2-[4-(3-Метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил]бензойной кислоты (Соединение 24) К раствору 2-(толуол-4-сульфонил)этилового эфира 3-[2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил]бензойной кислоты (Полупродукта 48, 1,5 г, 2,0 ммоль) в 15 мл CH3CN при комнатной температуре прибавляли раствор NaHCO3 (200 мг, 2,4 ммоль, 1,2 экв.) в 7 мл H2O и затем смесь нагревали при температуре 65°С в течение 2,5 часов. После этого удаляли основную часть CH3CN, смесь экстрагировали EtOAc и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в СН2Cl2 и смесь фильтровали от нерастворимых твердых частиц. Фильтрат концентрировали почти досуха. Маслянистый остаток растворяли приблизительно в 2 мл СН2Cl2 и прибавляли смесь EtOAc-эфир (1:1) до выпадения осадка белого цвета. Выпавший осадок отфильтровывали, промывали смесью EtOAc-эфир (1:1), сушили в вакууме и получали 700 мг (60%) натриевой соли 3-[2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил]бензойной кислоты (Соединение 24) в виде кристаллического вещества белого цвета. 1Н ЯМР (D2O, 400 МГц)
2-(n-Толилсульфонил)этиловый эфир 3-хлорсульфонил-4-метилбензойной кислоты Полупродукт 49) Гетерогенную смесь 3-хлорсульфонил-4-метилбензойной кислоты (11,7 г, 50,0 ммоль) в 18 мл SOCl2 кипятили с обратным холодильником в течение 3 часов. После отгонки избытка SOCl2 маслянистый остаток коричневого цвета растворяли в 50 мл СН3СН и прибавляли 2-(n-толилсульфонил)этанол (9,4 г, 47,0 ммоль, 0,95 экв.). Смесь нагревали при температуре кипения в течение 24 часов. После этого удаляли основную часть СН3CN, образовавшееся масло очищали хроматографией на короткой колонке (от CH2Cl2 до 1% МеОН в CH2Cl2) и получали масло светло-коричневого цвета, которое закристаллизовалось при стоянии. Кристаллы дополнительно сушили в вакууме и получали 19,5 г (99%) 2-(p-толилсульфонил)этилового эфира 3-хлорсульфонил-4-метилбензойной кислоты (Полупродукт 49). 1Н ЯМР (CDCl3, 400 МГц) Смесь 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 50) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 51) К гетерогенной смеси 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (омепразола, 1,0 г, 2,90 ммоль), 5 мл Et3N и приблизительно 1 г NaHCO3 в CH2Cl2 (15 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 49, 1,45 г, 3,48 ммоль, 1,2 экв.) в CH2Cl2 (5 мл) и затем смесь перемешивали в течение 0,5 часа. После этого добавляли воду, смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали. Масло очищали колоночной хроматографией (от СН2Cl2 до 1% МеОН в CH2Cl2) и получали 1,5 г (71%) смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 50) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 51) (отношение между 5- и 6-изомерами 1:1) в виде пены светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 25) и натриевой соли 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 26) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 50) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 51) (1,5 г, (2,0 ммоль) в 15 мл СН3CN при комнатной температуре прибавляли раствор NaHCO3 (210 мг, 2,40 ммоль, 1,2 экв.) в 7 мл Н2О и затем смесь нагревали при температуре 65°С в течение 2,5 часов. После этого удаляли основную часть СН3CN, смесь экстрагировали EtOAc и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в CH2Cl2, раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали почти досуха. Маслянистый остаток растворяли приблизительно в 2 мл CH2Cl2 и к раствору прибавляли EtOAc до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали EtOAc, сушили в вакууме и получали 840 мг (74%) смеси натриевой соли 3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 25) и натриевой соли 3-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 26) (отношение между 5- и 6-изомерами 1:1) в виде кристаллического вещества белого цвета. 1Н ЯМР (D2O, 400 МГц)
3-{2-[3-Метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойная кислота 2-(толуол-4-сульфонил)этиловый эфир (Полупродукт 52) К гетерогенной смеси 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (лансопразола, 700 мг, 1,89 ммоль), 3 мл Et3N и приблизительно 1 г NaHCO3 в CH2Cl2 (15 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 49, 1,03 г, 2,47 ммоль, 1,3 экв.) в СН2Cl2 (5 мл) и затем массу перемешивали в течение 2 часов. После этого добавляли воду, смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали. Полученное масло очищали колоночной хроматографией (от СН2Cl2 до 1% МеОН в СН2Cl2) и получали 1,1 г (78%) 2-(толуол-4-сульфонил)этилового эфира 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Полупродукт 52) в виде пены желтого цвета. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Соединение 27) К раствору 2-(толуол-4-сульфонил)этилового эфира 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Полупродукта 52, 1,1 г, 1,46 ммоль) в 8 мл CH3CN при комнатной температуре прибавляли раствор NaHCO3 (160 мг, 1,91 ммоль, 1,3 экв.) в 4 мл Н2O и затем смесь нагревали при температуре 65°С в течение 2 часов. После этого удаляли основную часть СН3CN, смесь экстрагировали EtOAc и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в CH2Cl2, после чего смесь фильтровали от нерастворимых примесей. Фильтрат концентрировали почти досуха. Маслянистый остаток растворяли приблизительно в 2 мл СН2Cl2 и к смеси прибавляли EtOAc до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали EtOAc, сушили в вакууме и получали 540 мг (62%) натриевой соли 3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Соединение 27) в виде кристаллов светло-коричневого цвета. 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Полупродукт 53) К раствору натриевой соли 2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]-1Н-бензимидазола (рабепразола, натриевой соли, 1,0 г, 2,62 ммоль), 5 мл Et3N и приблизительно 1 г NaHCO3 в CH2Cl2 (15 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 49, 1,30 г, 3,15 ммоль, 1,2 экв.) CH2Cl2 (5 мл), после чего смесь перемешивали в течение 0,5 часа. После этого добавляли воду, смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали. Маслянистый остаток очищали колоночной хроматографией (от СН2Cl2 до 1% МеОН в CH2Cl2) и получали 1,6 г (80%) 2-(толуол-4-сульфонил)этилового эфира 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Полупродукт 53) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Соединение 28) К раствору 2-(толуол-4-сульфонил)этилового эфира 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Полупродукта 53, 1,5 г, 1,97 ммоль) в 15 мл CH3CN при комнатной температуре прибавляли раствор NaHCO3 (200 мг, 2,36 ммоль, 1,2 экв.) в 7 мл H2O, после чего смесь нагревали при температуре 65°С в течение 2 часов. После этого основную часть СН3CN удаляли, смесь экстрагировали EtOAc и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в СН2Cl2 и раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали почти досуха. Маслянистый остаток растворяли приблизительно в 2 мл CH2Cl2 и прибавляли смесь EtOAc-гексан (7:1) до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали смесью EtOAc-гексан (7:1), сушили в вакууме и получали 950 мг натриевой соли (80%) 3-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}-4-метилбензойной кислоты (Соединение 28) в виде кристаллического вещества белого цвета. 1Н ЯМР (D2O, 400 МГц)
Смесь 2-(толуол-4-сульфонил)этилового эфира 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 54) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 55) К гетерогенной смеси натриевой соли 5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}-1Н-бензимидазола (лансопразола, натриевой соли, 1,0 г, 2,30 ммоль), 2 мл Et3N и приблизительно 1 г NaHCO3 в СН2Cl2 (15 мл) при комнатной температуре прибавляли сульфонилхлорид (Полупродукт 49, 1,15 г, 2,76 ммоль, 1,2 экв.) в СН2Cl2 (5 мл) и затем массу перемешивали в течение 2 часов. После этого добавляли воду, смесь экстрагировали CH2Cl2, органические слои сушили и концентрировали. Масло очищали колоночной хроматографией (от CH2Cl2 до 1% МеОН в СН2Cl2) и получали 1,5 г (85%) смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 54) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 55) в виде пены светло-желтого цвета (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 29) и натриевой соли 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 30) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродуктя 54) и 2-(толуол-4-сульфонил)этилового эфира 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Полупродукта 55) 1,5 г (1,96 ммоль) в 10 мл CH3CN при комнатной температуре прибавляли раствор NaHCO3 (200 мг, 2,36 ммоль, 1,2 экв.) в 5 мл H2O и затем смесь нагревали при температуре 65°С в течение 2 часов. После этого основную часть CH3CN удаляли, смесь экстрагировали EtOAc и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в CH2Cl2, после чего смесь фильтровали от нерастворимых примесей. Фильтрат концентрировали почти досуха. Маслянистый остаток растворяли приблизительно в 2 мл CH2Cl2 и к раствору прибавляли смесь EtOAc-эфир (1:1) до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали смесью EtOAc-эфир (1:1), сушили в вакууме и получали 850 мг (72%) смеси натриевой соли 3-[5-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 29) и натриевой соли 3-[6-дифторметокси-2-{(3,4-диметоксипиридин-2-ил)метансульфинил}бензимидазол-1-сульфонил]-4-метилбензойной кислоты (Соединения 30) в виде кристаллического вещества белого цвета (отношение между 5- и 6-изомерами 1:1). 1H ЯМР (D2O, 400 МГц)
Смесь натриевой соли 3,5-диметил-4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]феноксиуксусной кислоты (Соединения 31) и натриевой соли 3,5-диметил-4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]феноксиуксусной кислоты (Соединения 32) К гетерогенному раствору 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (омепразола, 4,0 г, 11,6 ммоль) и 15 мл Et3N в 80 мл CH2Cl2 прибавляли хлорсульфониловый эфир (Полупродукт 13, 6,94 г, 15,0 ммоль, 1,3 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли около 2 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 2 часов. После этого отгоняли растворитель, а маслянистый остаток для очистки от примесей пропускали через короткую колонку (силикагель, СН2Cl2) и получали смесь Полупродуктов 56 и 57 в виде масла коричневого цвета. Масло (Полупродукты 56 и 57, 9,5 г) растворяли в 65 мл CH3CN и прибавляли раствор NaHCO3 (1,07 г, 12,7 ммоль, 1,1 экв.) в 35 мл Н2O. Смесь нагревали при температуре 65°С в течение 2 часов. После этого в вакууме удаляли все летучие вещества, смесь промывали EtOAc (2x) и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 150 мл СН2Cl2 и раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали, полученное масло растворяли в 10 мл СН2Cl2. К смеси прибавляли EtOAc до появления осадка белого цвета. Осадок отфильтровывали, промывали EtOAc, сушили в вакууме и получали 5,1 г (72%) смеси натриевой соли 3,5-диметил-4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]феноксиуксусной кислоты (Соединения 31) и натриевой соли 3,5-диметил-4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]феноксиуксусной кислоты (Соединения 32) в виде кристаллического вещества белого цвета (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3,5-диметил-4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}феноксиуксусной кислоты (Полупродукт 58) К гетерогенному раствору натриевой соли 2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]-1Н-бензимидазола (рабепразола, натриевой соли, 1,0 г, 2,62 ммоль) и 5 мл Et3N в 20 мл CH2Cl2 прибавляли хлорсульфониловый эфир (Полупродукт 13, 1,57 г, 3,41 ммоль, 1,3 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли приблизительно 1 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 3 часов. После этого отгоняли растворитель, маслянистый остаток очищали колоночной хроматографией (силикагель, от СН2Cl2 до 1% МеОН в CH2Cl2) и получали 1,7 г (77%) 2-(толуол-4-сульфонил)этилового эфира 3,5-диметил-4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}феноксиуксусной кислоты (Полупродукт 58) в виде пены белого цвета. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 3,5-диметил-4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}феноксиуксусной кислоты (Соединение 33) К раствору 2-(толуол-4-сульфонил)этилового эфира 3,5-диметил-4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил} феноксиуксусной кислоты (Полупродукта 58, 1,6 г, 1,90 ммоль) в 10 мл CH3CN прибавляли раствор NaHCO3 (208 мг, 2,47 ммоль, 1,3 экв.) в 5 мл Н2O. Смесь нагревали при температуре 65°С в течение 3 часов. После этого в вакууме отгоняли летучие вещества, смесь промывали EtOAc (2x) и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 80 мл СН2Cl2 и затем раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали, маслянистый остаток растворяли в 5 мл СН2Cl2. К смеси прибавляли EtOAc до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали EtOAc, сушили в вакууме и получали 800 мг (62%) натриевой соли 3,5-диметил-4-{2-[4-(3-метоксипропокси)-3-метилпиридин-2-илметансульфинил]бензимидазол-1-сульфонил}феноксиуксусной кислоты (Соединение 33) в виде кристаллического вещества белого цвета. 1H ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3-(2-метоксифенил)пропионовой кислоты (Полупродукт 59) Смесь 3-(2-метоксифенил)пропионовой кислоты (5,0 г, 27,8 ммоль), 2-(п-толуолсульфонил)этанола (5,5 г, 27,8 ммоль) и гидрата п-толуолсульфоновой кислоты (п-TsOH·H2O) (0,5 г) в 100 мл толуола кипятили в течение 5 часов с насадкой Дина-Старка. Затем добавляли воду и смесь экстрагировали CH2Cl2. Объединенные органические слои промывали насыщенным раствором NaHCO3 (2х), сушили над MgSO4, концентрировали и получали 2-(толуол-4-сульфонил)этиловый эфир 3-(2-метоксифенил)пропионовой кислоты (Полупродукт 59, 5,5 г, 88%) в виде жидкости светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир 3-(5-хлорсульфонил-2-метоксифенил)пропионовой кислоты (Полупродукт 60) К смеси 2-(толуол-4-сульфонил)этилового эфира 3-(2-метоксифенил)пропионовой кислоты (Полупродукта 59, 5,0 г, 13,8 ммоль) и 5 мл CH2Cl2 при охлаждении на ледяной бане прибавляли по каплям хлорсульфоновую кислоту (8,0 г, 69,0 ммоль, 5,0 экв.). Массу перемешивали при 0°С в течение 30 минут. Полученное вязкое масло при интенсивном перемешивании выливали на колотый лед. Смесь экстрагировали EtOAc. Органический слой сушили над MgSO4, концентрировали при упаривании и получали вязкое масло, которое очищали колоночной хроматографией (CH2Cl2) и получали 2-(толуол-4-сульфонил)этиловый эфир 3-(5-хлорсульфонил-2-метоксифенил)пропионовой кислоты (Полупродукт 60, 4,7 г, 74%) в виде масла светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) Смесь 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 61) и 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 62) К гетерогенному раствору 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (омепразола, 1,7 г, 4,93 ммоль) и 6 мл Et3N в 20 мл СН2Cl2 прибавляли хлорсульфониловый эфир (Полупродукт 60, 2,8 г, 6,0 ммоль, 1,3 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли приблизительно 1 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 2 часов. После этого отгоняли растворитель и маслянистый остаток очищали колоночной хроматографией (силикагель, от CH2Cl2 до 2% МеОН в CH2Cl2) и получали смесь 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 61) и 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 62) (1,88 г, 50%) в виде пены белого цвета с желтоватым оттенком (отношение между 5-и 6-изомерами 1:1). 