Патент на изобретение №2292202
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБЫ ВВЕДЕНИЯ АНАЛОГОВ ЭПОТИЛОНА ПРИ ЛЕЧЕНИИ РАКА
(57) Реферат:
Изобретение относится к области фармакологии и касается способа приготовления состава на основе аналогов эпотилона путем растворения указанного аналога эпотилона в водном бутаноле, сушки полученного раствора в две стадии до получения лиофилизированного продукта, лиофилизированному аналогу эпотилона и фармацевтическому средству для лечения рака, содержащему лиофилизированный аналог эпотилона. Способ обеспечивает повышение растворимости полученного продукта. 6 н. и 14 з.п. ф-лы, 1 табл.
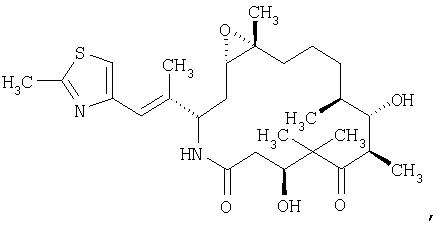
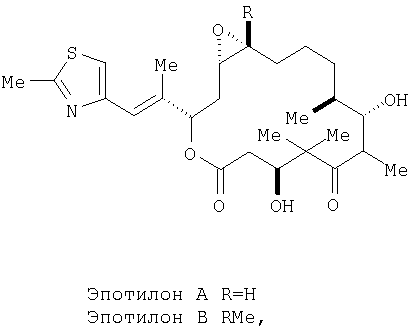
Область, к которой относится изобретение. Эпотилоны являются макролидными соединениями, используемыми в фармацевтической промышленности. Например, эпотилоны А и В, имеющие следующие структуры:
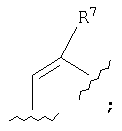
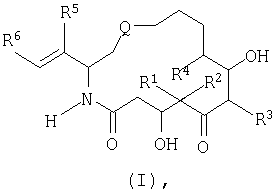
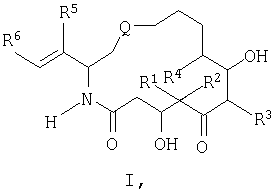
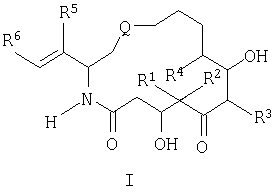
обладают стабилизирующим действием в отношении микротрубочек аналогично паклитакселу (TAXOL®) и, следовательно, цитотоксическим действием в отношении быстро пролиферирующих клеток, таких как клетки опухоли или другие гиперпролиферирующие клетки, см. Hofle et al., Angew. Chem. Int. Ed. Engl., Vol.35, №13/14, 1567-1569 (1996); заявка WO 93/10121, опубликованная 27 мая 1993 г.; заявка WO 97/19086, опубликованная 29 мая 1997г. Были синтезированы различные производные и аналоги эпотилонов А и В, которые могут быть использованы для лечения различных видов рака и других патологических пролиферативных заболеваний. Такие аналоги описаны Hofle et al.. Id., Nicolaou et al., Angew. Chem. Int. Ed. Engl., Vol.36, №19, 2097-2103 (1997); и Su et al., Angew. Chem. Int. Ed. Engl., Vol.36, №19, 2093-2097 (1997). Аналоги эпотилонов, обладающие большей активностью, описываются общей формулой I:
где различные радикалы указаны ниже. Хотя эти соединения имеют благоприятные терапевтические свойства, однако, получение препаратов на их основе затруднено вследствие некоторых свойств, которые будут описаны ниже. Согласно данному изобретению был разработан состав, при помощи которого вышеописанные аналоги эпотилона могут быть введены без труда путем инъекции без заметной потери их активности. Кроме того, многие противораковые лекарства обладают токсичностью. Действительно, терапевтический профиль многих активных противораковых препаратов ухудшается вследствие токсичности. Следовательно, существует необходимость в создании методов введения и схем дозировки, которые приводят к уменьшению или устранению токсичности противораковых агентов. Эти методы позволят применять активные противоопухолевые агенты, которые в других случаях нельзя использовать клинически. Сущность изобретения. Настоящее изобретение предусматривает новую схему дозировки эпотилоновых соединений, причем эта схема пригодна для лечения пациентов с твердыми опухолями, особенно, развившимися твердыми опухолями. Кроме того, способы по изобретению могут быть использованы для лечения и/или профилактики как метастазов, так и первичных опухолей. Согласно одному варианту изобретение предусматривает способ лечения пациентов, которые ранее подвергались радиотерапии и/или химиотерапии в отношении твердых опухолей. Было также установлено, что эпотилоновые соединения по изобретению, особенно предпочтительное соединение [1S-[1R*,3R*(Е),7R*,10S*,11R*,12R*,16S*]]-7,11-дигидрокси-8,8,10,12,16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17-оксабицикло[14.1.0]гептадекан-5,9-дион, могут быть использованы для лечения опухолей, резистентных к радиационной терапии или химиотерапии. Способы по изобретению пригодны для борьбы с раковыми клетками и, таким образом, опухолями, которые от природы нечувствительны или становятся нечувствительными к паклитакселу. Согласно одному варианту схема лечения согласно изобретению включает еженедельное введение эпотилона по изобретению, предпочтительно, в виде непрерывной инфузии в течение одного (1) часа каждую неделю. Согласно другому варианту введение осуществляют еженедельно в течение трех недель. Доза для еженедельной инфузии составляет от 1 мг/м2 до 30 мг/м2 и, более предпочтительно, 1 мг/м2 – 25 мг/м2. Согласно еще одному варианту схема лечения включает как пероральное, так и внутривенное введение этого же самого эпотилонового соединения. Например, еженедельному введению может предшествовать или следовать пероральное введение дозы 20 мг/м или большей дозы. Согласно особому способу схема введения включает трехнедельный цикл внутривенной инфузии один раз в неделю примерно в течение одного (1) часа, причем до этого цикла или после него вводят перорально определенную дозу один или несколько раз в неделю, предшествующую первому внутривенному введению, или в неделю, следующую после последнего внутривенного введения. Другие протоколы также охватываются данным изобретением, включая, но не ограничиваясь, следующими схемами: (а) ежедневное введение в течение 5-10 дней с последующим перерывом, по меньшей мере, в течение 3 дней; (б) еженедельное введение в течение 2-10 недель с последующим перерывом в течение, по меньшей мере, одной недели; (в) введение один раз каждые три недели с последующим перерывом в течение, по меньшей мере, одной недели. Изобретение также предусматривает применение H1 и Н2 антигистаминных агентов до, после и/или до и после цикла введения эпотилона. Точно также изобретение охватывает использование других химиотерапевтических агентов, особенно противоопухолевых агентов, вместе с одним эпотилоном или вместе с H1 и Н2 блокираторами и эпотилонами. Согласно другому варианту схема введения эпотилона применяется после проведения обычного курса с использованием паклитаксела. Способы согласно изобретению предусматривают лечение различных видов рака. Согласно предпочтительному варианту методы по изобретению используют для лечения твердых опухолей, включая, но не ограничиваясь этим, опухоли груди, головы и шеи, саркому, колоректальные опухоли, опухоли ИРТ, меланомы, опухоль пищевода, почек, шейки матки, щитовидной железы, ануса, яичников и ободочной кишки. Методы и композиции согласно данному изобретению предусматривают получение препарата и препарат на основе аналогов эпотилона, описываемых формулой:
где различные радикалы определены ниже. Согласно одному из вариантов аналог эпотилона вначале солюбилизируют смесью трет.бутанола и воды и затем лиофилизируют в оптимальных условиях. Лиофилизированное лекарство воссоздают путем добавления вначале смеси полиэтоксилированного касторового масла (сурфактанта) и безводного этанола и затем разбавления раствором Рингера с лактатом для инъекции до концентрации, подходящей для введения. Подробное описание изобретения. Согласно одному варианту данное изобретение предусматривает препарат для введения аналогов эпотилона формулы I:
В формуле I и далее в описании Q выбран из группы, состоящей из
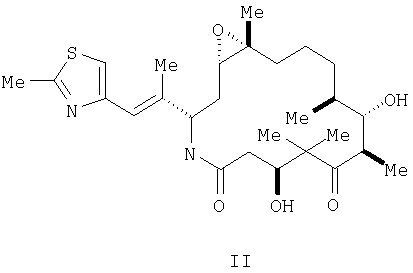