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 3-[2-метокси-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Соединения 34) и натриевой соли 3-[2-метокси-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Соединения 35) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 61) и 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 62), 1,7 г (2,21 ммоль), в 17 мл СН3CN прибавляли раствор NaHCO3 (205 мг, 2,43 ммоль, 1,1 экв.) в 8 мл Н2О. Смесь нагревали при температуре 65°С в течение 2 часов. После этого отгоняли в вакууме все летучие вещества, смесь промывали EtOAc (2x) и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 70 мл CH2Cl2, и раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали и маслянистый остаток растворяли в 3 мл CH2Cl2. К смеси прибавляли EtOAc до появления осадка белого цвета. Осадок отфильтровывали, промывали EtOAc, сушили в вакууме и получали 1,0 г (75%) смеси натриевой соли 3-[2-метокси-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Соединения 34) и натриевой соли 3-[2-метокси-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Соединения 35) в виде кристаллического вещества белого цвета (отношение между 5-и 6-изомерами 1:1). 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3-[2-метокси-5-{2-(3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукт 63) К гетерогенному раствору 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (лансопразола, 800 мг, 2,17 ммоль) и 3 мл Et3N в 20 мл СН2Cl2 прибавляли хлорсульфониловый эфир (Полупродукт 60, 1,2 г, 2,60 ммоль, 1,2 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли приблизительно 1 г твердого NaHCO3. Массу перемешивали при комнатной температуре 1 час.После этого отгоняли растворитель, маслянистый остаток очищали колоночной хроматографией (силикагель, от CH2Cl2 до 1% МеОН в CH2Cl2) и получали Полупродукт 63 (1,2 г, 70%) в виде пены белого цвета с желтоватым оттенком. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 3-[2-метокси-5-{2-(3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Соединение 36) К раствору 2-(толуол-4-сульфонил)этилового эфира 3-[2-метокси-5-{2-(3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Полупродукта 63, 1,0 г, 1,26 ммоль) в 10 мл CH3CN прибавляли раствор NaHCO3 (127 мг, 1,51 ммоль, 1,2 экв.) в 5 мл H2O. Смесь нагревали при температуре 65°С в течение 4 часов. После этого в вакууме отгоняли летучие вещества, смесь промывали Et2O-EtOAc (10:1) и далее водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 100 мл СН2Cl2, раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали и маслянистый остаток растворяли в 5 мл СН2Cl2. К смеси прибавляли Et2O до появления осадка белого цвета. Осадок отфильтровывали, промывали Et2O, сушили в вакууме и получали 730 мг (91%) натриевой соли 3-[2-метокси-5-{2-(3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил)бензимидазол-1-сульфонил}фенил]пропионовой кислоты (Соединение 36) в виде кристаллического вещества белого цвета. 1Н ЯМР (D2О, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 64) 3-Изопропил-5-метилфеноксиуксусную кислоту получали по известной методике следующим образом. 3-Изопропил-5-метилфенол (44,0 г, 0,29 моль) и натриевую соль хлоруксусной кислоты (35,0 г, 0,3 моль) растворяли в 180 мл Н2О и затем медленно прибавляли КОН (86%, 20 г, 0,3 моль). Реакционную массу кипятили с обратным холодильником в течение ночи и затем концентрировали дистилляцией. После охлаждения смесь подкисляли до рН 1 и экстрагировали EtOAc. Органический слой промывали H2O, сушили над MgSO4 и концентрировали. Осадок перекристаллизовывали из смеси 5% изопропанол-гексан и получали чистую 3-изопропил-5-метилфеноксиуксусную кислоту (38 г, 63%) в виде кристаллов белого цвета. Смесь 3-изопропил-5-метилфеноксиуксусной кислоты (полученной выше, 6,0 г, 28,8 ммоль), 2-(n-толуолсульфонил)этанола (5,77 г, 28,8 ммоль) и гидрата п-толуолсульфоновой кислоты (p-TsOH·H2O) (0,5 г) в 100 мл толуола кипятили с обратным холодильником и насадкой Дина-Старка в течение 3 часов. Затем добавляли воду и смесь экстрагировали СН2Cl2. Объединенные органические слои промывали насыщенным раствором NaHCO3 (2х), сушили над MgSO4, концентрировали и получали 2-(толуол-4-сульфонил)этиловый эфир 3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 64, 11,0 г, 98%) в виде масла светло-коричневого цвета. 1Н ЯМР (CDCl3, 400 МГц) 2-(Толуол-4-сульфонил)этиловый эфир 4-хлорсульфонил-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 65) К смеси 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукта 64, 5,5 г, 14,0 ммоль) и 4 мл СН2Cl2 при охлаждении на ледяной бане прибавляли по каплям хлорсульфоновую кислоту (8,2 г, 70,0 ммоль, 5,0 экв.). Массу перемешивали при 0°С в течение 3 часов. Полученное вязкое масло выливали при интенсивном перемешивании на колотый лед. Смесь экстрагировали EtOAc. Органический слой промывали 1N раствором NaHCO3, сушили над MgSO4, концентрировали при упаривании и получали технический 2-(толуол-4-сульфонил)этиловый эфир 4-хлорсульфонил-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 65, 6,0 г, 87%) в виде вязкого масла светло-коричневого цвета, которое очищали хроматографией на колонке с силикагелем. 1Н ЯМР (CDCl3, 400 МГц) Смесь 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-4-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Полупродукт 66) и 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-4-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Полупродукт 67) К гетерогенному раствору 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (омепразола, 2,9 г, 8,4 ммоль) и 10 мл Et3N в 60 мл CH2Cl2 прибавляли 2-(толуол-4-сульфонил)этиловый эфир 4-хлорсульфонил-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 65, 5,5 г, 11,3 ммоль, 1,3 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли около 2 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 2 часов. После этого отгоняли растворитель и маслянистый остаток очищали колоночной хроматографией (силикагель, от СН2Cl2 до 2% МеОН в CH2Cl2) и получали смесь 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-4-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Полупродукта 66) и 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-4-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Полупродукта 67), 4,1 г (61%), в виде пены белого цвета с желтоватым оттенком (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (CDCl3, 400 МГц) Смесь натриевой соли 3-изопропил-4-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Соединения 37) и натриевой соли 3-изопропил-4-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Соединения 38) К раствору смеси 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-4-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Полупродукта 66) и 2-(толуол-4-сульфонил)этилового эфира 3-изопропил-4-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Полупродукта 67), 4,0 г (5,02 ммоль), в 40 мл CH3CN прибавляли раствор NaHCO3 (500 мг, 6,02 ммоль, 1,2 экв.) в 20 мл H2O. Смесь нагревали при температуре 65°С в течение 4 часов. После этого в вакууме отгоняли все летучие вещества, смесь промывали эфиром (2х) и далее водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 80 мл EtOAc, раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали и маслянистый остаток растворяли в 10 мл EtOAc. К смеси прибавляли сначала диэтиловый эфир, а затем гексан до появления осадка белого цвета. Осадок отфильтровывали, промывали EtOAc, сушили в вакууме и получали 2,8 г (87%) смеси натриевой соли 3-изопропил-4-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Соединения 37) и натриевой соли 3-изопропил-4-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}-5-метилфеноксикислоты (Соединения 38) в виде кристаллического вещества белого цвета (отношение между 5- и 6-изомерами 1:1). 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 4-[2-{3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил}бензимидазол-1-сульфонил]-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 68) К гетерогенному раствору 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (лансопразола, 1,0 г, 2,71 ммоль) и 2 мл Et3N в 20 мл CH2Cl2 прибавляли 2-(толуол-4-сульфонил)этиловый эфир 4-хлорсульфонил-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 65, 1,6 г, 3,25 ммоль, 1,2 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли около 1 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 2 часов. После этого отгоняли растворитель, маслянистый остаток очищали колоночной хроматографией (силикагель, от CH2Cl2 до 1% МеОН в СН2Cl2) и получали 2-(толуол-4-сульфонил)этиловый эфир 4-[2-{3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил}бензимидазол-1-сульфонил]-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукт 68, 1,76 г, 79%) в виде пены белого цвета. 1Н ЯМР (CDCl3, 400 МГц) Натриевая соль 4-[2-{3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил}бензимидазол-1-сульфонил]-3-изопропил-5-метилфеноксиуксусной кислоты (Соединение 39) К раствору 2-(толуол-4-сульфонил)этилового эфира 4-[2-{3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил}бензимидазол-1-сульфонил]-3-изопропил-5-метилфеноксиуксусной кислоты (Полупродукта 68, 1,6 г, 1,95 ммоль) в 15 мл CH3CN прибавляли раствор NaHCO3 (200 мг, 2,34 ммоль, 1,2 экв.) в 8 мл Н2О. Смесь нагревали при температуре 65°С в течение 1,5 часов. После этого в вакууме отгоняли все летучие вещества, смесь промывали эфиром (2х) и далее водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 110 мл смеси эфир-EtOAc (10:1) и раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали и маслянистый остаток растворяли в 10 мл смеси эфир-EtOAc (10:1). К смеси прибавляли гексан до появления осадка белого цвета. Осадок отфильтровывали, промывали гексаном, сушили в вакууме и получали 1,0 г (77%) натриевой соли 4-[2-{3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил}бензимидазол-1-сульфонил]-3-изопропил-5-метилфеноксиуксусной кислоты (Соединение 39) в виде кристаллического вещества белого цвета. 1Н ЯМР (D2O, 400 МГц)
2-(Толуол-4-сульфонил)этиловый эфир 5-хлорсульфонил-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]бензойной кислоты (Полупродукт 69) 2-(Карбоксиметокси)-5-хлорсульфонилбензойную кислоту получали взаимодействием 2-карбоксиметоксибензойной кислоты с хлорсульфоновой кислотой по методике, аналогичной синтезу Полупродукта 6. Смесь 2-(карбоксиметокси)-5-хлорсульфонилбензойной кислоты (4,0 г, 13,5 ммоль) и хлористого тионила (10 мл) нагревали при температуре кипения в течение 2 часов, затем отгоняли избыток хлористого тионила. Маслянистый остаток растворяли в 15 мл CH3CN и прибавляли 2-(толилсульфонил)этанол (5,0 г, 25,0 ммоль). Смесь нагревали при температуре кипения в течение 40 часов, затем охлаждали и добавляли воду. Смесь экстрагировали EtOAc, органический слой промывали 1N NaHCO3, сушили, концентрировали и получали 7,7 г (93%) пены коричневого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц) Динатриевая соль 2-(карбоксиметокси)-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Соединение 40) К гетерогенному раствору 2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]-1Н-бензимидазола (лансопразола, 3,0 г, 8,13 ммоль) и 6 мл Et3N в 70 мл СН2Cl2 прибавляли 2-(толуол-4-сульфонил)этиловый эфир 5-хлорсульфонил-2-[2-(толуол-4-сульфонил)этоксикарбонилметокси]бензойной кислоты (Полупродукт 69, 7,0 г, 10,6 ммоль, 1,3 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли около 3 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 2 часов. После этого отгоняли растворитель и маслянистый остаток пропускали через короткую колонку (силикагель, от СН2Cl2 до 1% МеОН в СН2Cl2) для удаления окрашенных примесей и Et3N. После концентрирования элюента получали Полупродукт 70 (около 9,0 г) в виде пены белого цвета с желтоватым оттенком. К раствору [бис-{2-(толуол-4-сульфонил)этилового}эфира] 2-(карбоксиметокси)-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Полупродукта 70, 9,0 г, 8,13 ммоль) в 80 мл СН3CN прибавляли раствор NaHCO3 (1,70 г, 20,3 ммоль, 2,5 экв.) в 40 мл Н2O. Смесь нагревали при температуре 65°С в течение 5 часов. После этого в вакууме отгоняли все летучие вещества, смесь промывали EtOAc (2x) и далее водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 200 мл СН2Cl2, раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали и маслянистый остаток растворяли в 20 мл CH2Cl2. К смеси прибавляли EtOAc-эфир (1:1) до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали диэтиловым эфиром, сушили в вакууме и получали 4,5 г (82%) динатриевой соли 2-(карбоксиметокси)-5-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Соединение 40) в виде кристаллов светло-коричневого цвета. 1Н ЯМР (D2O, 400 МГц)
Смесь динатриевой соли 2-(карбоксиметокси)-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}бензойной кислоты (Соединение 41) и динатриевой соли 2-(карбоксиметокси)-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}бензойной кислоты (Соединение 42) К гетерогенному раствору 5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)-1Н-бензимидазола (омепразола, 1,0 г, 3,03 ммоль) и 4 мл Et3N в 20 мл CH2Cl2 прибавляли хлорсульфониловый эфир (Полупродукт 69, 2,6 г, 3,95 ммоль, 1,3 экв.). После полного растворения хлорсульфонилового эфира в реакционной массе прибавляли около 1 г твердого NaHCO3. Массу перемешивали при комнатной температуре в течение 2 часов. После этого добавляли воду и смесь экстрагировали EtOAc (2x). Объединенные органические слои промывали водой, сушили, концентрировали и получали смесь Полупродуктов 71 и 72 (около 2,9 г) в виде пены белого цвета с желтоватым оттенком, которую использовали без дополнительной очистки. К раствору смеси Полупродуктов 71 и 72 (около 2,9 г) в 30 мл CH3CN прибавляли раствор NaHCO3 (520 мг, 6,19 ммоль, 2,2 экв.) в 15 мл Н2О. Смесь нагревали при температуре 65°С в течение 3 часов. После этого в вакууме отгоняли все летучие вещества, смесь промывали EtOAc (2x) и водный слой лиофилизировали в течение ночи. Кристаллы растворяли в 150 мл СН2Cl2, раствор фильтровали от нерастворимых примесей. Фильтрат концентрировали и маслянистый остаток растворяли в 5 мл СН2Cl2. К смеси прибавляли EtOAc до появления осадка белого цвета. Выпавший осадок отфильтровывали, промывали эфиром, сушили в вакууме и получали 1,86 г (95%) смеси динатриевой соли 2-(карбоксиметокси)-5-{5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}бензойной кислоты (Соединения 41) и динатриевой соли 2-(карбоксиметокси)-5-{6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил}бензойной кислоты (Соединения 42) в виде кристаллов светло-коричневого цвета (отношение между 5-и 6-изомерами 1:1). 1Н ЯМР (D2O, 400 МГц) В следующих реакционных схемах и примерах, как и во всем описании, если только не указано иначе, реагенты и/или исходные вещества либо имеются в продаже, либо легко могут быть получены по химической научной или патентной литературе рядовым специалистом в данной области.
Метил-(4-хлорсульфонил-3,5-диметилфенокси)ацетат (Полупродукт А1) Метил-(3,5-диметилфенокси)ацетат (19,4 г, 0,10 моль) прибавляли по каплям в течение приблизительно 30 минут к быстро перемешиваемой хлорсульфоновой кислоте (58 г, 0,50 моль), охлажденной на бане с температурой -20°С. Еще через 20 минут температуру смеси медленно отпускали до комнатной. Через 2 часа массу медленно выливали в 400 мл смеси вода/лед. Полученную суспензию экстрагировали 400 мл дихлорметана, органический слой промывали водой и концентрировали. Остаток очищали флэш-хроматографией на силикагеле (гексан->дихлорметан) и получали 7,6 г (27%) требуемого соединения. Метил-[4-(4-метокси-2-нитрофенил)аминосульфонил-3,5-диметилфенокси]ацетат (Полупродукт А2) Смесь метил-(4-хлорсульфонил-3,5-диметилфенокси)ацетата (Полупродукт А1, 7,6 г, 0,026 моль), 4-метокси-2-нитроанилина (4,0 г, 0,024 моль) и пиридина (50 мл) перемешивали при комнатной температуре в течение 16 часов, на бане с температурой 90°С – в течение 5 часов и на бане с температурой 130°С – в течение 30 минут. Смесь охлаждали и перемешивали с толуолом в течение 10 минут. Полученную суспензию фильтровали, фильтрат концентрировали, разбавляли 150 мл толуола и 100 мл этилацетата, промывали 1 М соляной кислотой и водой и концентрировали приблизительно до 50 мл. Смесь выдерживали в течение ночи в морозильной камере. Осадок отфильтровывали и промывали несколькими порциями холодного толуола. После сушки его вес составил 8,2 г (74%). Дополнительную порцию 0,45 г требуемого соединения получали экстракцией водных фаз и кристаллизацией второй порции из толуольного фильтрата. Метил-[4-(2-амино-4-метоксифенил)аминосульфонил-3,5-диметилфенокси] ацетат (Полупродукт A3) Метил-[4-(4-метокси-2-нитрофенил)аминосульфонил-3,5-диметилфенокси] ацетат (Полупродукт А2, 8,1 г, 0,019 моль) в 200 мл этилацетата в течение 30 минут перемешивали с никелем Ренея (0,75 г) и фильтровали через слой целита с помощью дополнительных 100 мл этилацетата. Фильтрат смешивали с оксидом платины (0,40 г, 0,0018 моль), гидрировали и получали 7,8 г (100%) титульного соединения. Метил-{4-[(2-меркапто-5-метоксибензимидазолил)сульфонил]-3,5-диметилфенокси}ацетат (Полупродукт А4) Смешивали метил-[4-(2-амино-4-метоксифенил)аминосульфонил-3,5-диметилфенокси]ацетат (Полупродукт A3, 7,5 г, 0,019 моль), тиокарбонилдиимидазол (5,1 г, 0,029 моль) и пиридин (100 мл) и перемешивали их в течение 3 часов. Реакционную массу выливали в 1 л быстро перемешиваемой воды. Через 2 часа выпавший осадок отфильтровывали, промывали несколькими порциями воды, сушили на воздухе и получали 8,9 г (110%) требуемого титульного соединения с небольшой примесью пиридина. Неочищенный продукт использовали на стадии алкилирования. Гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина К раствору 4-метокси-3,5-диметилпиридинметанола (25,1 г, 0,15 моль) в дихлорметане (400 мл) в течение 30 минут при комнатной температуре под аргоном прибавляли раствор хлористого тионила (18,8 г, 0,158 моль) в дихлорметане (100 мл). После перемешивания в течение еще 30 минут при комнатной температуре растворитель удаляли при пониженном давлении. Твердый остаток суспендировали в гексане (200 мл) и фильтровали. Кристаллы промывали гексаном (50 мл), сушили на воздухе и получали 33,3 г (100%) титульного соединения в виде кристаллического вещества белого цвета. Метил-{4-[(5-метокси-2-{[4-метокси-3,5-диметил-(2-пиридил)метил]тио}бензимидазолил)сульфонил]-3,5-диметилфенокси}ацетат (Полупродукт А5) Гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина (4,4 г, 0,020 моль) прибавляли к смеси метил-{4-[(2-меркапто-5-метоксибензимидазолил)сульфонил]-3,5-диметилфенокси}ацетата (Полупродукта А4, 8,2 г, 0,019 моль), карбоната калия (5,7 г, 0,041 моль) и диметилформамида (80 мл). Массу перемешивали в течение 1,5 часов и затем выливали в 150 мл быстро перемешиваемой ледяной воды, содержащей 40 мл 1,2 М соляной кислоты. Выпавшие кристаллы отфильтровывали и промывали несколькими порциями воды. После частичной сушки на воздухе 10 г осадка растворяли в 100 мл дихлорметана. Полученный раствор промывали насыщенным водным раствором бикарбоната натрия, фильтровали через бумагу 1PS и концентрировали. Остаток смешивали с кипящим метанолом, фильтровали и получали 8,9 г (80%) титульного соединения. Метил-{4-[(5-метокси-2-{[4-метокси-3,5-диметил-(2-пиридил)метил]сульфинил}-бензимидазолил)сульфонил]-3,5-диметилфенокси}ацетат (Полупродукт А6) Растворы метил-{4-[(5-метокси-2-{[4-метокси-3,5-диметил-(2-пиридил)метил]тио}бензимидазолил)сульфонил]-3,5-диметилфенокси}ацетата (Полупродукт А5, 7,1 г, 0,12 моль) в 100 мл тетрагидрофурана и 3-хлорпероксибензойной кислоты (2,6 г, 0,15 моль) в 20 мл того же растворителя охлаждали в морозильной камере в течение 1 часа и затем смешивали. Полученный раствор оставляли в морозильной камере приблизительно на 16 часов, разбавляли этилацетатом, промывали по несколько раз 5% метабисульфитом натрия, 5% бикарбонатом натрия и насыщенным хлоридом натрия. Органическую фракцию концентрировали и затем очищали с помощью флэш-силикагеля (гексан->этилацетат->1% метанол в этилацетате). Извлеченное титульное соединение весило 3,3 г (46%). Натриевая соль 2-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]-3,5-диметилфенокси}уксусной кислоты (Соединение 31, изомерно чистая) Раствор метил-{4-[(5-метокси-2-{[4-метокси-3,5-диметил-(2-пиридил)метил]сульфинил}бензимидазолил)сульфонил]-3,5-диметилфенокси} ацетата (Полупродукта А6, 2,96 г, 0,493 моль) в 250 мл диметоксиэтана быстро перемешивали по мере прибавления 49 мл 0,100 N водного раствора гидроксида натрия. Смесь концентрировали при пониженном давлении (температура бани <22°С). Остаток повторно концентрировали из 200 мл диметоксиэтана, выдерживали непродолжительное время в глубоком вакууме и распределяли между этилацетатом и водой. Водный слой промывали несколькими порциями этилацетата и затем концентрировали в глубоком вакууме. Остаток распределяли между этилацетатом и водой при рН 3. Водный слой экстрагировали дополнительной порцией этилацетата, объединенные органические слои уменьшали в объеме и разбавляли гексаном. Полученные кристаллы отфильтровывали и объединяли со второй порцией, извлеченной из фильтрата. Продукт промывали смесью диэтиловый эфир-тетрагидрофуран, 10:1, растворяли в диметоксиэтане, нейтрализовали 0,0100 N раствором гидроксида натрия и промывали этилацетатом. Продукт, титульное соединение, весил 0,925 г (31%). ЯМР (300 МГц) (D2O)
Метил-2,2-диметил-3-(4-толилсульфонилокси)пропионат (Полупродукт А7) Смесь метил-2,2-диметил-3-гидроксипропионата (100 г, 0,76 моль), 4-толуолсульфонилхлорида (151 г, 0,80 моль), 4-диметиламинопиридина (4,6 г, 0,038 моль) и пиридина (200 мл) перемешивали в течение 20 часов, затем разбавляли 200 мл толуола, перемешивали в течение 30 минут и фильтровали. Фильтрат концентрировали до 250 мл при пониженном давлении, разбавляли 100 мл толуола, фильтровали и концентрировали. Остаток суспендировали в 200 мл гексана, при пониженном давлении отгоняли растворитель и получали титульное соединение (235 г, 100%), содержащее следы толуолсульфонилхлорида. Метил-3-(3,4-диметилфенилокси)-2,2-диметилпропионат (Полупродукт А8) В реакционную колбу загружали метил-2,2-диметил-3-тозилоксипропионат (Полупродукт А7, 100 г, 0,35 моль), 3,5-диметилфенол (45 г, 0,37 моль), карбонат калия (73 г, 0,53 моль) и диметилацетамид (300 мл). Смесь нагревали в течение 16 часов на паровой бане и в течение 6 часов – на бане с температурой 170°С. Охлажденную реакционную массу разбавляли 1 л воды и экстрагировали этилацетатом порциями 2×200 мл. Объединенные органические фракции промывали водой, 0,5 М гидроксидом натрия (дважды), водой и насыщенным водным раствором хлорида натрия. Растворитель удаляли при пониженном давлении и в глубоком вакууме. Остаток обрабатывали дихлорметаном и промывали несколькими порциями 1 М раствора гидроксида натрия, водой и насыщенным раствором хлорида натрия. Раствор концентрировали, остаток перегоняли при давлении 2 торр и получали 43,5 г (52%) титульного соединения. Метил-3-(4-хлорсульфонил-3,5-диметилфенилокси)-2,2-диметилпропионат (Полупродукт А9) Метил-3-(3,4-диметилфенилокси)-2,2-диметилпропионат (Полупродукт А8, 23,6 г, 0,100 моль) прибавляли по каплям в течение 30 минут к быстро перемешиваемой хлорсульфоновой кислоте (46,6 г, 0,400 моль), поддерживая температуру реакции -2°С. Затем реакционную массу выливали в перемешиваемую смесь 300 г льда и 300 мл дихлорметана. Водную фракцию экстрагировали дополнительной порцией дихлорметана, объединенные органические слои промывали водой, 0,5 М раствором бикарбоната натрия (x 2) и снова водой. Растворитель отгоняли при пониженном давлении, остаток очищали за несколько проходов с помощью флэш-хроматографии (силикагель; гексан->2% этилацетат в гексане) и получали 9,6 г (29%) титульного соединения. Метил-3-{4-[4-метокси-2-нитрофениламиносульфонил}-3,5-диметилфенокси]-2,2-диметилпропионат Смешивали метил-3-(4-хлорсульфонил-3,5-диметилфенилокси)-2,2-диметилпропионат (Полупродукт А9, 8,3 г, 0,025 моль), 3-метокси-2-нитроанилин (3,9 г, 0,023 моль) и пиридин (40 мл) и перемешивали их в течение 1,5 часов, нагревая на бане с температурой 100°С и в течение 4 дней при комнатной температуре. Смесь разбавляли 150 мл толуола и фильтровали. Фильтрат концентрировали при пониженном давлении, остаток распределяли между 1,2 М соляной кислотой и этилацетатом. Органический слой промывали водой и концентрировали. Пенистый остаток перемешивали с 50 мл смеси гексан-этилацет, 2:1. Выпавшие кристаллы отфильтровывали и промывали несколькими порциями того же растворителя. После сушки продукт, титульное соединение, весил 9,1 г (85%). 3-{4-[4-Метокси-2-нитрофенил]аминосульфонил}-3,5-диметилфенокси]-2,2-диметилпропионовая кислота (Полупродукт А10) Порцию 54 мл (0,054 моль) 0,100 N раствора гидроксида натрия прибавляли к раствору метил-3-({4-[4-метокси-2-нитрофенил]аминосульфонил}-3,5-диметилфенокси)-2,2-диметилпропионата (8,5 г, 0,018 моль) в диметоксиэтане. Приблизительно через 20 часов смесь разбавляли 60 мл 1,2 М соляной кислоты и 100 г льда. Образовавшуюся суспензию экстрагировали несколькими порциями этилацетата, объединенные органические фракции промывали насыщенным водным раствором хлорида натрия и концентрировали. Остаток перемешивали с гексаном в течение 1 часа и затем собирали с помощью дополнительного количества гексана и получали 8,2 г (100%) титульного соединения. 2-(4-Толилсульфонил)этил-3-{4-[4-метокси-2-нитрофениламиносульфонил]-3,5-диметилфенокси}-2,2-диметилпропионат (Полупродукт A11) Смесь 3-{4-[4-метокси-2-нитрофениламиносульфонил]-3,5-диметилфенокси}-2,2-диметилпропионовой кислоты (Полупродукта А10, 24,2 г, 0,054 моль) и карбонилдиимидазола (9,5 г, 0,059 моль) разбавляли 45 мл дихлорметана и нагревали на бане с температурой 36°С в течение 15 минут. Затем прибавляли 4-толилсульфонилэтанол, температуру бани повышали до 55°С и растворитель отдували в токе азота. Через 1,5 часа подачу азота прекращали и еще через 2,5 часа охлажденную реакционную массу распределяли между этилацетатом и 1,2 М соляной кислотой. Водный слой экстрагировали дополнительным количеством этилацетата, объединенные органические фракции промывали водой и насыщенным раствором хлорида натрия, концентрировали при пониженном давлении и в глубоком вакууме. Продукт, титульное соединение (34 г, 99%), получали без дополнительной очистки. 2-(4-Толилсульфонил)этил-3-({4-[2-амино-4-метоксифенил]аминосульфонил}-3,5-диметилфенокси)-2,2-диметилпропионат (Полупродукт А12) 2-(4-Толилсульфонил)этил-3-{4-[4-метокси-2-нитрофениламиносульфонил]-3,5-диметилфенокси}-2,2-диметилпропионат (Полупродукт A11, 34 г, 0,54 моль) гидрировали в этилацетате, используя оксид платины (1,7 г, 0,0075 моль) в качестве катализатора. Продукт, титульное соединение, получали без дополнительной очистки. 2-(4-Толилсульфонил)этил-3-(4-[{2-меркапто-5-метоксибензимидазолил}сульфонил]-3,5-диметилфенокси)-2,2-диметилпропионат (Полупродукт А13) 2-(4-Толилсульфонил)этил-3-({4-[2-амино-4-метоксифенил]аминосульфонил}-3,5-диметилфенокси)-2,2-диметилпропионат (Полупродукт А12, 4,1 г, 0,0068 моль) и тиокарбонилдиимидазол (1,8 г, 0,010 моль) растворяли в пиридине (40 мл). Приблизительно через 16 часов смесь выливали в 400 мл воды. Суспензию экстрагировали несколькими порциями этилацетата, объединенные органические фракции промывали 2 порциями 1,2 М соляной кислоты и концентрировали. Остаток повторно концентрировали из дихлорметана и получали 4,3 г (98%) титульного соединения. 2-(4-Толилсульфонил)этил-3-(4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метилтио}бензимидазолил)сульфонил]-3,5-диметилфенокси}-2,2-диметилпропионат 2-(4-Толилсульфонил)этил-3-(4-[{2-меркапто-5-метоксибензимидазолил}сульфонил]-3,5-диметилфенокси)-2,2-диметилпропионат (Полупродукт А13, 4,2 г, 0,0065 моль) и карбонат калия (1,97 г, 0,014 моль) смешивали в диметилформамиде (41 мл). Затем в один прием прибавляли гидрохлорид 2-хлорметил-4-метокси-3,5-диметилпиридиния (1,5 г, 0,0068 моль). Через 90 минут реакционную массу выливали в быстро перемешиваемую смесь 16 мл 1,2 М соляной кислоты и 45 г льда. Полученная маслянистая суспензия начала кристаллизоваться при интенсивном растирании стеклянной палочкой. После этого ее перемешивали в течение нескольких дней. Продукт, титульное соединение, отфильтровывали и промывали несколькими порциями воды. После сушки его вес составил 4,8 г (93%). 2-(4-Толилсульфонил)этил-3-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метилсульфинил}бензимидазолил)сульфонил]-3,5-диметилфенокси}-2,2-диметилпропионат (Полупродукт А14) 2-(4-Толилсульфонил)этил-3-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метилтио}бензимидазолил)сульфонил]-3,5-диметилфенокси}-2,2-диметилпропионат (4,63 г, 0,00582 моль) и 70% 3-хлорпероксибензойную кислоту (1,8 г, 0,00728 моль) по отдельности растворяли в 30 мл тетрагидрофурана и охлаждали в морозильной камере в течение 40 минут. Растворы смешивали и оставляли в морозильной камере на 16 часов. Затем реакционную массу выливали в смесь этилацетата и 5% метабисульфита натрия. Органический слой промывали дополнительной порцией раствора бисульфита, несколькими порциями 5% раствора бикарбоната натрия и насыщенным раствором хлорида натрия. После отгонки растворителя в вакууме остаток делили с помощью флэш-хроматографии (силикагель; 1:1, гексан-этилацетат->0,5% метанола в этилацетате) и получали 1,4 г (30%) титульного соединения. Натриевая соль 3-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метилсульфинил}бензимидазолил)сульфонил]-3,5-диметилфенокси}-2,2-диметилпропионовой кислоты (Соединение 43) 2-(4-Толилсульфонил)этил-3-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]-3,5-диметилфенокси}-2,2-диметилпропионат (Полупродукт А14, 1,4 г, 0,0017 моль), растворенный в ацетонитриле (9 мл), смешивали с изопропанолом (3 мл) и 3 мл воды, содержащей бикарбонат натрия (0,17 г, 0,0020 моль). Смесь нагревали на бане с температурой 70°С приблизительно в течение 6 часов и затем концентрировали сначала при пониженном давлении, а затем в глубоком вакууме. Остаток промывали этилацетатом, затем высаживали из раствора в смеси дихлорметан-изопропанол добавлением этилацетата и, наконец, концентрировали из воды при пониженном давлении. Остаток перерастворяли в воде и этилацетате. рН доводили до 4, органический слой фильтровали через слой сульфата натрия и концентрировали. После сушки в течение нескольких дней в глубоком вакууме полученную кислоту растворяли в диметоксиэтане и нейтрализовали стехиометрическим количеством 0,100N гидроксида натрия. Растворитель отгоняли сначала при пониженном давлении, затем в глубоком вакууме и остаток собирали с помощью диэтилового эфира. После сушки продукт, титульное соединение, весил 0,71 г (64%). ЯМР (300 МГц) (D2O)