М выбран из группы, состоящей из кислорода, серы, NR8 и CR9R10; каждый из R1, R2, R3, R4, R5, R7, R11, R12, R13, R14 и R15 независимо выбран из группы, состоящей из водорода, алкила, замещенного алкила, арила, замещенного арила, гетероциклила и, если R1 и R2 обозначают алкил, они могут вместе образовать циклоалкил; R6 выбран из группы, состоящей из водорода, алкила, замещенного алкила, арила, замещенного арила, циклоалкила, гетероциклила и замещенного гетероциклила; R8 выбран из группы, состоящей из водорода, алкила, замещенного алкила, R11C=O, R12OC=O и R13SO2; и каждый из R9 и R10 независимо выбран из группы, состоящей из водорода, галоида, алкила, замещенного алкила, арила, гетероциклила, гидрокси, R14C=O и R15OC=O. Ниже приведены определения различных терминов, использованных в данной заявке для описания данного изобретения. Эти определения относятся к терминам, используемым по всему тексту, если иное не оговорено в конкретных случаях при описании отдельных соединений или части большой группы. Термин «алкил» относится к возможно замещенным линейным или разветвленным насыщенным углеводородным группам, содержащим от 1 до примерно 20 атомов углерода, предпочтительно, от 1 до примерно 7 атомов углерода. Термин «низший алкил» относится к возможно замещенным алкильным группам, содержащим от 1 до примерно 4 атомов углерода. Термин «замещенный алкил» относится к алкилам, замещенньм, например, одним – четырьмя заместителями, такими как галоген, трифторметил, трифторметокси, гидрокси, алкокси, пиклоалкилокси, гетероциклокси, оксо, алканоил, арил, арилокси, аралкил, алканоилокси, амино, алкиламино, ариламино, аралкиламино, циклоалкиламино, гетероциклиламино, дизамещенная аминогруппа, в которой заместители выбраны из алкила, арила, аралкила, аралканоиламино, замещенной алканоиламиногруппы, замещенной ариламиногруппы, замещенной аралканоил-аминогруппы, алкилтио, арилтио, аралкилтио, циклоалкилтио, гетероциклилтио, алкилтионо, аралкилтионо, алкилсульфонила, арилсульфонила, аралкилсульфонила, сульфонамидо (например, SO2NH2), замещенной сульфонамидогруппы, нитро, циано, карбокси, карбамоила (например, CONH2), замещенного карбамоила (например, CONHалкила, CONHарила, CONHаралкила или случаев, когда имеются два заместителя у атома азота, выбранные из алкила, арила или аралкила), алкоксикарбонила, арила, замещенного арила, гуанидино и гетероциклилов, таких как индолил, имидазолил, фурил, тиенил, тиазолил, пирролидил, пиридил, пиримидил и т.п. В тех случаях, когда, как указано выше, заместители сами содержат заместители, последние выбирают из группы, состоящей из галогена, алкила, алкокси, арила и аралкила. Определения, приведенные для алкила и замещенного алкила, относятся также к алкильной части алкоксигрупп. Термин «галоген» или «галоид» относится к фтору, хлору, брому и йоду. Термин «кольцевая система» относится к возможно замещенной кольцевой системе, содержащей одно-три кольца и, по меньшей мере, одну углерод-углеродную двойную связь в, по меньшей мере, одном кольце. Примеры кольцевых систем включают, не ограничиваясь этим, арил или частично или полностью ненасыщенную гетероциклическую кольцевую систему, которая может содержать заместители. Термин «арил» относится к моноциклическим или бициклическим ароматическим углеводородным группам, содержащим примерно от 6 до примерно 12 атомов углерода в кольце, например, к фенилу, нафтилу, бифенилу и дифенилу, каждый из которых может иметь заместители. Термин «аралкил» относится к арилу, присоединенному к остальной части молекулы через алкильную группу, например к бензильной группе. Термин «замещенный арил» относится к арильной группе, замещенной, например, одним-четырьмя заместителями, такими как алкил, замещенный алкил, галоид, трифторметил, трифторметокси, гидрокси, алкокси, циклоалкилокси, гетероциклоокси, алканоил, алканоилокси, амино, алкиламино, диалкиламино, аралкиламино, циклоалкиламино, гетероциклиламино, алканоиламино, тиольная группа, алкилтио, циклоалкилтио, гетероциклилтио, уреидо, нитро, циано, карбокси, карбоксиалкил, карбамил, алкоксикарбонил, алкилтионо, арилтионо, алкилсульфонил, сульфонамидо, арилокси и т.п. Заместитель, в свою очередь, может быть замещен одним или несколькими заместителями, выбранными из группы, состоящей из галоида, гидрокси, алкила, алкокси, арила, замещенного алкила, замещенного арила и аралкила. Термин «циклоалкил» относится к возможно замещенным насыщенным циклическим кольцевым системам, предпочтительно, содержащим 1-3 кольца и 3-7 атомов углерода в одном кольце, которое может быть конденсированным с ненасыщенным С3-С7-карбоциклическим кольцом. Примеры таких групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил, циклододецил и адамантил. Примеры заместителей включают одну или несколько алкильных групп, описанных выше, или одну или несколько групп, описанных выше в качестве заместителей алкильных групп. Термины «гетероцикл», «гетероциклический» и «гетероциклил» относятся к возможно замещенной, ненасыщенной, частично насыщенной или полностью насыщенной ароматической или неароматической циклической группе, которая, например, является 4-7 членной моноциклической, 7-11-членной бициклической или 10-15-членной трициклической кольцевой системой, которая содержит, по меньшей мере, один гетероатом в, по меньшей мере, одном углеродсодержащем кольце. Каждое кольцо гетероциклической группы, содержащей гетероатом, может содержать 1, 2 или 3 гетероатома, выбранных из атомов азота, атомов кислорода, атомов серы, причем атомы азота и серы могут быть окислены и атомы азота могут быть также кватернизованы. Гетероциклическая группа может быть присоединена через любой гетероатом или атом углерода. Примерами моноциклических гетероциклических групп служат пирролидинил, пирролил, индолил, пиразолил, оксетанил, пиразолинил, имидазолил, имидазолинил, имидазолидинил, оксазолил, оксазолидинил, изоксазолинил, изоксазолил, тиазолил, тиадиазолил, тиазолидинил, изотиазолил, изотиазолидинил, фурил, тетрагидрофурил, тиенил, оксадиазолил, пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 2-оксазепинил, азепинил, 4-пиперидонил, пиридил, N-оксопиридил, пиразинил, пиримидинил, пиридазинил, тетрагидропиранил, тетрагидротиопиранил, тетра-гидротиопиранилсульфон, морфолинил, тиоморфолинил, тиоморфолинил-сульфоксид, тиоморфолинилсульфон, 1,3-диоксолан и тетрагидро-1,1-диоксо-тиенил, диоксанил, изотиазолидинил, тиетанил, тииранил, триазинил и триазолил и т.п. Примеры бициклических гетероциклических групп включают бензотиазолил, бензоксазолил, бензотиенил, хинуклидинил, хинолинил, хинолинил-N-оксид, тетрагидроизохинолинил, изохинолинил, бензимидазолил, бензопиранил, индолизинил, бензофурил, хромонил, кумаринил, циннолинил, хиноксалинил, индазолил, пирролопиридил, фуропиридинил (такой как фуро[2,3-с]пиридинил, фуро[3,1-b]пиридинил или фуро[2,3-b]пиридинил), дигидроизоиндолил, дигидрохиназолинил, (такой как 3,4-дигидро-4-оксо-хиназолинил), бензизотиазолил, бензизоксазолил, бензодиазинил, бензофуразинил, бензотиопиранил, бензотриазолил, бензопиразолил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, дигидробензотиопиранилсульфон, дигидробензопиранил, индолинил, изохроманил, изоиндолинил, нафтиридинил, фталазинил, пиперонил, пуринил, пиридопиридил, хиназолинил, тетрагидрохинолинил, тиенофурил, тиенопиридил, тиенотиенил и т.