2-Карбоксиметокси-4-хлорсульфонилфеноксиуксусная кислота (Полупродукт А15) 2-Карбоксиметоксифеноксиуксусную кислоту (15,3 г, 0,0675 моль) прибавляли порциями в течение 30 минут к перемешиваемой хлорсульфоновой кислоте (59,0 г, 0,506 моль) при охлаждении на бане лед/метанол. Смесь оставляли нагреваться до комнатной температуры на 2,5 часа и затем медленно вливали в перемешиваемую ледяную воду. Выпавшие кристаллы отфильтровывали, промывали водой и сушили в глубоком вакууме над гидроксидом натрия. Продукт, титульное соединение, весил 6,54 г (30%). 2-(4-Толилсульфонил)этил-4-хлорсульфонил-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А16, также известный как Полупродукт 36) 2-Карбоксиметокси-4-хлорсульфонилфеноксиуксусную кислоту (Полупродукт А15, 6,54 г, 0,0202 моль) кипятили с обратным холодильником с 15 мл (0,202 моль) хлористого тионила. Через 1 час смесь разбавляли 20 мл толуола и концентрировали досуха при пониженном давлении и в глубоком вакууме. Кристаллический остаток растворяли в 300 мл дихлорметана и смешивали с 2-(4-толилсульфонил)этанолом (8,89 г, 0,044 моль). После этого по каплям в течение 15 минут прибавляли пиридин (3,6 мл, 0,044 моль) в 100 мл дихлорметана. Еще через 1,75 часа реакционную смесь промывали 1 М серной кислотой, сушили над сульфатом магния и концентрировали. Остаток очищали на флэш-силикагеле (дихлорметан->7,5% этилацетата в дихлорметане) и получали 9,74 г (70%) титульного соединения. 2-(4-Толилсульфонил)этил-4-(4-метокси-2-нитрофенил)аминосульфонил-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А17) 4-Метокси-2-нитроанилин (2,3 г, 0,014 моль) и 2-(4-толилсульфонил)этил-4-хлорсульфонил-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А16, 9,6 г, 0,014 моль) растворяли в 40 мл пиридина и перемешивали при положительном давлении азота. Приблизительно через 16 часов смесь разбавляли 50 мл толуола и 20 мл дихлорметана и концентрировали при пониженном давлении. Остаток очищали на флэш-колонке с силикагелем (дихлорметан->5:1, дихлорметан-этилацетат) и получали 6,7 г (58%) титульного соединения. 2-(4-Толилсульфонил)этил-4-(2-амино-4-метоксифенил)аминосульфонил-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А18) 2-(4-Толилсульфонил)этил-4-(4-метокси-2-нитрофенил)аминосульфонил-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А17, 6,6 г, 0,0080 моль) гидрировали, используя оксид платины в качестве катализатора. Смесь фильтровали и концентрировали. После сушки в глубоком вакууме остаток, титульное соединение, весил 5,9 г (93%). 2-(4-Толилсульфонил)этил-4-[(2-меркапто-5-метоксибензимидазолил)сульфонил]-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А19) Смесь 2-(4-толилсульфонил)этил-4-(2-амино-4-метоксифенил)аминосульфонил-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетата (Полупродукт А18, 5,9 г, 0,0075 моль) и тиокарбонилдиимидазола (2,0 г, 0,011 моль) в 35 мл пиридина перемешивали под азотом в течение 4 часов. Затем смесь выливали в 400 мл быстро перемешиваемой воды. Образовавшуюся суспензию перемешивали приблизительно в течение 16 часов и затем экстрагировали толуолом, диэтиловым эфиром, дихлорметаном (2 порциями) и смесью дихлорметан-метанол, 1:1 (3 порциями). Экстракты промывали, объединяли, концентрировали и получали 5,9 г (96%) титульного соединения. 2-(4-Толилсульфонил)этил-4-[{5-метокси-([4-метокси-3,5-диметил-(2-пиридил)]метилтио)бензимидазолил}сульфонил]-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А20) 2-(4-Толилсульфонил)этил-4-[(2-меркапто-5-метоксибензимидазолил)сульфонил]-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А19, 5,9 г, 0,0072 моль) растворяли в 50 мл диметилформамида и затем смешивали с карбонатом калия (2,2 г, 0,016 моль) (в атмосфере N2). Далее в один прием прибавляли гидрохлорид 2-(хлорметил)-4-метокси-3,5-диметилпиридиния (1,7 г, 0,0076 моль). Приблизительно через 2,5 часа объем смеси уменьшали приблизительно до 20 мл в глубоком вакууме (температура бани <22°С). Оставшийся раствор вливали в быстро перемешиваемую смесь 10 мл 1,2 М соляной кислоты с 200 мл воды и льда. Суспензию перемешивали до гранулирования осадка. Осадок отфильтровывали, промывали водой и сушили на воздухе. Затем кристаллы в течение 14 часов перемешивали со смесью изопропанол-вода, 1:1. Декантировали надосадочную жидкость, осадок обрабатывали дихлорметаном и концентрировали досуха. Остаток очищали на флэш-колонке с силикагелем (4% этилацетата в дихлорметане->этилацетат) и получали 4,8 г (75%) титульного соединения. 2-(4-Толилсульфонил)этил-4-[{5-метокси-([4-метокси-3,5-диметил-(2-пиридил)]метилсульфинил)бензимидазолил}сульфонил]-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А21) 2-(4-Толилсульфонил)этил-4-[{5-метокси-([4-метокси-3,5-диметил-(2-пиридил)]метилтио)бензимидазолил}сульфонил]-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А20, 4,8 г, 0,0054 моль) растворяли в 20 мл тетрагидрофурана и раствор оставляли в морозильной камере на 40 минут. Раствор 3-хлорпероксибензойной кислоты (1,3 г, 0,0075 моль) в 6 мл тетрагидрофурана охлаждали в течение 30 минут. Оба раствора объединяли и оставляли в морозильной камере на 16 часов. Затем реакционную массу выливали в быстро перемешиваемую смесь 5% водного раствора метабисульфита натрия и этилацетата. Разделяли слои и органический слой промывали 2 порциями 2,5% водного раствора бикарбоната натрия и насыщенным водным раствором хлорида натрия. Раствор фильтровали через бумагу 1PS и концентрировали при пониженном давлении. Остаток делили на колонке с флэш-силикагелем (2:1, этилацетат-гексан->этилацетат) и получали 2,0 г (41%) титульного соединения. Динатриевая соль 2-карбоксиметокси-4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метилсульфинил}бензимидазолил)сульфонил]фенокси}уксусной кислоты (Соединение 14) 2-(4-Толилсульфонил)этил-4-[{5-метокси-([4-метокси-3,5-диметил-(2-пиридил)]метилсульфинил)бензимидазолил}сульфонил]-2-(2-[4-толилсульфонил]этоксикарбонилметокси)феноксиацетат (Полупродукт А21, 2,0 г, 0,0033 моль) растворяли в 15 мл ацетонитрила и 5 мл изопропанола. Полученный раствор смешивали с бикарбонатом натрия (0,61 г, 0,0072 моль) в 10 мл воды и нагревали при перемешивании на бане с температурой 70°С в течение 75 минут. При пониженном давлении отгоняли растворители (температура бани <22°С) и остаток выдерживали в течение ночи в глубоком вакууме. Затем его растирали с 2 порциями диэтилового эфира и 2 порциями этилацетата. Остаток растворяли в воде и промывали 3 порциями этилацетата. Далее водный раствор экстрагировали этилацетатом при рН 5, 4, 3 и, наконец, при рН 3 с повышением количества хлорида натрия. Объединенные органические экстракты концентрировали и продукт фильтровали с помощью смеси дихлорметан-этилацетат, 1:1. Фильтрат объединяли с аналогично полученным веществом и обрабатывали при помощи такой же нейтральной и кислой экстракции. Всего было получено 1,1 г (суммарный выход 49%) титульного соединения. ЯМР (300 МГц) (D2O)
Метил-2-[4-(хлорсульфонил)фенокси] ацетат (Полупродукт А22) Метилфеноксиацетат (99,9 г, 0,6 моль) прибавляли по каплям к хлорсульфоновой кислоте (279,6 г, 159,5 мл, 2,4 моль) при температуре -5°С с такой скоростью, чтобы внутренняя температура была в интервале от 0 до -5°С (прибавление заняло около 60 минут). В процессе прибавления образовалось некоторое количество осадка. Охлаждающую баню убирали, а реакционную массу перемешивали при комнатной температуре в течение еще 1,5 часов. Реакционную массу выливали при 0°С в интенсивно перемешиваемую смесь дихлорметана (900 мл) и метанола (100 мл). Через 15 минут охлаждающую баню удаляли и полученную смесь перемешивали в течение 1 часа при комнатной температуре. Полученную смесь промывали ледяной водой (2×250 мл). Объединенные водные фракции снова экстрагировали дихлорметаном (1×250 мл). Объединенные органические слои промывали солевым раствором (1×200 мл), сушили над безводным сульфатом магния (15 г), концентрировали при пониженном давлении и получали 132 г (83%) титульного соединения в виде кристаллического вещества белого цвета. 1Н ЯМР (60 МГц) (CDCl3) Метил-2-(3-([4-метокси-2-нитрофенил)амино]сульфонил)фенокси)ацетат (Полупродукт А23) Твердый метил-2-[4-(хлорсульфонил)фенокси]ацетат (Полупродукт А22, 63,5 г, 0,24 моль) прибавляли к раствору 4-метокси-2-нитроанилина (33,6 г, 0,2 моль) в пиридине (1 л) при комнатной температуре под аргоном. Полученную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную массу концентрировали при пониженном давлении. Остаток распределяли между 1,5 М соляной кислотой (1000 мл) и этилацетатом (500 мл). Отделяли водный слой и экстрагировали его этилацетатом (2×250 мл). Объединенные органические фракции промывали солевым раствором (1×200 мл), сушили над безводным сульфатом магния (15 г), концентрировали при пониженном давлении и получали осадок оранжевого цвета. Осадок растворяли в кипящем этилацетате (500 мл), выдерживали 1 час при комнатной температуре и 4 часа в морозильной камере. Отфильтровывали кристаллы желтого цвета и после сушки на воздухе получали 64,2 г (81%) титульного соединения. 1Н ЯМР (60 МГц) (CDCl3) Метил-2-(3-([2-амино-4-метоксифенил)амино]сульфонил)фенокси)ацетат (Полупродукт А24) Раствор метил-2-(3-([4-метокси-2-нитрофенил)амино]сульфонил)фенокси)ацетата (Полупродукта А23, 22,57 г, 0,057 моль) в этилацетате (500 мл) суспендировали с никелем Ренея (3 г), фильтровали и затем гидрировали над оксидом платины(IV) (0,5 г) до прекращения поглощения водорода. По окончании реакционная масса содержала некоторое количество твердого продукта. Кристаллы отфильтровывали, сушили на воздухе и получали 15,6 г технического продукта, содержащего 0,5 г катализатора. Фильтрат концентрировали при пониженном давлении и получали еще 3,1 г продукта. Общий вес продукта, титульного соединения, составил 18,2 г (87%). 1Н ЯМР (60 МГц) (d6-ДМСО) Метил-2-{4-[(5-метокси-2-сульфанилбензимидазолил)сульфонил}ацетат (Полупродукт А25) Технический метил-2-(3-([2-амино-4-метоксифенил)амино]сульфонил)фенокси)ацетат (Полупродукт А24, 17,93 г, приблизительно 0,05 моль) растворяли в пиридине (200 мл) и через фильтровальную бумагу со стекловолокном отфильтровывали катализатор с предыдущей стадии. В раствор загружали 1,1′-тиокарбонилдиимидазол (13,1 г, 0,0735 моль) и перемешивали при комнатной температуре в течение ночи под аргоном. Затем в реакционную массу добавляли воду (2,5 л) и продолжали перемешивание в течение еще 1,5 часов. Выпавшие кристаллы отфильтровывали, промывали 2 л воды, сушили на воздухе и получали 20 г (100%) титульного соединения. 1Н ЯМР (60 МГц) (d6-ДМСО) Метил-2-[4-({5-метокси-2-[(4-метокси-3,5-диметил-(2-пиридил)метилтио]-бензимидазолил}сульфонил)фенокси]ацетат (Полупродукт А26) Безводный карбонат калия (14,88 г, 0,108 моль) растирали в ступке, толкли и прибавляли к раствору метил-2-{4-[(5-метокси-2-сульфанилбензимидазолил)сульфонил}ацетата (Полупродукта А25, 19,99 г, 0,049 моль) в N,N-диметилформамиде (150 мл). Затем к смеси прибавляли гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина (11,42 г, 0,051 моль) и интенсивно перемешивали под аргоном при комнатной температуре. Через 2 часа анализ методом ВЭЖХ показал, что реакция завершилась. К реакционной массе прибавляли сначала смесь СН2Cl2-ИПС (75:25) (800 мл), затем воду (400 мл). После перемешивания в течение 10 минут отделяли органический слой, промывали его водой (400 мл), затем солевым раствором (400 мл) и сушили над безводным сульфатом магния. Растворитель отгоняли на роторном испарителе сначала при пониженном давлении, а затем в глубоком вакууме и получали осадок коричневого цвета. Этот коричневый осадок растирали в смеси 35% этилацетат-гексан (250 мл) и перемешивали в течение 1 часа. Кристаллы отфильтровывали, промывали смесью 35% этилацетат-гексан (100 мл), сушили на воздухе и получали 21 г (77%) титульного соединения. 1Н ЯМР (60 МГц) (CDCl3) Метил-2-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]феноксиацетат (Полупродукт А27) Раствор 3-хлорпероксибензойной кислоты (12,64 г 70%, 0,051 моль) в ТГФ (150 мл) прибавляли к холодному раствору сульфида (Полупродукта А26, 22,84 г, 0,041 моль) в ТГФ (500 мл). Полученную смесь выдерживали в морозильной камере в течение ночи (19 часов). Анализ методом ВЭЖХ показал наличие 58% метилового эфира сульфоксида, 6% непрореагировавшего исходного сульфида и присутствие двух других неидентифицированных примесей (8 и 24%). Реакционную массу разбавляли этилацетатом (750 мл) и промывали 5% метабисульфитом натрия (2×200 мл), 5% бикарбонатом натрия (2×200 мл) и солевым раствором (2×200 мл). Полученный раствор фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали 26 г пены. Пену очищали с помощью флэш-хроматографии (силикагель, от этилацетата до 1% МеОН-этилацетат) и получали 16,2 г технического продукта в виде пены. Эту пену растирали со смесью 10% этилацетат-гексан (100 мл). Отфильтровывали осадок белого цвета, сушили на воздухе и получали 15,25 г (65%) титульного соединения. 1Н ЯМР (60 МГц) (CDCl3) 2-{4-[(5-Метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфонил}бензимидазолил)сульфонил]фенокси}уксусная кислота (Соединение 9) В 3-литровую 3-горлую колбу, оборудованную механической мешалкой, загружали метил-2-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфонил}бензимидазолил)сульфонил]фенокси}ацетат (Полупродукт А27, 13,75 г, 0,024 моль) в глиме (1200 мл). К этому интенсивно перемешиваемому раствору прибавляли 0,1N раствор NaOH (300 мл). Полученную смесь концентрировали при пониженном давлении и температуре 47°С. Остаток растирали со свежим этилацетатом (100 мл) в общей сложности четыре раза. Остаток выдерживали в течение 1 часа в глубоком вакууме и получали 17 г смолистых кристаллов. В литровую 3-горлую колбу, оборудованную механической мешалкой, помещали 16 г полученных кристаллов (эквивалент 13,3 г натриевой соли, 0,0226 моль) в воде (250 мл). Раствор промывали перемешиванием с этилацетатом (250 мл) в течение 1 часа и затем разделяли слои. Анализ методом ВЭЖХ этилацетатного слоя показал наличие 64% омепразола и 33% непрореагировавшего метилового эфира сульфоксида. К водному слою прибавляли 250 мл свежего этилацетата. Полученную смесь перемешивали и подкисляли до рН 3-4 приблизительно 40 мл 0,5N соляной кислоты. Поскольку продукт начинал кристаллизоваться, быстро отделяли этилацетатный слой и выдерживали его в течение ночи в морозильной камере. Кристаллы отфильтровывали, сушили на воздухе и получали 6,7 г (53%) метил-2-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфонил}бензимидазолил)сульфонил]фенокси}ацетата. Натриевая соль 2-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфонил}бензимидазолил)сульфонил]фенокси}уксусной кислоты (Соединение 9) Сульфоксид кислоты (4,81 г, 0,0086 моль) суспендировали в глиме (500 мл). Полученную смесь перемешивали на магнитной мешалке и в течение 20 минут прибавляли 1N гидроксид натрия (8,6 мл, 0,0086 моль). Отфильтровывали нерастворившуюся часть, фильтрат концентрировали в вакууме и получали кристаллы рыжевато-коричневого цвета. Эти кристаллы суспендировали в этилацетате (100 мл) и перемешивали в течение 1 часа. Осадок отфильтровывали, сушили на воздухе и получали 4,1 г титульного соединения. 1Н ЯМР (60 МГц) (CDCl3, d6-ДМСО)