п. Примеры заместителей у «кольцевой системы», «гетероцикла», «гетероциклического» радикала и «гетероциклила» включают один или несколько заместителей, описанных выше в качестве заместителей для замещенного алкила или замещенного арила, и гетероциклы меньшего размера, такие как эпоксиды, азиридины и т.п. Термин «алканоил» относится к радикалу -С(O)-алкил. Термин «замещенный алканоил» относится к -С(O)-замещенному алкилу. Термин «гетероатомы» включает кислород, серу и азот. Соединения формулы I образуют соли с различными органическими и неорганическими кислотами. Такие соли включают соли, образованные с хлористоводородной кислотой, бромистоводородной кислотой, метансульфокислотой, гидроксиэтансульфокислотой, серной кислотой, уксусной кислотой, трифторуксусной кислотой, малеиновой кислотой, бензолсульфокислотой, толуолсульфокислотой и различными другими кислотами, известными специалисту в области фармакологии. Такие соли получают взаимодействием соединения формулы I с эквивалентным количеством кислоты в среде, в которой соль осаждается, или в водной среде с последующим выпариванием раствора. Кроме того, могут быть получены цвиттер-ионы («внутренние соли»), они также охватываются термином «соли», используемым в данном описании. Особенно предпочтительным аналогом эпотилона среди соединений формулы I является [1S-[1R*,3R*(Е),7R*,10S*,11R*,12R*,16S*]]-7,11-дигидрокси-8,8,10,12,-16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17-оксабицикло-[14.1.0]гептадекан-5,9-дион, описываемый формулой II:
Соединения, описываемые вышеуказанными формулами I и II, называются также в данном описании «эпотилоновыми соединениями по изобретению», их получение описано в заявке США №09/170582, поданной 13 октября 1998 г., и в заявке 09/280191, поданной 29 марта 1999 г. Соединения, представленные формулами I и II, могут быть в виде многочисленных оптических, геометрических и стереоизомеров. Хотя соединения, описанные в данной заявке, представлены в одной оптической ориентации, в объем данного изобретения входят все изомеры и их смеси. Соединения, представленные формулами I и II выше, являются агентами, стабилизирующими микротрубочки. Поэтому они пригодны при лечении различных раковых заболеваний и других пролиферативных болезней, включая, без ограничения, следующие заболевания: карциному, в том числе мочевого пузыря, груди, ободочной кишки, почек, печени, легкого, яичника, поджелудочной железы, желудка, шеи, щитовидной железы и кожи, включая карциному сквамозных клеток; гемалоетические опухоли лимфоидной системы, включая лейкемию, острую лимфоцитарную лейкемию, острую лимфобластную лейкемию, лимфому В-клеток, лимфому Т-клеток, лимфому Ходжкинса, лимфому не-Ходжкинская, лимфому волосатых клеток и лимфому Беркитта; гемапоетические опухоли миелоидной системы, включая острую и хроническую миелогенную лейкемию и промиелоцитарную лейкемию; опухоли мезенхимного происхождения, включая фибросаркому и рабдомиосаркому; другие опухоли, включая меланому, семиному, тератокарциному, нейробластому и глиому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванномы; опухоли мезенхимного происхождения, включая фибросаркому, рабдомиосаркому и остеосаркому и другие опухоли, включая меланому, ксеродерму пигментную, кератоакантому, семиному, тироидный фолликулярный рак и тератокарциному. Соединения, представленные формулами I и II, пригодны для лечения пациентов, которых уже лечили от рака ранее, а также тех, которых ранее не подвергали такому лечению. Действительно методы и композиции согласно данному изобретению можно использовать в терапии второй линии при лечении рака. Кроме того, соединения, представленные формулами I и II, пригодны для лечения резистентного рака. Соединения, представленные формулами I и II, также ингибируют ангиогенез, влияя при этом на рост опухолей и обеспечивая лечение опухолей и связанных с опухолями нарушений. Такие антиангиогенные свойства соединений формул I и II полезны также при лечении других состояний, восприимчивых к антиангиогенным агентам, включая, без ограничений, определенные формы слепоты, связанные с ретинальной васкуляризацией, артрит, особенно воспалительный артрит, рассеянный склероз, рестеноз и псориаз. Соединения, представленные формулами I и II, индуцируют или ингибируют апоптоз, физиологический процесс гибели клеток, критический для нормального развития и гомеостаза. Изменения апоптозных путей влияют на патогенез различных болезней у людей. Соединения, представленные формулами I и II, в качестве модуляторов апоптоза пригодны для лечения различных болезней у людей с аберрацией апоптоза, включая, без ограничения, раковые заболевания, предраковые поражения, заболевания, связанные с иммунным ответом, вирусные инфекции, дегенеративные заболевания мышечно-скелетной системы и заболевания почек. Каждое из соединений, представленных формулами I и II, может содержаться в препарате или вводиться вместе с другими терапевтическими агентами, которые выбирают вследствие их особой полезности для комбинированной терапии вышеуказанных состояний. Например, каждое из соединений формул I и II могут вводить в препарат вместе с агентами для предотвращения тошноты, гиперчувствительности, раздражения желудка, такими как противорвотные средства, H1 и Н2 антигистаминные средства. Вышеуказанные терапевтические агенты при использовании их в сочетании с соединениями формул I и II могут быть использованы в количествах, указанных в Physicians’ Desk Reference (PDR) или определяемых специалистом в данной области. Далее, соединения формул I и II могут вводиться в комбинации с другими противораковыми и цитотоксическими агентами и методами лечения, используемыми при лечении рака или других пролиферативных заболеваний. Особенно полезными являются комбинации противораковых и питотоксических лекарств, когда второе выбранное лекарство действует по другому механизму или в другой фазе клеточного цикла, например S-фазе, по сравнению с соединением формулы I и II, которые проявляют свое действие в G2-M-фазе. Примеры таких классов противораковых и цитотоксических агентов включают, без ограничения, алкилирующие агенты, такие как азотистые аналоги иприта, алкилсульфонаты, нитрозомочевины, этиленимины и триазены; антиметаболиты, такие как антагонисты фолатов, аналоги пурина и аналоги пиримидина, антибиотики, такие как антрациклины, блеомицины, митомипин, дактиномицин и пликамицин, ферменты, такие как L-аспарагиназа, ингибиторы фарнезилпротеинтрансферазы, гормональные агенты, например глюкокортикоиды, эстрогены/антиэстрогены, андрогены/антиандрогены, прогестины и антагонисты лютеинизирующий гормон высвобождающего гормона, ацетат октреотида, агенты, разрушающие микротрубочки, такие как эктеинасцидины или их аналоги и производные, агенты, стабилизирующие микротрубочки, такие как паклитаксел (Taxol®), доцетаксел (Taxotere®); продукты растительного происхождения, такие как алкалоиды семейства Vinca, эпиподофиллотоксины, таксаны и ингибиторы топоизомеразы, ингибиторы пренилпротеинтрансферазы и смешанные агенты, такие как гидроксимочевина, прокарбазин, митотан, гексаметилмеламин, координационные комплексы платины, такие как цисплатин и карбоплатин, и другие агенты, используемые как противораковые и цитотоксические вещества, такие как модификаторы биологического ответа, факторы роста, иммуномодуляторы и моноклональные антитела. Соединения, представленные формулами I и II, могут также применяться в сочетании с радиационной терапией. Примеры этой группы противораковых и цитотоксических агентов включают, без ограничения, мехлорэтамина гидрохлорид, циклофосфамид, хлорамбуцил, мелфалан, ифосфамид, бусульфан, кармустин, ломустин, семустин, стрептозоцин, тиотепу, дакарбазин, метотрексат, тиогуанин, меркаптопурин, флударабин, пентастатин, кладрибин, цитарабин, флуороурацил, доксорубицина гидрохлорид, даунорубицин, идарубицин, блеомицина сульфат, митомицин С, актиномицин D, сафрамицины, хинокарцины, дискодермолиды, винкристин, винбластин, винорелбина тартрат, этопозид, тенипозид, паклитаксел, тамоксифен, эстрамустин, эстрамустинфосфат натрия, флутамид, бусерелин, лейпролид, птеридины, диины, левамизол, афлакон, интерферон, интерлейцины, алдеслейкин, филграстим, сарграмостим, ритуксимаб, BCG, третиноин, иринотекана гидрохлорид, бетаметозон, гемцитабина гидрохлорид, алтретамин и топотеку и любые их аналоги и производные. Предпочтительными представителями этих соединений являются, без ограничения, паклитаксел, цисплатин, карбоплатин, доксорубицин, карминомицин, даунорубицин, аминоптерин, метотрексат, метоптерин, митомицин С, эктеинасцидин 743, порфиромицин, 5-флуороурацил, 6-меркаптопурин, гемцитабин, цитозинарабинозид, подофиллотоксин или производные подофиллотоксина, такие как этопозид, этопозида фосфат или тенипозид, мелфалан, винбластин, винкристин, лейросидин, виндезин и лейрозин. Примеры противораковых и других цитотоксических агентов включают следующие соединения: ингибиторы циклинзависимой киназы, такие как описанные в заявке WO 99/24416, и ингибиторы пренилпротеинтрансферазы, описанные в заявках WO 97/30992 и WO 98/54966. Соединения можно также вводить вместе с противораковыми и цитотоксическими агентами, являющимися нейротоксическими, то есть губительными для нервной системы или после этих агентов. Не ограничиваясь какой-либо теорией, рассматривающей механизм или морфологию, соединения, представленные формулами I и II, можно также использовать для лечения заболеваний, отличных от рака или других пролиферативных заболеваний. Такие болезни включают, без ограничения, вирусные инфекции, вызванные, например, вирусом герпеса, поксвирусом, вирусом Эпштейна-Барра, вирусом Синдбиса и аденовирусом; аутоиммунные заболевания, такие как системная красная волчанка, иммунный гломерулонефрит, ревматоидный артрит, псориаз, воспалительные заболевания кишечника и аутоиммунный сахарный диабет; нейродегенеративные заболевания, такие как болезнь Альцгеймера, связанная со СПИД’ом деменция, болезнь Паркинсона, боковой амиотрофический склероз, пигментный ретинит, мышечная атрофия спинного мозга и церебральная дегенерация, СПИД, миелодиспластические синдромы, апластическую анемию, инфаркты миокарда, связанные с ишемическими повреждениями, мозговой удар и повреждения при реперфузии, рестеноз, аритмию, атеросклероз, заболевания печени, вызванные токсинами или алкоголем, гематологические заболевания, такие как хроническая анемия и апластическая анемия, дегенеративные повреждения скелетно-мышечной системы, такие как остеопороз или артрит, риносинусит, восприимчивый к аспирину, кистозный фиброз, рассеянный склероз, заболевания почек и боль при раковых заболеваниях. Соединения, представленные формулами I и II, особенно последние, трудно поддаются получению рецептур на их основе, вследствие того, что они имеют очень низкую растворимость в водной среде, быстро разлагаются при контакте с водной средой, чувствительны к низкому рН, находясь в растворе, чувствительны к действию света, являются цитотоксическими (класс D) и чрезвычайно плохо смачиваются. Любая одна или две из этих характеристик могут быть компенсированы при получении фармацевтического препарата для внутривенного введения, но комбинация всех этих недостатков представляет затруднения для специалиста при получении препаратов. С учетом того, что все вещества, которые должны быть использованы при получении состава для внутривенного введения, должны отвечать требованиям, предъявляемым к таким компонентам для внутривенного введения, оказалось неожиданным то, что состав, полученный согласно данному изобретению, позволил преодолеть все недостатки аналогов эпотилона, указанные выше, которые затрудняют получение рецептур на их основе. Вначале вследствие того, что указанные аналоги эпотилона плохо растворяются в водной среде и быстро разлагаются при контакте с этой средой, было решено использовать лиофилизированную форму этих соединений. Было обнаружено, что подходящей средой для получения раствора указанных соединений для лиофилизации является смесь трет.бутанола и воды для инъекций. Эта смесь должна содержать, по меньшей мере, около 50% об./об., предпочтительно, от примерно 50% до примерно 80% об./об. третичного бутанола для предотвращения разложения вышеуказанных аналогов эпотилона. Кроме того, вследствие очень плохой смачиваемости указанных аналогов эпотилона начальный раствор должен быть получен с использованием смеси из, по меньшей мере, примерно 60% об./об., предпочтительно, примерно 60-95% об./об. трет.бутанола и воды. Как только раствор получен, можно добавить требующееся количество воды или смеси трет.бутанол – вода для достижения конечной концентрации раствора для лиофилизации, указанной выше. Неожиданно было установлено, что стабильность указанных аналогов эпотилона может быть значительно повышена при осуществлении получения раствора при примерно 15°С, более предпочтительно, при температуре около 5°С. Далее, оба процесса – получение раствора и последующая лиофилизация – должны проводиться в таких сосудах, чтобы аналоги эпотилона были защищены от воздействия света. Выгодно проводить лиофилизацию сравнительно небольшими партиями, чтобы аналоги эпотилона подвергались действию водной среды в течение минимального промежутка времени. Первая стадия сушки при лиофилизации раствора, полученного, как описано выше, осуществляется при температурах от примерно -10°С до примерно -40°С, предпочтительно, при температуре около -25°С, в условиях высокого вакуума, то есть примерно от 50 мторр до примерно 300 мторр, предпочтительно, около 200 мторр, в течение продолжительного периода времени, а именно, примерно от 24 часов до примерно 96 часов, предпочтительно, около 48 часов. Лиофилизация при указанном температурном режиме приводит к получению аморфного продукта, который является желательным для внутривенного препарата. Специалистам очевидно, что обычные методы, такие как порошковая дифракция Х-лучей, могут быть использованы для подтверждения аморфной природы лиофилизированного продукта. Остаточные растворители в полученном продукте удаляют на второй стадии сушки, которую проводят при сравнительно низких температурах, а именно от примерно 10°С до примерно 30°С, предпочтительно, при температуре около 25°С, в условиях высокого вакуума, а именно, при примерно от 50 мторр до примерно 300 мторр, предпочтительно, при примерно 150 мторр, в течение продолжительного периода времени, а именно, в течение от примерно 24 часов до примерно 96 часов, предпочтительно, в течение примерно 48 часов. Неожиданно было установлено, что стабильность лиофилизированных аналогов эпотилона, описанных в данной заявке, не повышается при использовании эксципиентов, обычно используемых для этих целей, таких как лактоза, маннит, декстрин и т.