3-(Хлорсульфонил)-4-метоксибензойная кислота (Полупродукт А28) Твердую 4-метоксибензойную кислоту (45,6 г, 0,3 моль) при комнатной температуре в несколько приемов прибавляли к хлорсульфоновой кислоте (139,8 г, 80 мл, 1,2 моль). После прекращения основного газовыделения реакционную смесь нагревали при температуре 80°С в течение 1 часа. Затем реакционную массу выливали в интенсивно перемешиваемый колотый лед (500 г) и добавляли воду (500 мл). Через 30 минут выпавшие кристаллы белого цвета отфильтровывали, промывали водой (2 л), сушили и получали 50 г (66%) титульного соединения. 2-(Толуол-4-сульфонил)этиловый эфир 3-хлорсульфонил-4-метоксибензойной кислоты (Полупродукт А29) Смесь 3-(хлорсульфонил)-4-метоксибензойной кислоты (Полупродукта А28, 13,78 г, 0,055 моль) и хлористого тионила (39,27 г, 25 мл, 0,33 моль) кипятили с обратным холодильником в течение 1 часа. Избыток хлористого тионила отгоняли сначала при атмосферном давлении, затем при пониженном давлении и получали 15 г хлорангидрида кислоты в виде кристаллов светло-коричневого цвета. К смеси хлорангидрида кислоты (15 г) и 2-(п-толилсульфонил)этанола (10,46 г, 0,0522 моль) в дихлорметане (100 мл) при комнатной температуре прибавляли триэтиламин (5,56 г, 0,055 моль). Анализ реакционной смеси методом ТСХ (в этилацетате) показал, что реакция полностью завершилась через час. Реакционную массу промывали водой (2×100 мл) и солевым раствором (1×100 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло. Масло растирали со смесью 15% этилацетат-гексан (100 мл), осадок отфильтровывали, сушили и получали 19,4 г, (81%) титульного соединения, эфира сульфонилхлорида, в виде кристаллического вещества белого цвета. 2-(Толуол-4-сульфонил)этиловый эфир 4-метокси-3-(4-метокси-2-нитрофенилсульфамоил)бензойной кислоты (Полупродукт А30) Смешивали сульфонилхлорид (Полупродукт А29, 18,17 г, 0,042 моль), 3-метокси-2-нитроанилин (7,06 г, 0,042 моль) и пиридин (100 мл) и перемешивали их в течение ночи при комнатной температуре под аргоном. Реакционную массу концентрировали при пониженном давлении. Остаток растворяли в этилацетате (250 мл), промывали 3 М соляной кислотой (3×100 мл) и солевым раствором (1×100 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло оранжево-красного цвета. Масло очищали с помощью флэш-хроматографии (силикагель, от 20% этилацетата в гексане до 50% этилацетата в гексане) и получали 13,8 г (57%) титульного соединения, нитросульфонамида, в виде пены. 2-(Толуол-4-сульфонил)этиловый эфир 3-(2-амино-4-метоксифенилсульфамоил)-4-метоксибензойной кислоты (Полупродукт А31) Раствор нитросульфонамида (Полупродукта А30, 13,54 г, 0,024 моль) в этилацетате (400 мл) перемешивали в течение 15 минут с никелем Ренея (3 г), фильтровали через слой целита и гидрировали над оксидом платины(IV) (1 г) в течение ночи. Реакционную массу фильтровали через бумажный фильтр со стекловолокном, концентрировали при пониженном давлении и получали 11,8 г (92%) титульного соединения, амина, в виде пены. 2-(Толуол-4-сульфонил)этиловый эфир 4-(2-меркапто-5-метоксибензимидазол-1-сульфонил)-3-метоксибензойной кислоты (Полупродукт А32) К раствору аминосоединения (Полупродукта А31, 11,75 г, 0,022 моль) в пиридине (150 мл) прибавляли 1,1′-тиокарбонилдиимидазол (5,88 г, 0,033 моль). Реакционную массу перемешивали при комнатной температуре в течение 2 часов и затем добавляли воду (600 мл). После перемешивания в течение 1,5 часов осадок отфильтровывали, промывали водой (1 л), сушили и получали 10,4 г (82%) титульного соединения, бензимидазолтиола. 2-(Толуол-4-сульфонил)этиловый эфир4-метокси-3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметилсульфанил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукт А33) Безводный карбонат калия (5,38 г, 0,039 моль) растирали в ступке, толкли и прибавляли к раствору бензимидазолтиола (Полупродукта А32, 10,2 г, 0,0177 моль) в N,N-диметилформамиде (100 мл). К этой смеси прибавляли гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина (4,13 г, 0,186 моль). Через 2 часа в реакцию добавляли 150 мл 25% изопропилового спирта в дихлорметане. Реакционную массу промывали водой (2×75 мл) и солевым раствором (1×75 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло коричневого цвета. Масло очищали флэш-хроматографией (силикагель, от 40% этилацетата в гексане до этилацетата) и получали 11 г (96%) титульного соединения, сульфида, в виде пены. 2-(Толуол-4-сульфонил)этиловый эфир 4-метокси-3-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]бензойной кислоты (Полупродукт А34) Сульфид (Полупродукт А33, 10,77 г, 0,0166 моль) растворяли в тетрагидрофуране (200 мл) и выдерживали в течение 1 часа в морозильной камере. После добавления холодного раствора м-хлорпероксибензойной кислоты (5,1 г, 70%, 0,0207 моль) в тетрагидрофуране (100 мл) реакционную массу возвращали в холодильник и оставляли там в течение ночи. Реакционную смесь разбавляли этилацетатом (300 мл) и промывали 5% метабисульфитом натрия (3×100 мл), насыщенным бикарбонатом натрия (2×100 мл) и солевым раствором (1×100 мл). Органическую фазу фильтровали через фильтровальную бумагу 1PS и концентрировали при пониженном давлении. Концентрат очищали флэш-хроматографией (силикагель, от 50% этилацетата в гексане до 4% метанола в этилацетате) и получали 5,7 г (52%) титульного соединения, сульфоксида, в виде пены. Натриевая соль 4-метокси-3-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]бензойной кислоты (Соединение 44) К раствору сульфоксида (Полупродукта А34, 5,52 г, 0,0083 моль) в смеси ацетонитрила (45 мл), изопропилового спирта (15 мл), воды (30 мл) прибавляли бикарбонат натрия (0,837 г, 0,00996 моль). Полученную массу нагревали при температуре 78°С в течение 45 минут. Смесь концентрировали при пониженном давлении и получали пену. Пену растирали со смесью 2% метанол-дихлорметан и получали 4,5 г кристаллического вещества. Кристаллы растворяли в воде (150 мл), затем прибавляли смесь 4% метанол-хлороформ (100 мл) и подкисляли 3 М соляной кислотой (3 мл). Водный слой экстрагировали смесью 4% метанол-хлороформ (2×100 мл). Объединенные органические слои промывали водой (1×100 мл), концентрировали при пониженном давлении до постоянного веса и получали 3,6 г пены. Пену растворяли в глиме (100 мл) и добавляли 0,2N раствор гидроксида натрия (7,45 мл). Полученную смесь концентрировали при пониженном давлении, растирали с эфиром (50 мл) и после сушки получали 2,45 г твердого вещества. Это кристаллическое вещество растворяли в горячем дихлорметане, фильтровали горячим, концентрировали при пониженном давлении и получали 1,8 г (43%) титульного соединения 1Н ЯМР (300 МГц)
Метил-2,2-диметил-3-(4-толилсульфонилокси)пропионат (Полупродукт А35) Смесь метил-2,2-диметил-3-гидроксипропионата (100 г, 0,76 моль), 4-толуолсульфонилхлорида (151 г, 0,80 моль), 4-диметиламинопиридина (4,6 г, 0,038 моль) и пиридина (200 мл) перемешивали в течение 20 часов, затем разбавляли 200 мл толуола, перемешивали в течение еще 30 минут и фильтровали. Фильтрат концентрировали при пониженном давлении до 250 мл, разбавляли 100 мл толуола, фильтровали и концентрировали. Остаток суспендировали в 200 мл гексана, растворитель отгоняли при пониженном давлении и получали титульное соединение, требуемый продукт (235 г, 100%), содержащий следы 4-толуолсульфонилхлорида. Метил-2,2-диметил-3-феноксипропионат (Полупродукт А36) Смесь метил-2,2-диметил-3-(4-толуолсульфонилокси)пропионата (Полупродукта А35, 80 г, 0,28 моль), фенола (28 г, 0,029 моль), карбоната калия (58 г, 0,42 моль) и 250 мл N,N-диметилацетамида перемешивали и кипятили с обратным холодильником в течение 4 часов. Реакционную массу охлаждали, разбавляли этилацетатом и выливали в воду. Органическую фракцию промывали водой и концентрировали при пониженном давлении. Остаток растворяли в гексане и промывали водой, 1 М раствором гидроксида натрия и дополнительно водой. Концентрированный раствор перегоняли при давлении 15 торр (34-37°С) и получали 25 г (42%) метил-2,2-диметил-3-феноксипропионата. Метиловый эфир 3-(4-хлорсульфонилфенокси)-2,2-диметилпропионовой кислоты (Полупродукт А37) Метил-2,2-диметил-3-феноксипропионат (Полупродукт А36, 36 г, 0,17 моль) прибавляли по каплям в течение 60 минут к быстро перемешиваемой хлорсульфоновой кислоте, поддерживая температуру -5°С. Смесь нагревали до комнатной температуры, перемешивали еще 90 минут и вливали в охлажденную быстро перемешиваемую смесь дихлорметана (250 мл) и метанола (30 мл). Реакционную массу перемешивали в течение 30 минут при охлаждении и в течение еще 60 минут при комнатной температуре. Затем ее промывали несколькими порциями ледяной воды. Объединенные водные слои экстрагировали небольшой порцией дихлорметана. Объединенные органические фракции промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Растворитель отгоняли при пониженном давлении, остаток перемешивали с гексаном, фильтровали и получали 19 г (36%) титульного соединения, сульфонилхлорида. Метиловый эфир 3-[4-(4-метокси-2-нитрофенилсульфамоил)фенокси]-2,2-диметилпропионовой кислоты (Полупродукт А38) Смешивали хлорсульфонильное соединение (Полупродукт А37, 18,4 г, 0,06 моль), 3-метокси-2-нитроанилин (8,4 г, 0,05 моль) и пиридин (500 мл) и перемешивали их в течение ночи при комнатной температуре под аргоном. Реакционную массу концентрировали при пониженном давлении и остаток распределяли между 1,5 М соляной кислотой (300 мл) и этилацетатом (300 мл). Отделяли водный слой и экстрагировали его этилацетатом (2×150 мл). Объединенные органические фракции промывали солевым раствором (2×150 мл), сушили над безводным сульфатом магния, концентрировали при пониженном давлении и получали маслянистый остаток оранжевого цвета. Масло очищали флэш-хроматографией (силикагель, от гексана до 35% этилацетата в гексане) и получали 20,8 г (95%) титульного соединения, нитросульфонамида, в виде кремообразного масла. Метиловый эфир 3-[4-(2-амино-4-метоксифенилсульфамоил)фенокси]-2,2-диметилпропионовой кислоты (Полупродукт А39) Раствор нитросульфонамида (Полупродукт А38, 20,6 г, 0,047 моль) в этилацетате (300 мл) перемешивали в течение 15 минут с никелем Ренея (1,5 г), фильтровали через слой целита и гидрировали над оксидом платины(IV) (0,5 г) в течение ночи. Реакционную массу фильтровали через бумажный фильтр со стекловолокном, концентрировали при пониженном давлении и получали 16,3 г (85%) титульного соединения, амина, в виде пены. Метиловый эфир 3-[4-(2-меркапто-5-метоксибензимидазол-1-сульфонил)фенокси]-2,2-диметилпропионовой кислоты (Полупродукт А40) К раствору метилового эфира 3-[4-(2-меркапто-5-метоксибензимидазол-1-сульфонил)фенокси]-2,2-диметилпропионовой кислоты (Полупродукта А39, 16,1 г, 0,0395 моль) в пиридине (200 мл) прибавляли 1,1′-тиокарбонилдиимидазол (10,56 г, 0,0592 моль). Реакционную массу перемешивали при комнатной температуре в течение ночи и затем добавляли воду (2,5 л). После перемешивания в течение 1 часа осадок отфильтровывали, промывали водой (3 л), сушили и получали 16,5 г (93%) титульного соединения, бензимидазолтиола, в виде кристаллов рыжевато-коричневого цвета. Метиловый эфир 3-{4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметилсульфанил)бензимидазол-1-сульфонил]фенокси}-2,2-диметилпропионовой кислоты (Полупродукт А41) К раствору бензимидазолтиола (Полупродукта А40, 16,2 г, 0,036 моль) в N,N-диметилформамиде (150 мл) прибавляли карбонат калия (10,93 г, 0,079 моль). К этой смеси прибавляли твердый гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина (8,39 г, 0,0378 моль). Через час в реакционную массу добавляли 800 мл смеси 25% изопропилового спирта в дихлорметане. Далее реакционную массу промывали водой (2×400 мл). Объединенные водные фракции снова экстрагировали 100 мл 25% изопропилового спирта в дихлорметане. Объединенные органические слои промывали солевым раствором (1×400 мл), сушили над безводным сульфатом магния (20 г), концентрировали при пониженном давлении и получали масло. Масло закристаллизовалось при стоянии и его растирали со смесью 25% этилацетат-гексан (200 мл). Продукт, титульное соединение (сульфид), отфильтровывали и сушили (20,2 г (93%)). Литиевая соль 3-{4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметилсульфанил)бензимидазол-1-сульфонил]фенокси}-2,2-диметилпропионовой кислоты (Полупродукт А42) К раствору сульфида (Полупродукта А41, 9,58 г, 0,016 моль) в глиме (800 мл) в течение 30 минут прибавляли 1N раствор гидроксида натрия (240 мл). Через 3,5 часа добавляли уксусную кислоту (28,8 г) приблизительно до рН 6,5. Основную часть глима отгоняли при пониженном давлении. К остатку добавляли воду (250 мл) и смесь экстрагировали дихлорметаном (2×125 мл). Объединенные органические фракции промывали водой (1×100 мл) и солевым раствором (1×100 мл), фильтровали через фильтровальную бумагу 1PS и концентрировали при пониженном давлении. К раствору остатка (10 г) в дихлорметане прибавляли сначала 0,5 М раствор гидроксида лития (100 мл), а затем солевой раствор (100 мл). Полученную смесь перемешивали в течение 30 минут, после чего осадок отфильтровывали, промывали водой (50 мл), сушили и получали 4,1 г (43%) литиевой соли титульного соединения, сульфида. 3-{4-[5-Метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}-2,2-диметилпропионовая кислота (Полупродукт А43) Литиевую соль сульфида (Полупродукта А42, 4,02 г, 0,0068 моль) растворяли в тетрагидрофуране (250 мл) и охлаждали в морозильной камере в течение 1 часа. Прибавляли 3-хлорпероксибензойную кислоту (5,03 г, 0,0204 моль), полученную смесь возвращали в холодильник и выдерживали там в течение ночи. Реакционную массу разбавляли этилацетатом (600 мл), промывали 5% раствором метабисульфита натрия (3×150 мл), водой (2×150 мл), солевым раствором (2×150 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали кристаллический остаток. Кристаллы очищали флэш-хроматографией (силикагель, этилацетат) и получали 1,3 г (29%) титульного соединения, сульфоксида, в виде пены. Натриевая соль 3-{4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}-2,2-диметилпропионовой кислоты (Соединение 45) 1N раствор гидроксида натрия (2 мл) прибавляли к раствору сульфоксида (Полупродукта А43, 1,2 г, 0,002 моль) в глиме (25 мл). Полученную смесь концентрировали при пониженном давлении и получали кристаллический остаток. Этот кристаллический остаток растирали с этилацетатом (25 мл). Продукт отфильтровывали, сушили и получали 1,05 г (85%) титульного соединения в виде кристаллов белого цвета с желтоватым оттенком. 1Н ЯМР (300 МГц) (d6-ДМСО)
Методика 1 для получения нитросульфонамида (Полупродукта А45) Метиловый эфир (4-сульфамоилфенокси)уксусной кислоты (Полупродукт А44) В 2-литровую 3-горлую колбу, оборудованную механической мешалкой и термометром, помещали сульфонилхлорид (Полупродукт А22, 68,77 г, 0,26 моль) в метаноле (650 мл) и нагревали до температуры 30°С для растворения. К полученному раствору прибавляли метанольный раствор амммиака (159 мл, 4,9 М). Реакционную массу перемешивали в течение 2 часов при комнатной температуре и затем выдерживали в морозильной камере в течение 4 часов. Осадок отфильтровывали, промывали метанолом (100 мл), гексаном (300 мл), сушили и получали 55 г (86%) титульного соединения, сульфонамида, в виде кристаллического вещества белого цвета. 3-Фтор-4-нитроанизол Раствор 3-фтор-4-нитрофенола (75 г, 0,48 моль) в ацетоне (700 мл) охлаждали на ледяной бане. Затем в течение приблизительно 5 минут прибавляли 1,8-диазабицикло[5,4,0]ундек-7-ен (145 г, 0,96 моль). После этого в течение 10 минут прибавляли иодистый метил (135 г, 0,96 моль). Массу перемешивали при комнатной температуре в течение 16 часов. Прибавляли дополнительное количество 1,8-диазабицикло[5,4,0]ундек-7-ена (73 г, 0,48 моль) и иодистого метила (68 г, 0,48 моль) и смесь нагревали при температуре 50°С в течение 1 часа. Осадок отфильтровывали, фильтрат концентрировали и смешивали с этилацетатом и 1 М соляной кислотой. Органический слой промывали дополнительным количеством соляной кислоты и водным раствором бикарбоната натрия, концентрировали и перемешивали со смесью 1% этилацетат-гексан. Выпавшие кристаллы отфильтровывали, сушили и получали 72 г (88%) титульного соединения. Метиловый эфир [4-(5-метокси-2-нитрофенилсульфамоил)фенокси]уксусной кислоты (Полупродукт А45) Сульфонамид (Полупродукт А44, 44,6 г, 0,18 моль), карбонат калия (41,4 г, 0,3 моль), 3-фтор-4-нитроанизол (25,65 г, 0,15 моль) и N,N-диметилацетамид (250 мл) кипятили с обратным холодильником в течение 2,5 часов. Реакционную массу выливали в смесь колотого льда (800 г) и воды (400 мл). Полученную смесь экстрагировали этилацетатом (3×500 мл). Объединенные органические слои промывали солевым раствором (1×200 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло. Масло очищали флэш-хроматографией (силикагель, от гексана до 50% этилацетата в гексане) и получали 9,4 г (16%) титульного соединения, нитросульфонамида, в виде кристаллов желтого цвета. Методика 2 для получения нитросульфонамида (Полупродукта А45) Метиловый эфир [4-(3-метоксифенилсульфамоил)фенокси]уксусной кислоты (Полупродукт А46) Твердый сульфонилхлорид (Полупродукт А22, 29,1 г, 0,11 моль) в несколько приемов прибавляли к раствору м-анизидина (12,3 г, 0,1 моль) в пиридине (250 мл). Полученную массу перемешивали в течение ночи при комнатной температуре под аргоном. Реакционную смесь выливали в холодный раствор 3 М соляной кислоты (1 л) и перемешивали в течение 30 минут. Отфильтровывали кристаллы розового цвета, промывали водой, сушили и получали 33 г (94%) сульфонамида. Метиловый эфир [4-(5-метокси-2-нитрофенилсульфамоил)фенокси]уксусной кислоты (Полупродукт А45) Холодную смесь азотной кислоты (90 мл, 70%) и уксусной кислоты (140 мл) прибавляли к раствору сульфонамида (Полупродукт А46, 31,59 г, 0,09 моль) в дихлорметане (750 