п. Некоторые из этих эксципиентов в действительности могут оказывать отрицательное влияние на стабильность лиофилизированного продукта (лиофилизата). Следовательно, аналоги эпотилона, препаративные формы которых получают согласно данному изобретению, лиофилизируются в чистом виде, то есть без какого-либо эксципиента. Лиофилизированные аналоги эпотилона, представленные формулами I и II, воссоздаются при получении смеси равных объемных частей дегидратированного спирта, Dehydrated Alcohol, USP, и неионного поверхностно-активного вещества, предпочтительно, полиэтоксилированного касторового масла, производимого GAF Corporation, Mount Olive, New Jersey, под товарным знаком Cremophor EL. Лиофилизированный продукт и носитель для воссоздания упаковывают по отдельности в защищенные от света флаконы. Для минимизации количества поверхностно-активного вещества в воссозданном растворе при получении раствора добавляют только достаточное для получения раствора количество носителя с получением раствора, имеющего концентрацию аналога эпотилона от примерно 2 до примерно 4 мг/мл. Как только лекарство растворится, полученный раствор разбавляют перед инъекцией подходящим для парентерального введения разбавителем. Такие разбавители хорошо известны специалистам. Эти разбавители обычно доступны в клиниках. В рамках данного изобретения предусмотрено также наличие в упаковке аналогов эпотилона третьего флакона, содержащего достаточное количество парентерального разбавителя для достижения конечной концентрации. Предпочтительный разбавитель представляет собой раствор Рингера с лактатом. Конечная концентрация раствора для введения предпочтительно составляет от примерно 0,1 мг/мл до примерно 0,9 мг/мл. Окончательное разбавление воссозданного аналога эпотилона в составе по изобретению можно осуществлять, используя другие пригодные препараты, например 5% Dextrose Injection, раствор Рингера с лактатом и декстрозой, стерильную воду для инъекций и т.п. Однако из-за узкого интервала рН, рН=6,0-7,5, предпочтительным является раствор Рингера с лактатом. Раствор Рингера с лактатом содержит в 100 мл: хлорид натрия UPS 0,6 г, лактат натрия 0,31 г, хлорид калия UPS 0,03 г и хлорид кальция × 2Н2O UPS 0,02 г. Осмолярность равна 275 мОсмол/л, что очень близко к изотоническому раствору. Полученный препарат согласно данному изобретению, а именно раствор аналога эпотилона в носителе, представляющем собой спирт и поверхностно-активное вещество, может храниться до примерно 24 часов перед дальнейшим разбавлением при введении. Было установлено, что возникновение аллергических реакций, обусловленных наличием поверхностно-активного вещества в составе, сводится к минимуму при поддержании минимально необходимой для растворения аналога эпотилона концентрации. Кроме того, возникновение таких реакций происходит в такой же степени, что и при парентеральном введении других лекарственных веществ, таких как циклоспорин. Этот наблюдаемый уровень проявления аллергических реакций при применении состава по изобретению значительно ниже, чем при применении некоторых других онкологических агентов, таких как Паклитаксел. Данное изобретение предусматривает также способы лечения рака и других гиперпролиферативных заболеваний у пациентов, включающий введение пациенту терапевтически эффективного количества одного или нескольких соединений, представленных формулами I и II. Соединения формулы I и II можно вводить внутривенно или перорально, предпочтительно, и перорально, и внутривенно. Предпочтительно вводить соединения формул I и II с одним или несколькими дополнительными агентами для предотвращения тошноты, гиперчувствительности или раздражения желудочного тракта, такими как антиеметики или H1 или Н2 антигистаминные соединения. Количество соединения формул I и II, вводимого при каждой IV инфузии или перорально или обоими способами, может быть определено специалистом в данной области, например, для человека используют дозу от примерно 0,01 мг/кг/дн до примерно 200 мг/кг/дн, которую можно ввести один раз или в виде разделенных доз, например, 1-4 раза в день. Предпочтительно вводить соединения в дозе менее примерно 100 мг/кг/дн и, более предпочтительно, менее 25 мг/кг/дн один раз или в виде примерно 2-4 разделенных доз. Следует иметь в виду, что конкретные дозы и частота приема для любого конкретного субъекта могут меняться и зависят от ряда факторов, включающих активность конкретного используемого соединения, метаболическую стабильность и длительность действия этого соединения, вид субъекта, возраст, вес, общее состояние, пол и диету, способ и время введения, скорость выведения, комбинацию лекарств и степень развития заболевания. Предпочтительными субъектами, подвергающимися лечению, являются животные, наиболее предпочтительны млекопитающие, такие как люди и домашние животные, такие как собаки, кошки и т.п., страдающие от вышеперечисленных болезней. Обычно соединения формул I и II вводят до тех пор, пока у пациента не возникает реакция, например уменьшение размера опухоли, или пока не достигается доза, ограничивающая токсичность. Специалисту будет ясно, когда пациент обнаружит признаки реакции или когда будет достигнута доза, определяющая токсичность. Обычно доза, определяющая токсичность, связанную с соединениями формул I и II, проявляется, но не ограничивается этим, в усталости, артральгии/миалгии, анорексии, гиперчувствительности, нейропении, тромбоцитопении и нейротоксичности. При внутривенном введении соединения формул I и II, предпочтительно, вводятся в виде составов по изобретению. Обычно соединения формул I и II вводят при IV инфузии в течение от примерно 10 минут до примерно 3 часов, предпочтительно, в течение от примерно 30 минут до примерно 2 часов, более предпочтительно, в течение примерно 45-90 минут и, наиболее предпочтительно, в течение примерно 1 часа. Обычно соединения вводят внутривенно в дозе от примерно 0,5 мг/м2 до 65 мг/м2, предпочтительно, от примерно 1 мг/м2 до 50 мг/м2, более предпочтительно, от примерно 2,5 мг/м2 до 30 мг/м2 и, наиболее предпочтительно, в дозе примерно 25 мг/м2. Специалистам в данной области известно, как пересчитать дозы из мг/кг в мг/м2 При пероральном методе введения соединения формул I и II, предпочтительно, вводят в комбинации с фармацевтически приемлемым буфером, нейтрализующим кислоту. Буфер нейтрализует кислоту в желудке пациента, и скорость разложения соединений формул I и II значительно уменьшается, и они остаются в желудочно-кишечном тракте в течение срока, достаточного для их абсорбции. Соединения формул I и II можно вводить вместе с антацидом, таким как гидроокиси алюминия и магния, карбонаты, например карбонат натрия и карбонат кальция, силикаты и фосфаты, для нейтрализации кислоты в желудке до, во время и после введения соединений формул I и II. Используемый в данном описании термин «фармацевтически приемлемый буфер, нейтрализующий кислоту», относится к сочетанию фармацевтически приемлемой нетоксичной кислоты и фармацевтически приемлемой нетоксичной соли кислоты, которые при добавлении к раствору обеспечивают получение раствора, который более стоек к изменению рН по сравнению с раствором без буфера, когда кислоту или щелочь добавляют к раствору. Термин «фармацевтически приемлемый буфер, нейтрализующий кислоту», включает также соединения, такие как основные соединения, которые при добавлении к кислому раствору нейтрализуют кислоту и увеличивают рН раствора. Согласно одному варианту изобретения соединения формул I и II и фармацевтически приемлемый буфер, нейтрализующий кислоту, вводят перорально в одной дозированной форме одновременно. Композиция, содержащая комбинацию соединений формул I и II, может быть введена перорально в виде твердой дозированной формы (например, таблетки, капсулы или порошка) или жидкой дозированной формы (например, раствора, суспензии или эликсира). Раствор или суспензия могут быть приготовлены перед самым введением с применением подходящих растворителей или сорастворителей для растворения эпотилона и компонентов буфера. Например, соединения формул I и II и фармацевтически приемлемый буфер, нейтрализующий кислоту, могут быть введены одновременно перорально в виде раствора эпотилона формулы I или II, растворенного в жидкости, содержащей пропиленгликоль:этанол:фосфатный буфер (например, 1М, рН около 8) в соотношении 58:12:30 соответственно. Соединения формул I и II и фармацевтически приемлемый буфер, нейтрализующий кислоту, могут быть также в виде раздельных фармацевтических композиций, которые вводят по отдельности. Каждая из этих композиций вводится в виде твердой дозированной формы или жидкой дозированной формы перорально. Если соединения формул I и II и фармацевтически приемлемый буфер, нейтрализующий кислоту, вводят по отдельности, фармацевтически приемлемый буфер, нейтрализующий кислоту, может вводиться перорально до, после или до и после соединений формул I и II. Предпочтительно, фармацевтически приемлемый буфер, нейтрализующий кислоту, вводить и до, и после перорального приема соединений формул I и II в количестве, достаточном для нейтрализации кислоты в желудке. Когда фармацевтически приемлемый буфер, нейтрализующий кислоту, вводят перед приемом соединений формул I и II, он вводится за 5 часов, предпочтительно, примерно за 3 часа, более предпочтительно, примерно за 1 час и, наиболее предпочтительно, примерно за 10 минут до введения соединений формул I и II. Когда фармацевтически приемлемый буфер, нейтрализующий кислоту, вводят после введения соединений формулы I и II, его вводят через примерно 5 часов, предпочтительно, примерно через 3 часа, более предпочтительно, примерно через 1 час и, наиболее предпочтительно, примерно через 10 минут после введения соединений формул I и II. Соединения формул I и II можно вводить в виде пилюли или капсулы с энтеропокрытием для замедления высвобождения эпотилона до тех пор, пока не будет введен фармацевтически приемлемый буфер, нейтрализующий кислоту. Таблетки и капсулы с энтеропокрытием представляют собой указанные формы с покрытием из веществ, которые стойки к желудочному соку, но размельчаются в интестине. Обычно фармацевтически приемлемый буфер, нейтрализующий кислоту, вводят в количестве, достаточном для доставки, по меньшей мере, около 20 мэкв нейтрализатора кислоты, предпочтительно, по меньшей мере, около 30 мэкв нейтрализатора кислоты, более предпочтительно, по меньшей мере, около 40 мэкв нейтрализатора кислоты и, наиболее предпочтительно, около 50 мэкв нейтрализатора кислоты. Обычно фармацевтически приемлемый буфер, нейтрализующий кислоту, вводят в виде водного раствора, имеющего рН между 5 и 9, предпочтительно, около 6-8,5 и, более предпочтительно, около 7-8. При осуществлении способов по изобретению можно применять любой фармацевтически приемлемый буфер, нейтрализующий кислоту, который обеспечивает получение раствора с рН в желательном интервале. Предпочтительно применять фармацевтически приемлемый буфер, нейтрализующий кислоту, представляющий собой двухосновный фосфат – моноосновный фосфат или двухосновный фосфат – лимонная кислота – цитрат. Например, пероральный способ введения соединений формул I и II может включать первое пероральное введение пациенту фармацевтически приемлемого буфера, нейтрализующего кислоту, в виде 150 мл водного раствора, содержащего безводный двухосновный фосфат натрия (около 0,2 М), дигидрат цитрата натрия (около 0,07 М) и безводную лимонную кислоту (около 0,008 М) при рН около 7,4 с последующим пероральным введением соединений формул I и II в виде жидкой дозированной формы в смеси пропиленгликоль:этанол с отношением 80:20 с дальнейшим введением перорально другой порции (около 150 мл) водного раствора, содержащего безводный двухосновный фосфат натрия (около 0,2 М), дигидрат цитрата натрия (около 0,07 М) и безводную лимонную кислоту (около 0,008 М) при рН около 7,4. Как указано выше, соединения формул I и II можно вводить перорально, внутривенно или перорально/внутривенно. В частности, способы по изобретению включают протоколы введения, такие как один раз в день в течение 2-10 дней, предпочтительно, в течение 3-9 дней, более предпочтительно, в течение 4-8 дней и, наиболее предпочтительно, в течение 5 дней. Согласно одному из вариантов время введения включает период от 3 дней до 5 недель, предпочтительно, от 4 дней до 4 недель, более предпочтительно, от 5 дней до 3 недель и, наиболее предпочтительно, от 1 до 2 недель, между циклами, когда лечение не производится. Согласно другому варианту соединения формул I и II можно вводить перорально, внутривенно или перорально/внутривенно один раз в день в течение 3 дней с перерывом в 1-3 недели между циклами, когда лечение не производится. Согласно еще одному варианту соединения формул I и II можно вводить перорально и/или внутривенно один раз в течение 5 дней с перерывом, предпочтительно, в течение 1-3 недель, когда лечение не проводится. Согласно предпочтительному варианту цикл введения соединений формул I или II включает ежедневное введение один раз в день в течение 5 дней с перерывом между циклами лечения от 2 до 10 дней, предпочтительно, в течение одной недели. Соединения формул I и II можно вводить перорально и/или внутривенно один раз каждую неделю в течение 1-10 недель, предпочтительно, в течение 2-8 недель, более предпочтительно, в течение 3-6 недель, еще более предпочтительно, в течение 3 недель. Согласно другому способу по изобретению соединения формул I и II вводят в течение 28-дневного цикла, когда соединение формулы I или II вводят внутривенно в 1-ый, на 7-ой и на 14-ый день и вводят перорально на 21-ый день. Иначе можно вводить соединения формул I и II в течение 28-дневного цикла, при этом соединения формул I и II вводят перорально в 1-ый день и вводят внутривенно на 7-ой, 14-ый и 28-ой день. Согласно способам по изобретению соединения формул I и II вводят до тех пор, пока у пациента не возникнет реакция, например, произойдет уменьшение размера опухоли или до тех пор, пока не будет достигнут предел токсичности. Многие противораковые агенты являются нейротоксичными, например, известно, что они вызывают побочные эффекты, поражая центральную и периферическую нервную систему. Данное изобретение далее охватывает применение соединений формул I и II для лечения пациентов, у которых наблюдалась нейротоксичность после использования других противораковых агентов. Хотя соединения по изобретению могут быть также нейротоксичными в некоторых дозах, способы по изобретению можно применять для уменьшения или устранения такой токсичности. Следующие примеры служат для иллюстрации данного изобретения, не ограничивая его объем. Пример 1: IV дозированная форма. [1S-[1R*,3R*(Е),7R*,10S*,11R*,12R*,16S*]]-7,11-дигидрокси-8,8,10,12,16-пента-метил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17-оксабицикло[14.1.0]-гептадекан-5,9-дион, 9,86 г смачивали/частично растворяли в 600 мл смеси 9:1 трет.