мл) при температуре -10°С в течение 15 минут. Через 3,5 часа в реакционную массу добавляли воду (1 л). Органический слой отделяли, промывали водой (1×300 мл), насыщенным бикарбонатом натрия (1×300 мл), солевым раствором (1×300 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали 40 г масла темно-коричневого цвета. Масло очищали флэш-хроматографией (силикагель, от 30% этилацетата в гексане до этилацетата) и получали 19,2 г остатка, который после растирания с этилацетатом давал 7,5 г (21%) титульного соединения, нитросульфонамида. Метиловый эфир [4-(2-амино-5-метоксифенилсульфамоил)фенокси]уксусной кислоты (Полупродукт А47) Раствор нитросульфонамида (Полупродукта А45, 17,5 г, 0,0044 моль) в тетрагидрофуране (700 мл) обрабатывали никелем Ренея (2 г) в течение 15 минут, фильтровали через слой целита и гидрировали над оксидом платины(IV) (1,5 г) до прекращения поглощения водорода. Реакционную массу фильтровали через бумажный фильтр со стекловолокном, концентрировали при пониженном давлении и получали 16,2 г (100%) титульного соединения, амина, в виде кристаллов рыжевато-коричневого цвета. Метиловый эфир [4-(2-меркапто-6-метоксибензимидазол-1-сульфонил)фенокси]уксусной кислоты (Полупродукт А48) Амин (Полупродукт А47, 16 г, 0,0437 моль) растворяли в пиридине (200 мл). К полученному раствору прибавляли 1,1′-тиокарбонилдиимидазол (11,68 г, 0,0655 моль) и перемешивали при комнатной температуре в течение ночи под аргоном. К реакционной массе добавляли воду (400 мл) и перемешивали в течение 1 часа. Выпавшие кристаллы отфильтровывали, промывали водой (600 мл), сушили и получали 16,9 г (95%) титульного соединения, бензимидазолтиола. Метиловый эфир {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметилсульфанил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Полупродукт А49) Безводный карбонат калия (12,45 г, 0,09 моль) растирали в ступке, толкли и прибавляли к раствору тиола (Полупродукта А48, 16,7 г, 0,041 моль) в N,N-диметилформамиде (275 мл). К реакционной смеси прибавляли гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина (9,56 г, 0,043 моль). Через час добавляли воду (1 л) и перемешивали при комнатной температуре еще 15 минут. Осадок отфильтровывали, промывали водой (1 л), сушили и получали 20,3 г (89%) титульного соединения, сульфида. Метиловый эфир {4-[6-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенокси}уксусной кислоты (Полупродукт А50) Сульфид (Полупродукт А49, 20,05 г, 0,036 моль) растворяли в тетрагидрофуране (350 мл) и выдерживали в морозильной камере в течение 1 часа. После прибавления холодного раствора м-хлорпероксибензойной кислоты (11,6 г, 70%, 0,047 моль) в тетрагидрофуране (150 мл) реакционную смесь возвращали в холодильник и оставляли там в течение ночи. Затем реакционную массу разбавляли этилацетатом (750 мл), промывали 5% метабисульфитом натрия (2×200 мл), насыщенным бикарбонатом натрия (2×200 мл) и солевым раствором (2×200 мл), фильтровали через фильтровальную бумагу 1PS и концентрировали при пониженном давлении. Концентрат очищали флэш-хроматографией (силикагель, от гексанадо этилацетата) и получали 14,5 г пены. Пену растирали со смесью 35% этилацетат-гексан и получали 13 г (63%) титульного соединения, сульфоксида, в виде кристаллического вещества белого цвета. Натриевая соль 2-{4-[(6-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]фенокси}уксусной кислоты (Соединение 46) 0,1N раствор гидроксида натрия (2 мл) прибавляли к раствору сульфоксида (Полупродукта А50, 11,46 г, 0,02 моль) в глиме (750 мл). Полученную смесь экстрагировали этилацетатом (2×400 мл). Объединенные органические фракции повторно экстрагировали водно-солевым раствором, 1:1 (200 мл). Объединенные водные слои подкисляли 3 М соляной кислотой (20 мл). Добавляли солевой раствор (200 мл) и полученную смесь экстрагировали этилацетатом (5×250 мл). Объединенные органические слои фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали кристаллический остаток. Кристаллический остаток растирали с этилацетатом (100 мл). Продукт отфильтровывали, сушили и получали 4,5 г (40%) 2-{4-[(6-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]фенокси}уксусной кислоты (сульфоксид кислоты). К суспензии сульфоксида кислоты (4,47 г, 0,008 моль) в глиме (75 мл) прибавляли раствор бикарбоната натрия (0,672 г, 0,008 моль) в воде (50 мл). Реакционную массу экстрагировали этилацетатом (4×50 мл). Объединенные органические слои повторно экстрагировали водой (2×25 мл). Объединенные водные слои лиофилизировали и получали 4,1 г (88%) титульного соединения. 1Н ЯМР (300 МГц) (D2O)
3-[4-(Хлорсульфонил)фенил]пропионовая кислота (Полупродукт А51) Твердую 3-фенилпропионовую кислоту (45 г, 0,3 моль) при температуре от -5 до 0°С в течение 45 минут порциями прибавляли к хлорсульфоновой кислоте (174,75 г, 100 мл, 1,5 моль). Через час реакционную массу выливали в интенсивно перемешиваемый колотый лед (500 г). Через 30 минут отфильтровывали кристаллы белого цвета, промывали водой (1,5 л), сушили и получали 12 г смеси двух изомеров. Целевой изомер отделяли с помощью флэш-хроматографии (силикагель, от 50% этилацетата в гексане до 1% метанола в этилацетате) и получали 9 г (12%) титульного соединения. 2-(Толуол-4-сульфонил)этиловый эфир 3-(4-хлорсульфонилфенил)пропионовой кислоты (Полупродукт А52) Смесь 3-[4-(хлорсульфонил)фенил]пропионовой кислоты (Полупродукта А51, 8,2 г, 0,033 моль) и хлористого тионила (23,56 г, 0,198 моль) кипятили с обратным холодильником в течение 1 часа. Избыток хлористого тионила отгоняли сначала при атмосферном давлении, а затем при пониженном и получали 9 г хлорангидрида кислоты в виде масла. К смеси хлорангидрида кислоты (9 г) и 2-(п-толилсульфонил)этанола (6,28 г, 0,0313 моль) в дихлорметане (25 мл) при комнатной температуре прибавляли триэтиламин (3,33 г, 0,033 моль). Анализ методом ТСХ (этилацетат) показал, что реакция полностью завершилась через 1 час. Реакционную массу промывали водой (2×50 мл) и солевым раствором (1×50 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло. Масло очищали флэш-хроматографией (силикагель, от гексана до 50% этилацетата в гексане) и получали 7,5 г (56%) титульного соединения, эфира сульфонилхлорида, в виде кристаллического вещества белого цвета. 2-(Толуол-4-сульфонил)этиловый эфир 3-[4-(4-метокси-2-нитрофенилсульфамоил)фенил]пропионовой кислоты (Полупродукт А53) Смешивали сульфонилхлорид (Полупродукт А52, 7,75 г, 0,018 моль), 3-метокси-2-нитроанилин (3,02 г, 0,018 моль) и пиридин (60 мл) и перемешивали их при комнатной температуре в течение ночи под аргоном. Реакционную массу концентрировали при пониженном давлении. Остаток растворяли в этилацетате (100 мл), промывали 3 М соляной кислотой (2×75 мл) и солевым раствором (1×100 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло красного цвета. Масло очищали флэш-хроматографией (силикагель, от 15% этилацетата в гексане до этилацетата) и получали 7,7 г (76%) титульного соединения, нитросульфонамида, в виде вязкого масла красного цвета. 2-(Толуол-4-сульфонил)этиловый эфир 3-[4-(2-амино-4-метоксифенилсульфамоил)фенил]пропионовой кислоты (Полупродукт А54) Раствор нитросульфонамида (Полупродукта А53, 7,47 г, 0,0133 моль) в этилацетате (100 мл) перемешивали в течение 15 минут с никелем Ренея (3 г), фильтровали через слой целита и гидрировали над оксидом платины(IV) (0,25 г) в течение ночи. Реакционную массу фильтровали через бумажный фильтр со стекловолокном, концентрировали при пониженном давлении и получали 6,85 г (97%) титульного соединения, амина, в виде пены светло-желтого цвета. 2-(Толуол-4-сульфонил)этиловый эфир 3-[4-(2-меркапто-5-метоксибензимидазол-1-сульфонил)фенил]пропионовой кислоты (Полупродукт А55) К раствору амина (Полупродукта А54, 6,7 г, 0,0126 моль) в пиридине (100 мл) прибавляли 1,1′-тиокарбонилдиимидазол (3,37 г, 0,0189 моль). Реакционную массу перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь разбавляли водой (150 мл) и экстрагировали смесью дихлорметан-изопропиловый спирт, 3:1 (2×100 мл). Объединенные органические слои промывали солевым раствором (1×100 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали 8 г вязкого масла. Масло очищали флэш-хроматографией (силикагель, от 20% этилацетата в гексане до 75% этилацетата в гексане) и получали 5,8 г (80%) титульного соединения, бензимидазолтиола, в виде пены светло-желтого цвета. 2-(Толуол-4-сульфонил)этиловый эфир 3-{4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметилсульфанил)бензимидазол-1-сульфонил]фенил}пропионовой кислоты (Полупродукт А56) Безводный карбонат калия (3,04 г, 0,022 моль) растирали в ступке, толкли и прибавляли к раствору тиола (Полупродукта А55, 5,74 г, 0,01 моль) в N,N-диметилформамиде (50 мл). Прибавляли гидрохлорид 2-хлорметил-3,5-диметил-4-метоксипиридина (2,33 г, 0,0105 моль). Через 2 часа в реакционную массу добавляли 100 мл смеси 25% изопропилового спирта в дихлорметане. Затем реакционную массу промывали водой (2×50 мл) и солевым раствором (1×50 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали масло светло-коричневого цвета. Масло очищали флэш-хроматографией (силикагель, от 20% этилацетата в гексане до 75% этилацетата) и получали 6,47 г (100%) титульного соединения, сульфида, в виде пены. 2-(Толуол-4-сульфонил)этиловый эфир 3-{4-[5-метокси-2-(4-метокси-3,5-диметилпиридин-2-илметансульфинил)бензимидазол-1-сульфонил]фенил}пропионовой кислоты (Полупродукт А57) Сульфид (Полупродукт А56, 6,47 г, 0,01 моль) растворяли в тетрагидрофуране (100 мл) и выдерживали в морозильной камере в течение 1 часа. После добавления холодного раствора м-хлорпероксибензойной кислоты (3,08 г, 70%, 0,0125 моль) в тетрагидрофуране (50 мл) реакционную массу возвращали в морозильную камеру и оставляли там в течение ночи. Затем реакционную массу разбавляли этилацетатом (250 мл) и промывали 5% метабисульфитом натрия (2×100 мл), насыщенным бикарбонатом натрия (2×100 мл) и солевым раствором (1×100 мл). Органический слой фильтровали через фильтровальную бумагу 1PS и концентрировали при пониженном давлении. Концентрат очищали флэш-хроматографией (силикагель, от 25% этилацетата в гексане до 1% метанола в этилацетате) и получали 3,8 г (57%) титульного соединения, сульфоксида, в виде пены белого цвета. Натриевая соль 3-{4-[(5-метокси-2-{[(4-метокси-3,5-диметил-(2-пиридил))метил]сульфинил}бензимидазолил)сульфонил]фенил}пропионовой кислоты (Соединение 47) К раствору сульфоксида (Полупродукта А57, 3,71 г, 0,005 моль) в ацетонитриле (30 мл), изопропиловом спирте (10 мл), воде (20 мл) прибавляли бикарбонат натрия (0,5 г, 0,006 моль). Полученную смесь нагревали при температуре 78°С в течение 60 минут. Образовавшуюся массу концентрировали при пониженном давлении и получали стекловидные кристаллы. Продукт растворяли в смеси 2% метанол-дихлорметан (75 мл), отфильтровывали нерастворившуюся часть, концентрировали при пониженном давлении и получали 34,5 г пены рыжевато-коричневого цвета. Эту пену растворяли в воде (25 мл) и экстрагировали этилацетатом (2×25 мл). Водный слой концентрировали при пониженном давлении и получали 1,35 г (54%) титульного соединения в виде кристаллов рыжевато-коричневого цвета. 1Н ЯМР (300 МГц) (D2O)
3-(Хлорсульфонил)бензойная кислота (Полупродукт 73) Твердую бензойную кислоту (50 г, 0,41 моль) при комнатной температуре порциями прибавляли к хлорсульфоновой кислоте (348,7 г, 200 мл, 3 моль). Реакционную смесь нагревали при температуре 125-130°С в течение 2 часов и затем выливали в интенсивно перемешиваемый колотый лед (1000 г). Через 30 минут отфильтровывали осадок белого цвета, промывали водой (1 л), сушили на воздухе и получали 72,5 г (80%) титульного соединения. 1Н ЯМР (CDCl3: d4-MeOH, 4:1, 60 МГц) 2-[(4-Метилфенил)сульфонил]этил-3-(хлорсульфонил)бензоат (Полупродукт 43) Смесь 3-(хлорсульфонил)бензойной кислоты (Полупродукт 73, 5,51 г, 0,025 моль) и хлористого тионила (17,85 г, 11 мл, 0,15 моль) кипятили с обратным холодильником в течение 1 часа. Избыток хлористого тионила отгоняли сначала при атмосферном давлении, а затем при пониженном и получали технический хлорангидрид кислоты. К смеси технического хлорангидрида кислоты и 2-(п-толилсульфонил)этанола (4,8 г, 0,024 моль) в дихлорметане (50 мл) при комнатной температуре прибавляли триэтиламин (2,52 г, 0,025 моль). Анализ реакционной смеси методом ТСХ показал, что реакция полностью завершилась через 1 час. Реакционную массу промывали водой (2×25 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали 10 г вязкого масла. Масло очищали флэш-хроматографией (силикагель, от 25 до 50% этилацетата в гексане) и получали 6,1 г (63%) титульного соединения в виде кристаллического вещества белого цвета. 1Н ЯМР (CDCl3, 60 МГц) 2-[(4-Метилфенил)сульфонил]этил-3-{[2-({[3-метил-4-(2,2,2-трифторэтокси)(2-пиридил)]метил}сульфинил)бензимидазолил]сульфонил}бензоат (Полупродукт 74) К суспензии лансопразола (4,62 г, 0,0125 моль) и 2-[(4-метилфенил)сульфонил]этил-3-(хлорсульфонил)бензоата (Полупродукта 43, 6,04 г, 0,015 моль) в дихлорметане (50 мл) прибавляли триэтиламин (3,03 г, 0,03 моль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную массу промывали водой (2×25 мл) и солевым раствором (1×25 мл), фильтровали через фильтровальную бумагу 1PS, концентрировали при пониженном давлении и получали 12 г пены желтого цвета. Пену очищали флэш-хроматографией (силикагель, этилацетат) и получали 8 г (87%) титульного соединения в виде пены желтого цвета. 1Н ЯМР (CDCl3, 60 МГц) Натриевая соль 3-{[2-({[3-метил-4-(2,2,2-трифторэтокси)-2-пиридил]метил}сульфинил)бензимидазолил]сульфонил}бензойной кислоты (Соединение 48) К раствору эфира (Полупродукта 74, 4,78 г, 0,0065 моль) в смеси ацетонитрила (30 мл), изопропилового спирта (10 мл), воды (20 мл) прибавляли бикарбонат натрия (0,655 г, 0,0078 моль). После нагревания при температуре 70°С в течение 2 часов реакционную массу концентрировали при пониженном давлении и получали кристаллы рыжевато-коричневого цвета. К этим кристаллам рыжевато-коричневого цвета прибавляли этилацетат (50 мл) и перемешивали в течение 15 минут. Полученную смесь фильтровали от нерастворимых примесей и фильтрат экстрагировали водой (2×10 мл). Объединенные водные фракции повторно промывали этилацетатом (1×10 мл), концентрировали при пониженном давлении и получали 2 г (53%) титульного соединения в виде кристаллов рыжевато-коричневого цвета. 1Н ЯМР (300 МГц) (d6-ДМСО)
Натриевая соль (3-изопропил-4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)уксусной кислоты (Соединение 49) Соединение 49 получали, как показано на Реакционной схеме 41. ЯМР (d6-ДМСО) Масс-спектр (отрицательный ион) m/z для С27Н25F3N3NaO7S2. Вычислено 647,62. Найдено 624 (М + минус Na).
Натриевая соль 4-метокси-3-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}бензойной кислоты (Соединение 50) Соединение 50 получали, как показано на Реакционной схеме 42. ЯМР (d6-ДМСО) Масс-спектр (отрицательный ион) m/z для С24Н19F3N3NaO7S2. Вычислено 605. Найдено 582 (М + минус Na).
Натриевая соль 3-(4-{2-[3-Метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенил)пропионовой кислоты (Соединение 51) Соединение 51 получали, как показано на Реакционной схеме 43. ЯМР (d6-ДМСО) Масс-спектр (отрицательный ион) m/z для C25H21F3N3NaO6S2. Вычислено 623. Найдено 580 (М + минус Na).
Натриевая соль 2,2-диметил-3-(4-{2-[3-метил-4-(2,2,2-трифторэтокси)пиридин-2-илметансульфинил]бензимидазол-1-сульфонил}фенокси)пропионовой кислоты (Соединение 52) Соединение 52 получали, как показано на Реакционной схеме 44. ЯМР (d6-ДМСО) Масс-спектр (отрицательный ион) m/z для C27H25F3N3NaO7S2. Вычислено 647,62. Найдено 648 (М+), 625 (М + минус Na). Пример фармацевтической композиции по изобретению, мг:
Готовят крахмальный клейстер из указанного количества крахмала и воды, смешивают клейстер с соединением 2 и соединением формулы w. Готовят гранулы, опудривают их стеаратом магния и прессуют в таблетки массой 500 мг.
Формула изобретения
1. Соединение формулы 1, формулы 2, формулы 3 или формулы 4
или изомеры соединений формул 2 и 3, в которых группы ОСН3 и HF2CO, соответственно, находятся в 6-положении бензимидазольного кольца, и где R представляет собой группу (i)
пунктирная линия означает связь, соединяющую группу R с группой SO2, R1 и R2 независимо означают Н, прямой или разветвленный насыщенный углеводород, содержащий не более 12 атомов углерода, включающую одну или две группы R5, и, необязательно, дополнительно включающую от одной до трех групп X, где Х независимо выбран из группы, состоящей из -О-, -NHCO-, -CONH-; или группа R5 без участия групп R1 или R2 напрямую присоединена к ароматическому кольцу; R3 и R4 независимо представляют собой Н, алкил, содержащий от одного до трех атомов углерода, или O-алкил, содержащий от одного до трех атомов углерода, R5 независимо означает Н или СООН; при условии, что: по меньшей мере, одна из групп R1 и R2 не является Н, и по меньшей мере, один заместитель R5 не является Н, и не более чем две группы R5 представляют собой СООН, так что соединение содержит, по меньшей мере, одну, но не более двух групп СООН; или фармацевтически приемлемая соль указанного соединения. 2. Соединение по п.1, имеющее структуру формулы 1. 3. Соединение по п.1, имеющее структуру формулы 2. 4. Соединение по п.1, имеющее структуру формулы 3. 5. Соединение по п.1, имеющее структуру формулы 4. 6. Соединение по п.1, где формула включает, по меньшей мере, одну группу X. 7. Соединение по п.1, где, по меньшей мере, один из заместителей Х означает О. 8. Соединение по п.1 где, по меньшей мере, один из заместителей Х означает CONH. 9. Соединение по п.1, содержащее две группы R5, означающие СООН, или фармацевтически приемлемая соль указанного соединения. 10. Соединение по п.1, где формула 1 имеет структуру формулы 1а, формула 2 имеет структуру формулы 2а, формула 3 имеет структуру формулы 3а и формула 4 имеет структуру формулы 4а
или изомеры соединений формул 2а и 3а, в которых группы ОСН3 и HF2С соответственно находятся в 6-положении бензимидазольного кольца, и R1 и R2 независимо означают Н, прямую или разветвленную алкильную группу, содержащую от 1 до 12 атомов углерода, включающую одну или две группы R5, и, необязательно, дополнительно включающую от одной до трех групп X, где Х независимо выбран из группы, состоящей из -О-, -NHCO-, -CONH-, или группа R5 без участия групп R1 или R2 напрямую присоединена к ароматическому кольцу; R5 независимо означает Н или СООН; при условии, что: по меньшей мере, одна из групп R1 и R2 не является Н, и по меньшей мере, один заместитель R5 не является H и не более чем две группы R5 представляют собой СООН, так что соединение содержит, по меньшей мере, одну, но не более двух групп СООН; или фармацевтически приемлемая соль указанного соединения. 11. Соединение по п.10, имеющее формулу 1а. 12. Соединение по п.10, имеющее формулу 2а. 13. Соединение по п.12, в котором группа СН3О находится в 5-положении бензимидазольного фрагмента. 14. Соединение по п.10, имеющее формулу 3а. 15. Соединение по п.12, в котором группа HF2СО находится в 5-положении бензимидазольного фрагмента. 16. Соединение по п.10, имеющее формулу 4а. 17. Соединение по п.10, содержащее только одну группу СООН, или его фармацевтически приемлемая соль. 18. Соединение по п.10, содержащее только две группы СООН, или его фармацевтически приемлемая соль. 19. Соединение по п.10, в котором заместители R2, R3 и R4 представляют собой водород, a R1 представляет собой группу ОСН2СООН, присоединенную к 4-положению фенильного кольца относительно сульфонильной группы, или его фармацевтически приемлемая соль. 20. Соединение по п.1, где формула 1, формула 2, формула 3 или формула 4 дополнительно определены как показано ниже
или изомеры соединений формул 2 и 3, в которых группы ОСН3 и HF2CO соответственно находятся в 6-положении бензимидазольного кольца, и где R означает группы, выбранные из формул (а)-(s), пунктирная линия представляет собой связь, соединяющую группу R с группой SO2,
или фармацевтически приемлемая соль указанного соединения. 21. Соединение по п.20 формулы 1. 22. Соединение по п.20 формулы 2. 23. Соединение по п.22, в котором группа СН3О находится в 5-положении бензимидазольного фрагмента. 24. Соединение по п.20 формулы 3. 25. Соединение по п.24, в котором группа HF2О находится в 5-положении бензимидазольного фрагмента. 26. Соединение по п.20 формулы 4. 27. Соединение по п.20, имеющее формулу
или фармацевтически приемлемая соль указанного соединения. 28. Соединение по п.20, имеющее формулу
или его фармацевтически приемлемая соль. 29. Соединение по п.20, имеющее формулу
или фармацевтически приемлемая соль указанного соединения. 30. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении секреции желудочной кислоты, включающая фармацевтически приемлемый наполнитель и соединение по п.1. 31. Фармацевтическая композиция по п.30, обладающая ингибирующей активностью в отношении секреции желудочной кислоты, включающая фармацевтически приемлемый наполнитель и соединение по п.10. 32. Фармацевтическая композиция по п.30, обладающая ингибирующей активностью в отношении секреции желудочной кислоты, включающая фармацевтически приемлемый наполнитель и соединение по п.20. 33. Фармацевтическая композиция по п.30, обладающая ингибирующей активностью в отношении секреции желудочной кислоты, включающая фармацевтически приемлемый наполнитель и соединение по п.27. 34. Фармацевтическая композиция по п.30, обладающая ингибирующей активностью в отношении секреции желудочной кислоты, включающая фармацевтически приемлемый наполнитель и соединение по п.28. 35. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении секреции желудочной кислоты, включающая фармацевтически приемлемый наполнитель, соединение по пп.1, 10, 20, 27, 28 или 29 и лекарственное средство – ингибитор протонного насоса, выбранный из группы, состоящей из формул (w), (x), (y) и (z):
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
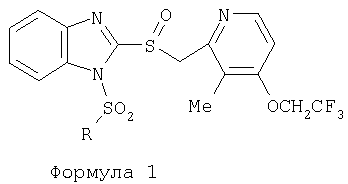
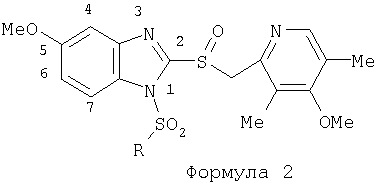
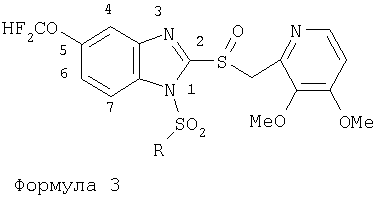
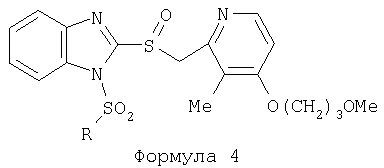
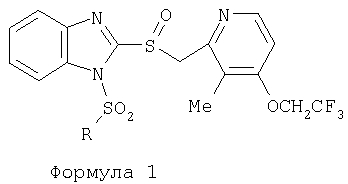
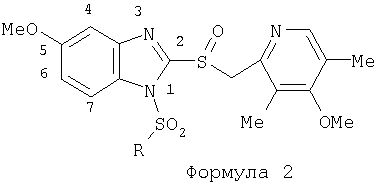
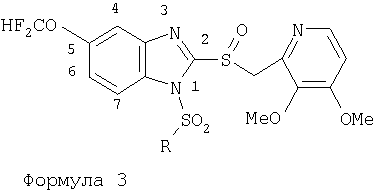
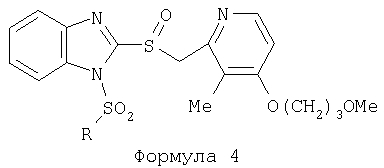
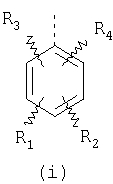
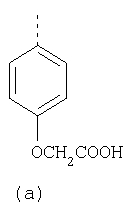
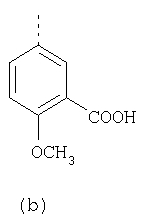
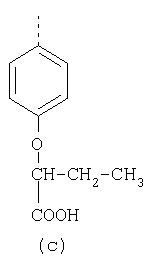
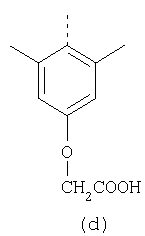
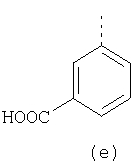
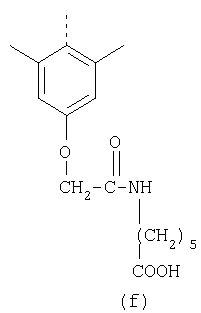
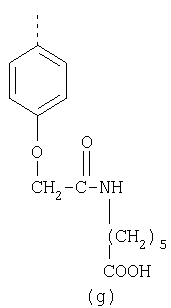
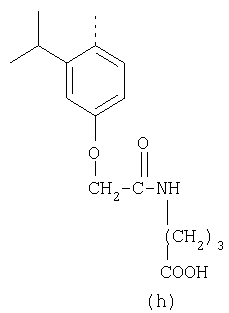
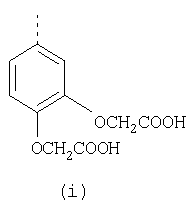
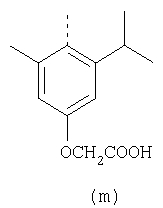
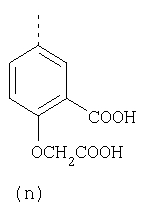
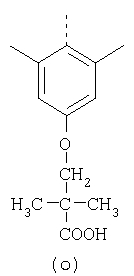
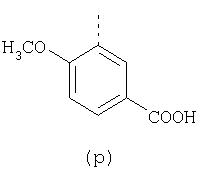
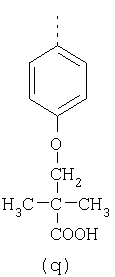
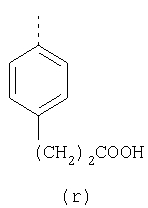
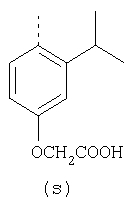
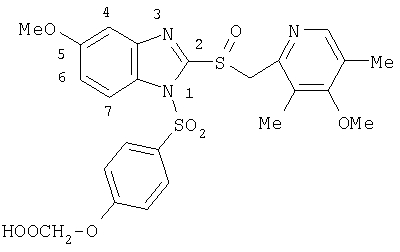
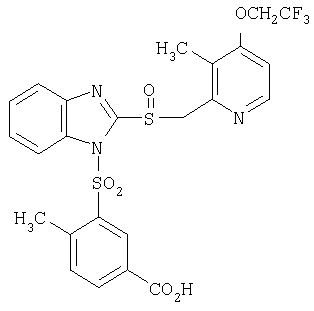
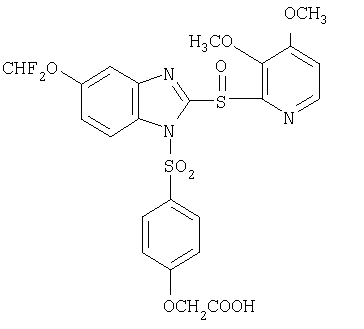
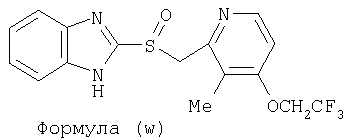
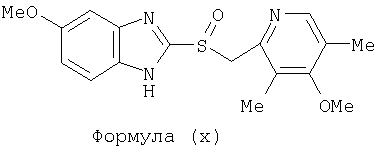
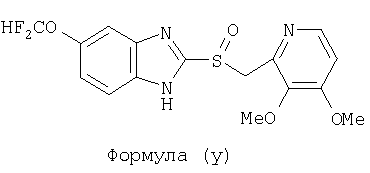
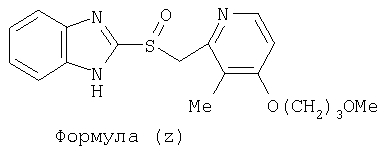
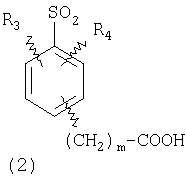
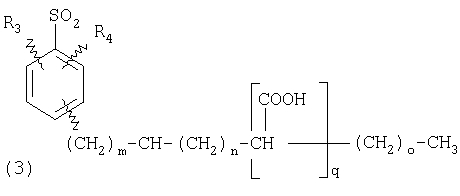
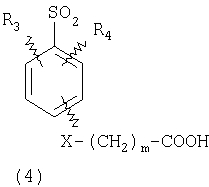
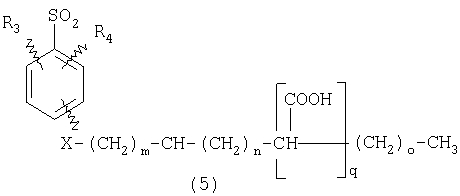
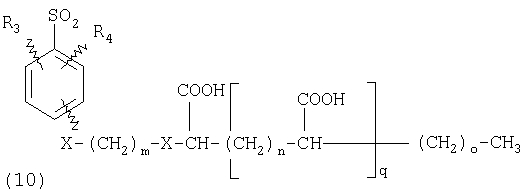
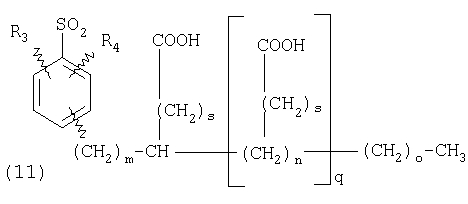
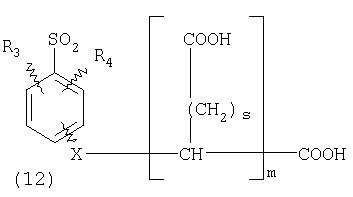
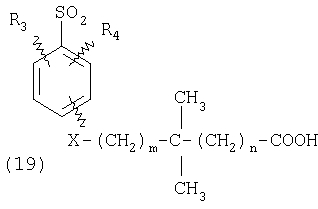
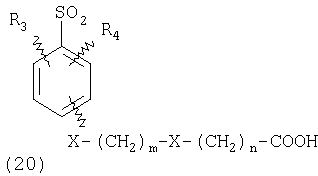
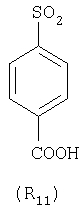
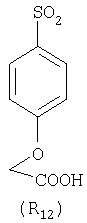
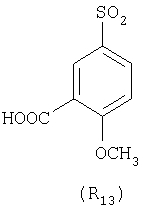
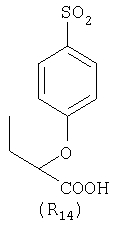
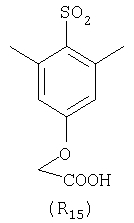
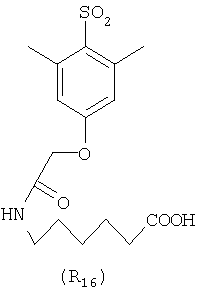
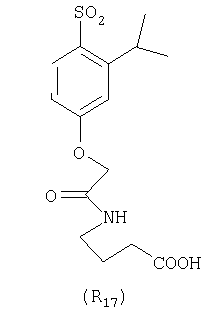
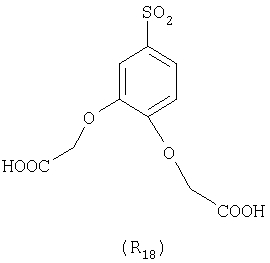
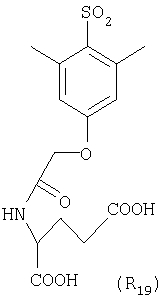
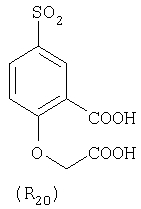
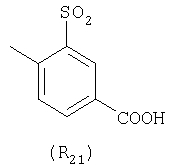
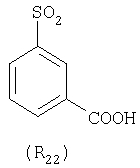
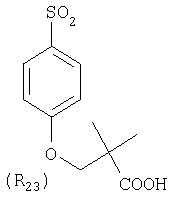
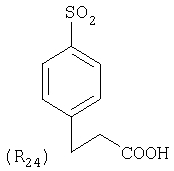
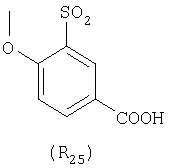
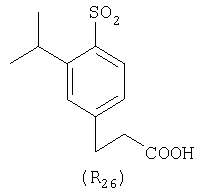
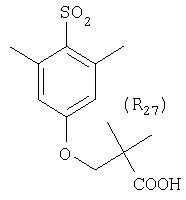
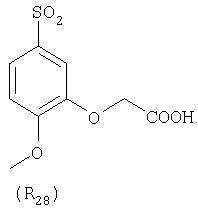
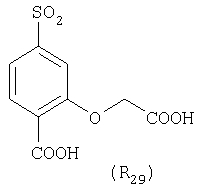
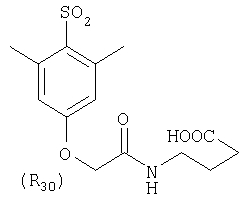
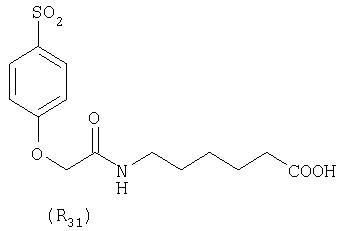
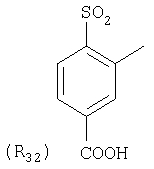
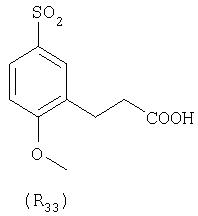
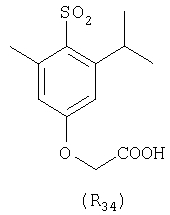
 означает заместитель или заместители, присутствующие в пиридиновой части этих препаратов – ингибиторов протонного насоса. Соединение Формулы 5 взаимодействует с хлорсульфонильным соединением Формулы 6 в присутствии основания, такого как гидрид натрия, триэтиламин и ди(изопропил)метиламин, либо другого подходящего основания, в апротонном растворителе, таком как СН2Cl2. Соединение Формулы 6 содержит фрагмент замещенного или незамещенного фенилсульфонилэтилового эфира карбоновой кислоты, включенного в соединения изобретения.