бутанола и воды для инъекции USP, которая была предварительно охлаждена до 5°С. Как только было достигнуто полное смачивание порошка лекарства, завершали его растворение путем добавления 600 мл смеси 9:1 трет.бутанола и воды для инъекций и 766 мл смеси 1:1 трет.бутанола и воды для инъекций, которая также была предварительно охлаждена до 5°С, с получением конечного раствора, представляющего собой смесь 1:1. Растворение осуществляли в условиях защиты от света. Полученный выше раствор подвергали лиофилизации в лиофилизаторе Virtis INOTOP при -16°С в условиях защиты от света в течение 48 часов. Полученный лиофилизированный продукт (лиофилизат) затем подвергали сушке при 15°С в условиях высокого вакуума в течение 48 часов. Во время осуществления этих процедур не наблюдалось заметного разложения лекарства. Лиофилизат упаковывали в стерильных условиях во флаконы объемом 30 мл, каждый флакон содержал 10 мг лекарства и стандартный избыток для возмещения потери лекарства во флаконе/шприце. Лиофилизат воссоздавали при помощи 5,5 мл смеси 1:1 по объему Dehydrated Alcohol USP и Cremophor EL®, которая обычно поставляется вместе с лекарством в отдельном флаконе, для получения конечной концентрации лекарства, равной 2 мг/мл. Как только растворение при аккуратном вращении флакона завершится, полученный раствор разбавляют до достижения концентрации 0,2 мг/мл, добавляя 9 мл раствора Рингера с лактатом на каждый миллилитр воссозданного лекарственного препарата. Пример 2: IV Введение соединения II. Группа, состоящая из 24 больных раком (12 мужчин и 12 женщин), принимала соединение II при IV инфузии для того, чтобы определить максимально толерантную дозу (MTD), дозу, ограничивающую токсичность (DLT), фармакокинетику и фармакодинамику и оценить противоопухолевую активность соединения II. Средний возраст (интервал) пациентов был равен 57 (34-74). У пяти пациентов был рак груди, у 5 – рак головы и шеи, у 2-х пациентов была саркома, у 2-х – рак прямой кишки, у 2-х – рак ИТР, у 2-х пациентов – меланома, еще у 2-х – рак пищевода, у одного пациента был рак почек, у одного – цервикальный рак, еще у одного – рак щитовидной железы и у одного – рак заднего прохода. 21 пациент прошел ранее курс химиотерапии (18 принимали нейротоксические агенты и 18 прошли курс радиотерапии). Среднее число линий химиотерапии, включающих адъювант, равнялось 2 (1-3). Пациенты принимали перорально соединение II в 1-й день (в дозах 20 мг/м2 и выше) с последующей 30-минутной IV инфузией соединения II каждую неделю, начиная с седьмого дня. Пациентам вводили соединение II в дозах 1, 2,5, 5, 10, 20, 25 и 30 мг/м2. Проводился мониторинг пациентов во время курса лечения для определения дозы, ограничивающей токсичность (DLT). Результаты исследования показывают, что соединение II можно вводить еженедельно в дозах 30 мг/м2 без проявления заметной токсичности. В процессе второго исследования 12 пациентам, больным раком (5-ти мужчинам и 7-ми женщинам), перорально вводили соединение II в первый день с последующей 30-минутной IV инфузией соединения II каждую неделю, начиная с 7-го дня, в дозе 25 мг/м2 для оценки нейротоксичности соединения II. Средний возраст больных равнялся 51 году (30-65). У 4-х пациентов был рак прямой кишки, у 3-х – рак груди, у 2-х пациентов была меланома, у одного – рак почек, у одного – саркома и у одного – рак яичников. 10 пациентов прошли ранее курс химиотерапии (6 пациентов принимали нейротоксичные агенты и 18 пациентов прошли курс радиотерапии). Среднее число линий химиотерапии, включающих адъювант, равнялось 2 (0-3). Это исследование показало, что соединение II можно вводить пациентам, которые прошли ранее курсы химиотерапии с применением нейротоксичных противораковых агентов. Однако, предпочтительно, чтобы у пациентов, прошедших курсы химиотерапии с применением нейротоксичных противораковых агентов, кумулятивная доза соединения II не превышала примерно 200 мг/м на один цикл. Исследования далее показали, что опухоли груди и ободочной кишки у пациентов, прошедших ранее курс химиотерапии, поддавались лечению соединением формулы II. В частности, пациенты с раком груди, которые ранее прошли курсы лечения адриамицином и таксотером вместе с циклофосфамидом, 5-флуорацилом, метотрексатом; адриамицином и таксотером вместе с циклофосфамидом, 5-флуорацилом, метотрексатом или адриамицином, циклофосфамидом, 5-флуорацилом для лечения метастазного рака, поддавались лечению соединением II. Пациенты, страдающие от метастазного рака ободочной кишки, прошедшие ранее курс лечения таксолом и карбоплатином; 5-флуорацилом и лейковорином или иринотеканом, поддавались лечению соединением II. Пример 3: Фармакокинетика соединения II, вводившегося перорально раковым больным. Пациентам с развившимися злокачественными опухолями вводили еженедельно соединение формулы II в виде 30-минутной инфузии (курс = 3 внутривенных еженедельных введения). Пациенты получали дозы 1, 2,5, 5, 10, 20, 25 или 30 мг/м2. Начиная с дозы, равной 20 мг/м2, пациенты принимали одну пероральную дозу соединения II на 6-ой день в носителе, представляющем собой 80% пропиленгликоля и 20% этанола (об./об.) с последующим введением цитрат/фосфатного буфера (22,5 г) перед курсом 1 для оценки абсолютной биодоступности соединения II. Доза соединения II, введенная перорально на 6-ой день, соответствовала дозе IV соединения II, введенной в первый день. Отбирали образцы плазмы на 6-ой день и в первый день курса 1 для оценки фармакокинетики при помощи системы LC/MS/MS. Образцы анализировали, добавляя внутренний эталон к 0,2 мл образца плазмы, осаждая ацетатом и затем экстрагируя надосадочную жидкость 1-хлорбутаном. Органический слой удаляли и выпаривали досуха. Остаток воссоздавали и вводили в систему LC/MS/MS. производили хроматографическое разделение на колонке YMC ODS-AQ (4,6×50 мм, 3:m) с подвижной фазой ацетонитрил:0,01 М ацетата аммония, рН 5,0 (65:35). Детектирование осуществляли методом тандем-масс-спектрометрии с ионизацией электрораспылением. Стандартная кривая в интервале от 2 до 500 нг/мл для всех аналитов соответствовала модели 1/х-взвешенной квадратичной регрессии. Соединение II для перорального введения в количестве 25 мг/флакон содержалось как «лекарство во флаконе» («drug in bottle»). Носитель (буфер) для воссоздания соединения II, 25 мг/флакон, представлял, собой смесь 80% пропиленгликоля и 20% этанола (об./об.). Смесь пропиленгликоль/этанол готовили смешением 80 ч. По объему пропиленгликоля и 20 ч. по объему этанола в подходящем сосуде, при этом сосуд аккуратно вращали до полного смешения компонентов раствора. Цитратно-фосфатный буфер для перорального введения после приема соединения II находился в отдельном флаконе. Буфер для соединения II воссоздавали при помощи воды для инъекций (WFI). Соединение II готовили для введения пациентам, используя подходящий шприц для медленной инъекции 2,5, 5 или 10 мл смеси пропиленгликоль/этанол во флакон объемом 20 см3, содержащий 25 мг/флакон соединения II, с получением растворов с концентрацией 10, 5 или 2,5 мг/мл соответственно, в зависимости от дозы, которую нужно ввести пациенту. Иглу шприца вынимали и энергично встряхивали флакон в течение 10 секунд. Флакон помещали в баню с воздействием ультразвука, и это воздействие осуществляли до получения прозрачного раствора. Флаконы группировали в зависимости от дозы. Буфер для введения вместе с соединением II поставлялся в прозрачном стеклянном флаконе (8 унций), его воссоздавали при помощи воды для инъекций (WFI). Пробку вынимали из флакона и добавляли около 140 мл воды для инъекций (WFI). Флакон энергично встряхивали или подвергали воздействию ультразвука и встряхивали до получения прозрачного раствора. После перорального введения на 6-ой день были отобраны образцы крови, 7 мл, в пробирки Becton Dickinson Vacutainer с применением в качестве антикоагулянта K3EDTA (верхняя часть цвета лаванды) согласно следующей схеме (выражено в часах:минутах от начала перорального введения): перед введением дозы, 00:15, 00:30, 00:45, 1:00, 1:30, 2:00, 3:00, 4:00, 6:00, 8:00, 24:00, 48:00 и 72:00. После IV инфузии в 1-ый день отбирали образцы (7 мл) крови в пробирки Becton Dickinson Vacutainer с применением в качестве антикоагулянта K3EDTA (верхняя часть цвета лаванды) согласно следующей схеме (выражено в часах:минутах от начала IV инфузии): перед введением дозы, 00:15, 00:30 (конец инфузии), 00:45, 1:00,1:30, 2:00, 3:00, 4:00, 6:00, 8:00, 24:00, 48:00 и 72:00. Сразу же после отбора крови пробирки Vacutainer несколько раз переворачивали, чтобы обеспечить смешение с антикоагулянтом, и затем сразу же помещали в измельченный лед. Через 30 минут после отбора образцы центрифугировали в течение 5 минут со скоростью около 2000 × г и при температуре от 0 до 5°С. Затем помещали плазму в отдельные полипропиленовые пробирки с завинчивающимися пробками и с этикетками и хранили их при -70°С до проведения биоанализа. Концентрации соединения II в плазме определяли методом LC/MS/MS. Концентрацию в плазме в зависимости от времени определяли, используя некомпартментальный анализ. Фармакокинетические параметры, определенные для соединения II, включали максимальную концентрацию в плазме (Смах), время для достижения Смах (Тмах), площадь под кривой зависимости концентрации от времени от момента времени 0 до момента отбора последнего образца Т (AUC(0-T)). 18 пациентов принимали соединение II перорально в виде раствора на 6-ой день и путем IV в 1-ый день. Фармакокинетические параметры, полученные у этих пациентов, представлены в Таблице 1.
Варианты изобретения, описанные выше, предназначены для простой иллюстрации, и специалисты в данной области могут, не используя ничего, кроме рутинных экспериментов, осуществить получение многочисленных эквивалентных соединений, материалов и способов. Все такие эквиваленты находятся в рамках данного изобретения, объем которого определяется формулой изобретения.
Формула изобретения
1. Способ приготовления состава на основе аналога эпотилона, представленного формулой
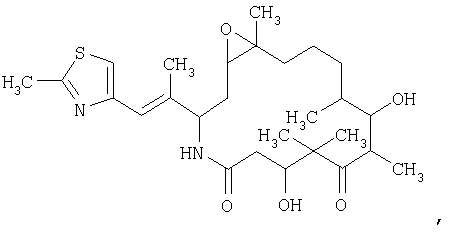
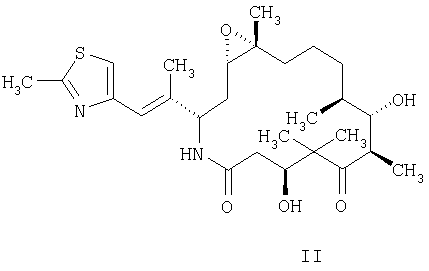
и/или его геометрических, оптических или стереоизомеров, включающий а) растворение указанного аналога эпотилона в смеси, содержащей, по меньшей мере, около 50 об.% трет.бутанола в воде, с получением раствора; б) первую стадию сушки указанного раствора в условиях высокого вакуума до примерно 300 мторр в течение промежутка времени, достаточного для получения первичного лиофилизированного продукта в) вторую стадию сушки первичного лиофилизированного продукта в условиях вакуума в течение промежутка времени, достаточного для получения лиофилизированного продукта аналога эпотилона. 2. Способ по п.1, отличающийся тем, что указанный аналог эпотилона представлен формулой II
3. Способ по п.1 или 2, отличающийся тем, что на стадии а) указанный аналог эпотилона вначале смачивают смесью, по меньшей мере, примерно 60% трет.бутанола с водой и затем к полученному продукту добавляют воду или смесь трет.бутанола и воды в количестве, достаточном для того, чтобы полученный раствор содержал от примерно 2 мг/мл до примерно 30 мг/мл указанного аналога в смеси, содержащей от примерно 50 об.% до примерно 80 об.% трет.бутанола в воде. 4. Способ по п.3, отличающийся тем, что на стадии а) указанный аналог вначале смачивают смесью от примерно 60% до примерно 95% по объему трет.бутанола и воды. 5. Способ по п.1 или 2, отличающийся тем, что первую стадию сушки проводят при температуре около -16°С и давлении от 200 до 300 мторр. 6. Способ по п.1 или 2, отличающийся тем, что вторую стадию сушки проводят при температуре около 16°С и давлении от 150 до 300 мторр. 7. Способ по п.1 или 2, отличающийся тем, что по меньшей мере один из этапов проводят в темноте. 8. Способ по п.1, или 2, или 7, дополнительно включающий упаковку лиофилизированного продукта аналога эпотилона в первый флакон и упаковку во второй флакон средства для восстановления лиофилизированного продукта. 9. Способ по п.8, отличающийся тем, что средство для восстановления содержит дегидратированный спирт и неионное поверхностно-активное вещество. 10. Способ по п.9, отличающийся тем, что средство для восстановления содержит в равных частях по объему дегидратированный спирт и полиэтоксилированное касторовое масло. 11. Способ по п.1 или 2, отличающийся тем, что этап а) растворения аналога эпотилона проводят при температуре ниже температуры окружающей среды. 12. Способ по п.11, отличающийся тем, что на стадии а) указанный аналог эпотилона вначале смачивают смесью, по меньшей мере, примерно 60% трет.бутанола с водой и затем к полученному продукту добавляют воду или смесь трет.бутанола и воды в количестве, достаточном для того, чтобы полученный раствор содержал по меньшей мере 50 об.% трет.бутанола в воде. 13. Способ по п.11 или 12, отличающийся тем, что растворение аналога эпотилона проводят в отсутствие наполнителя. 14. Способ по любому из пп.11-13, отличающийся тем, что первую стадию сушки проводят при температуре около -16°С и давлении от 200 до 300 мторр в течение примерно 96 ч. 15. Способ по любому из пп.11-13, отличающийся тем, что вторую стадию сушки проводят при температуре около 16°С и давлении от 150 до 300 мторр в течение примерно 96 ч. 16. Способ лиофилизации аналога эпотилона, заключающийся в том, что аналог эпотилона формулы (II)
и/или его геометрический, оптический или стереоизомер растворяют в смеси трет.бутанола в воде, и затем полученный раствор сушат под вакуумом до получения лиофилизированного эпотилона аналога. 17. Лиофилизированный аналог эпотилона, полученный способом по п.16. 18. Фармацевтическое средство для лечения рака, содержащее лиофилизированный аналог эпотилона, приготовленный способом по любому из пп.1-7 или 11-16. 19. Фармацевтическое средство для лечения рака, полученное способом по любому из пп.8-10. 20. Фармацевтическое средство для лечения рака, содержащее, по меньшей мере, один флакон с лиофилизированным аналогом эпотилона, приготовленным способом по любому из пп.1-7 или 11-16 и второй флакон со средством для восстановления лиофилизированного аналога эпотилона.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 и
и