означает заместитель или заместители, присутствующие в пиридиновой части этих препаратов – ингибиторов протонного насоса. Соединение Формулы 5 взаимодействует с хлорсульфонильным соединением Формулы 6 в присутствии основания, такого как гидрид натрия, триэтиламин и ди(изопропил)метиламин, либо другого подходящего основания, в апротонном растворителе, таком как СН2Cl2. Соединение Формулы 6 содержит фрагмент замещенного или незамещенного фенилсульфонилэтилового эфира карбоновой кислоты, включенного в соединения изобретения.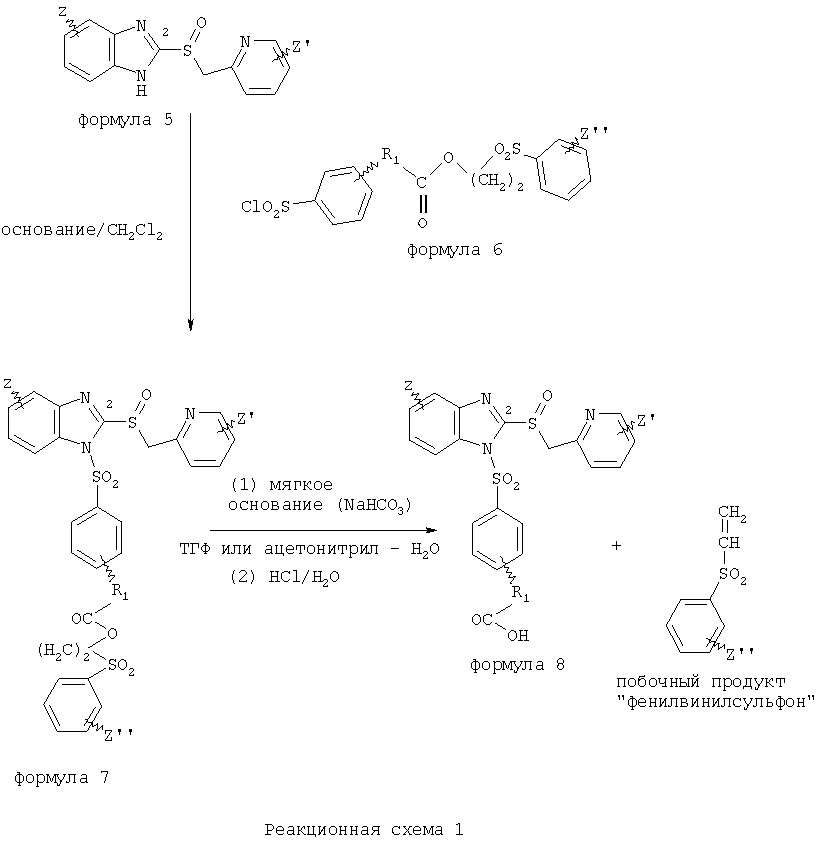
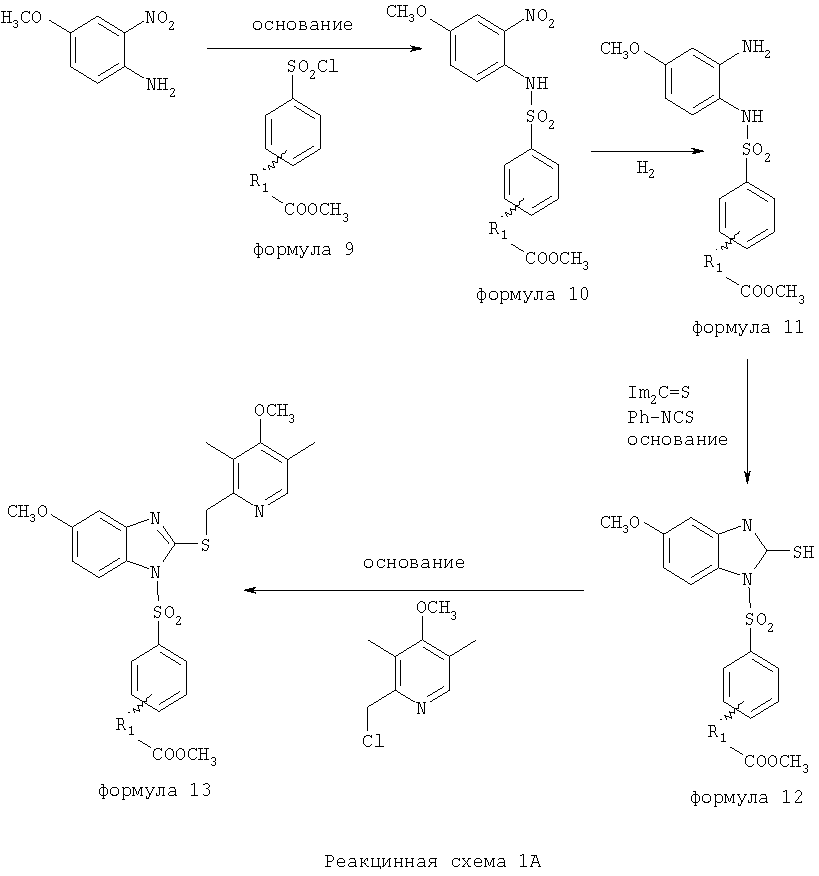
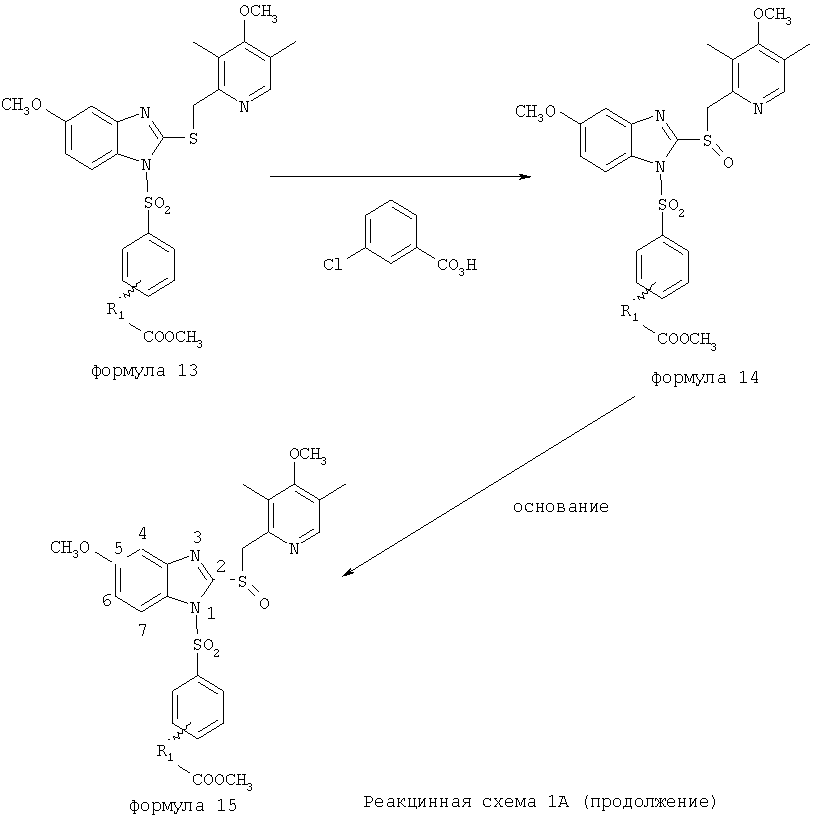
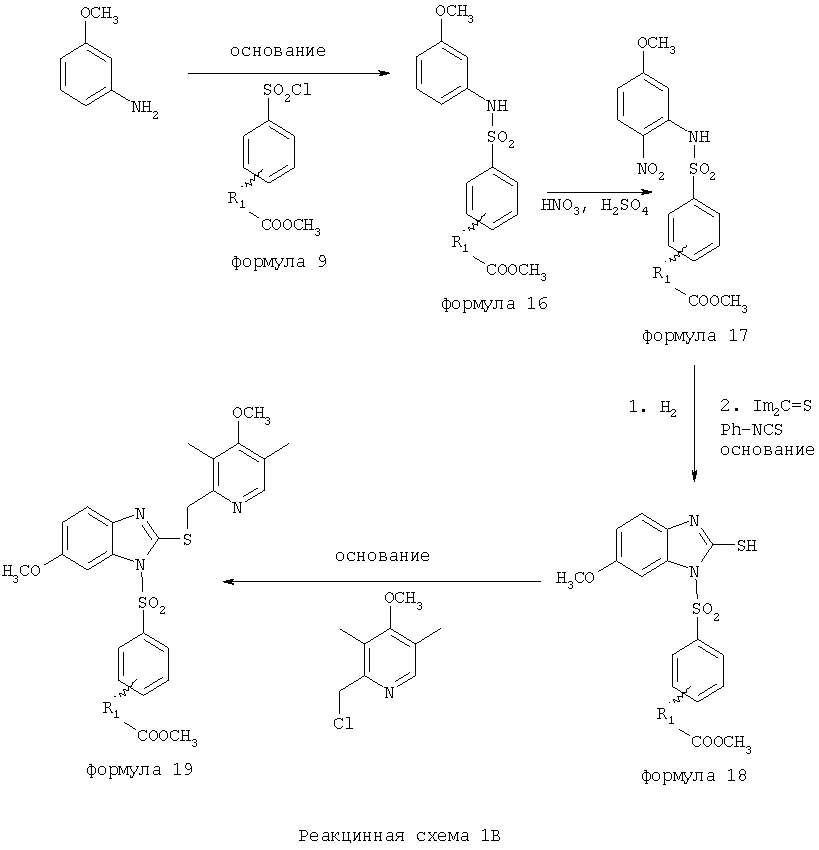
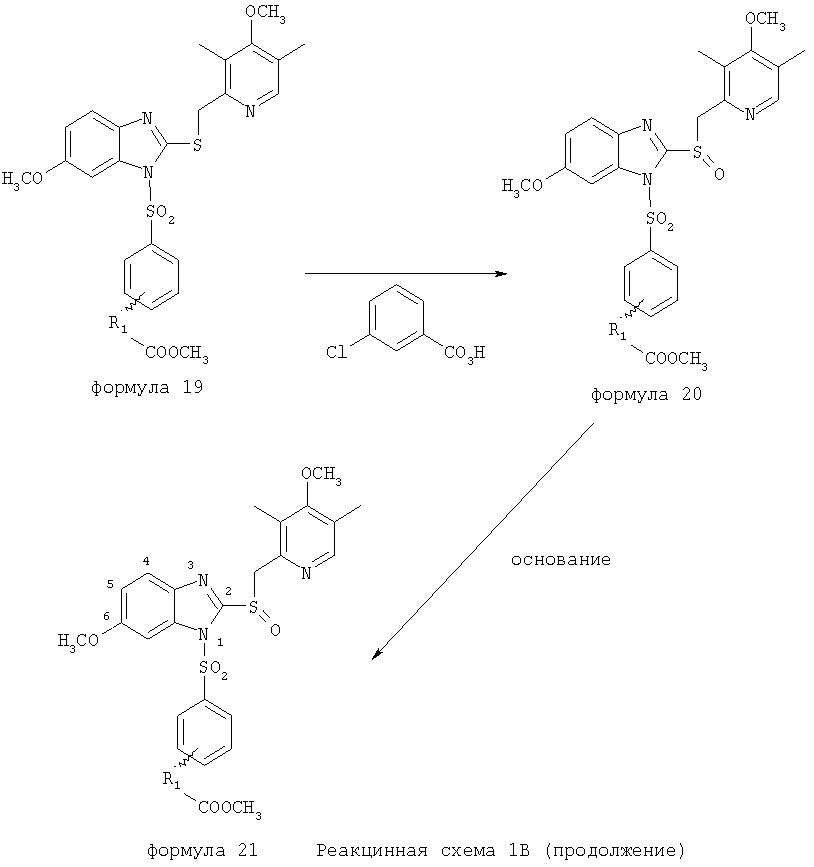
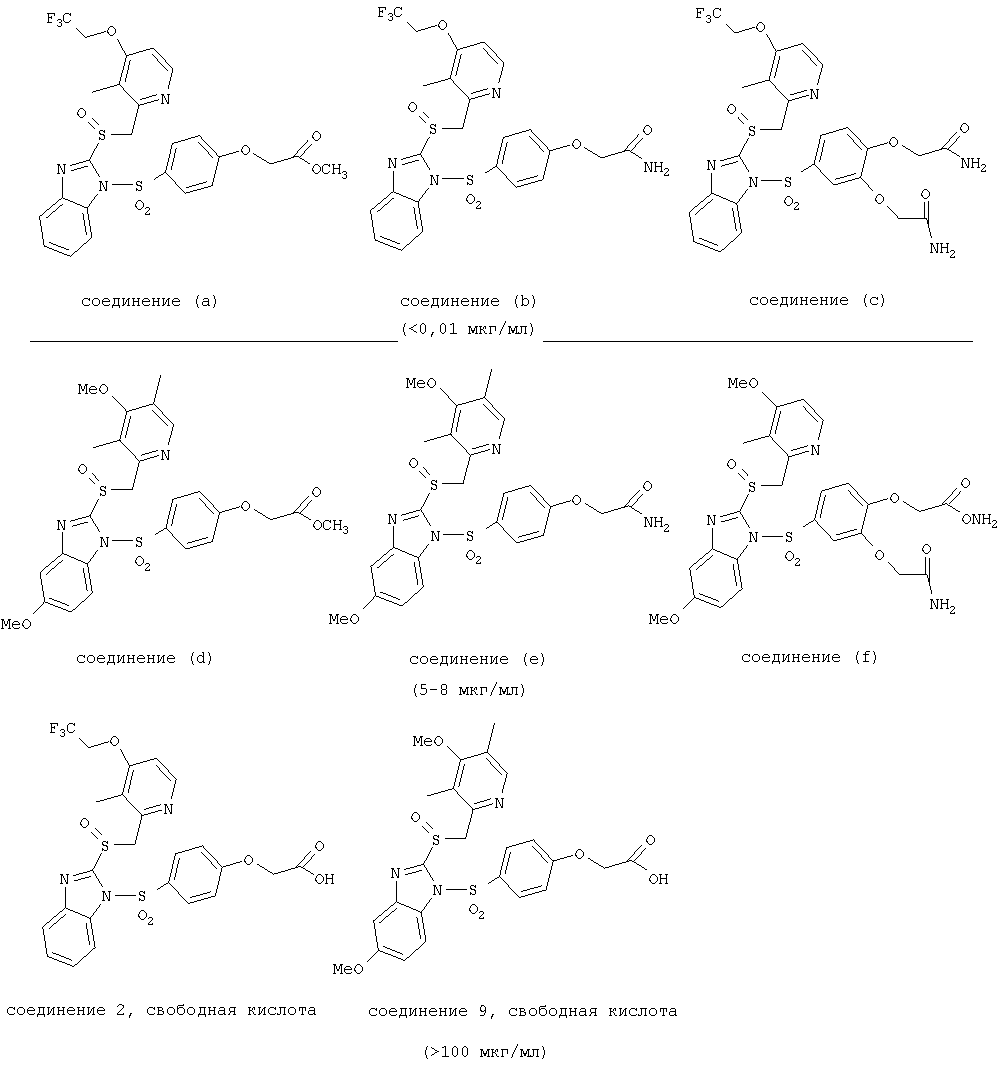
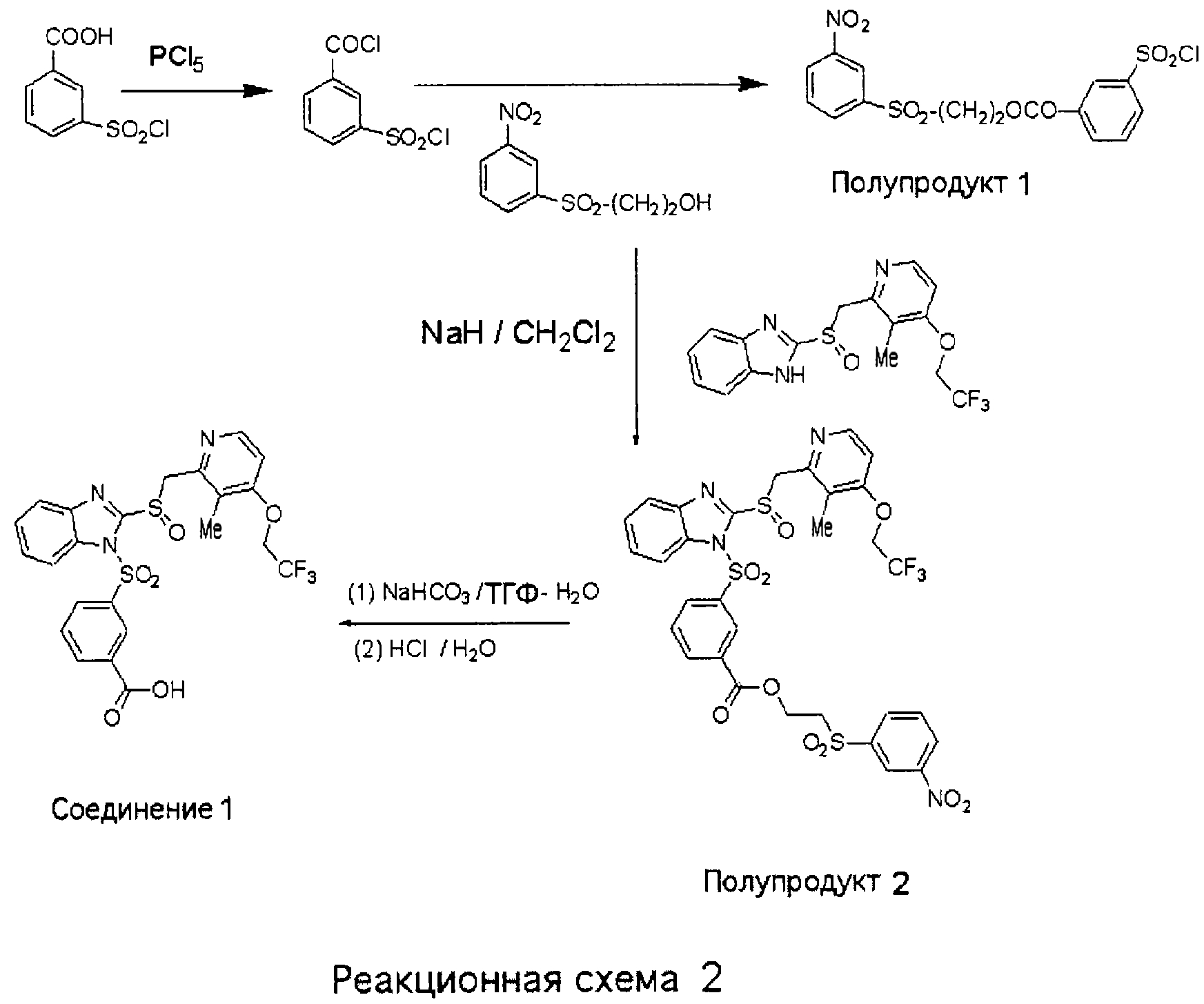
 4,08 (т., 2Н), 4,58 (т., 2Н), 7,33 (т., 1Н), 7,40 (д., 1Н), 7,77 (д., 1Н), 7,84 (м., 2Н), 8,32 (д., 1Н), 8,40 (д., 1Н), 8,56 (с., 1Н).
4,08 (т., 2Н), 4,58 (т., 2Н), 7,33 (т., 1Н), 7,40 (д., 1Н), 7,77 (д., 1Н), 7,84 (м., 2Н), 8,32 (д., 1Н), 8,40 (д., 1Н), 8,56 (с., 1Н).