Патент на изобретение №2292036
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОДГОТОВКИ ПРОБ ДЛЯ ОПРЕДЕЛЕНИЯ БОРА В ГЕКСАФТОРИДЕ УРАНА МЕТОДОМ АТОМНО-ЭМИССИОННОЙ СПЕКТРОСКОПИИ
(57) Реферат:
Изобретение относится к области аналитической химии. Способ включает смешивание соединения урана и активной добавки. В качестве универсальной активной добавки используют мочевину (карбамид C(NH2)2О) при следующих соотношениях компонентов, мас.%: солевой остаток после выпаривания гексафторида урана 45-49%; оксид алюминия 45-49%; мочевина 2-10%. Мочевину добавляют на стадии смешивания пробы с порошком оксида алюминия. Технический результат – повышение чувствительности анализа и снижение предела обнаружения бора. 2 ил., 1 табл.
Изобретение относится к области аналитической химии, а именно к способам подготовки проб для определения примесных элементов в соединениях урана. Изобретение может быть использовано в промышленных и научно-исследовательских лабораториях для определения бора в гексафториде урана спектральным методом. В настоящее время основным методом определения бора в уране и его соединениях является атомно-эмиссионный спектральный анализ с использованием дуги постоянного или переменного тока в качестве источника возбуждения спектров и графитовых электродов. При этом объектом для анализа является одно из химических соединений урана, как правило, в виде порошка. Бор имеет относительно большое эффективное сечение захвата тепловых нейтронов, поэтому его содержание в урановых материалах, предназначенных для ядерной энергетики, лимитируется на довольно низком уровне. Так, например, максимальное содержание бора в гексафториде урана (ГФУ) не должно превышать 1·10-4%. Существующие методики позволяют определять бор на уровне 5·10-5%, что является недостаточным, например в случае анализа основы для приготовления стандартных образцов (содержание бора в основе не должно превышать 1·10-5%). Определение малых количеств бора методом АЭСА с использованием графитовых электродов связано с определенными трудностями. Во-первых: при температурах 2500-2600°С бор взаимодействует с углеродом и образует карбид тетрабора В4С – чрезвычайно устойчивое и малолетучее соединение даже при температуре порядка 3500°С (температура поверхности анода дуги постоянного тока). Вследствие малой летучести карбида бор в ряду летучести элементов в угольной дуге занимает последнее место после вольфрама и молибдена. Поэтому при определении бора необходимо создание специальных условий для его испарения из всего объема образца. Во-вторых: известно, что в зависимости от формы соединений, в виде которой бор входит в состав стандартного образца состава (СОС), скорость его поступления в плазму разряда различна. Это приводит к ошибке анализа, если бор в пробах и СОС содержится в виде разных химических соединений. Для исключения этой ошибки приходится применять СОС, приготовленные так, чтобы их состав был согласован с составом проб. Форму, в которой бор находится в пробах, устанавливают химическими методами, что осложняет проведение анализов. Для снижения пределов обнаружения и повышения чувствительности спектральных методик обнаружения бора применяют различные способы. Наиболее распространенными являются способы введения в анализируемую пробу активных добавок (носителей, буферов). В качестве носителей обычно используют оксиды и галогениды металлов. Известен, например способ подготовки пробы для определения бора и других примесей спектральным методом в многокомпонентных урановых сплавах, включающий смешивание окисленной пробы с буферными смесями, в качестве которых применяют оксид индия, фтористый натрий и угольный порошок, составляющих 3,7-3,9; 0,25-0,40 и 11,0-14,0% массовых, соответственно, от общей массы смеси с окисленной пробой (авт. св. SU №945723, МПК G 01 N 1/28, оп. 23.07.82 г.) [1] Однако известные способы предназначены для подготовки окисленных проб в виде порошков и не могут быть прямо использованы для анализа гексафторида урана спектральным методом, потому что это соединение при нормальной температуре в замкнутом объеме представляет собой кристаллическое вещество, легко переходящее на открытом воздухе в газообразное состояние, и устойчивую форму при нормальных условиях может иметь только в гидролизованном виде (в виде раствора нитрата уранила и фтороводородной кислоты). Наиболее близким по технической сущности к предлагаемому является способ подготовки пробы для определения бора в урановых соединениях спектральным методом на основе гидролизованного гексафторида урана, включающий упаривание раствора гидролизованного гексафторида урана до образования солевого остатка и смешивание полученного остатка в равных долях с оксидом алюминия. Предел обнаружения бора этим методом составляет 5·10-5% [2]. Недостатком известного способа подготовки пробы является невозможность определения содержания бора в гидролизованном гексафториде урана на уровне ниже 5·10-5%. Задачей изобретения является создание такого способа подготовки пробы, который позволил бы повысить чувствительность анализа и снизить предел обнаружения бора до уровня 1·10-5%. Поставленная задача решена тем, что в известном способе подготовки пробы для атомно-эмиссионного спектрального определения бора в гексафториде урана, включающем упаривание раствора гидролизованного гексафторида урана до образования солевого остатка и смешивание солевого остатка с оксидом алюминия, в смесь добавляют мочевину (карбамид C(NH2)2О) при следующих соотношениях ингредиентов в мас.%: солевой остаток после выпаривания гексафторида урана 45-49%; оксид алюминия 45-49%; мочевина 2-10%, при этом мочевину добавляют на стадии смешивания пробы с порошком оксида алюминия. Определение оптимального количества добавляемой мочевины осуществлялось следующим образом. Пробу на основе гидролизованного ГФУ с содержанием бора на уровне 1·10-4% и образцы для градуировки, приготовленные также на основе гидролизованного ГФУ и содержащие бор в диапазоне от 3·10-5 до 1·10-3%, упаривали до образования солей и высушивали. Затем контрольную пробу и каждый образец для градуировки смешивали в равных пропорциях с оксидом алюминия. От смеси контрольной пробы с оксидом алюминия отбирали несколько порций и на их основе готовили смеси с разным содержанием мочевины. Навески смесей с мочевиной для анализа отбирали таким образом, чтобы масса смеси солевого остатка с оксидом алюминия за вычетом добавки мочевины составляла 40 мг. Съемку спектров проводили при следующих условиях: источник возбуждения спектров – дуга переменного тока, сила тока 18-20 А, графитовые электроды типа “рюмка” с глубиной отверстия 7 мм и диаметром 4 мм, экспозиция 15 с, межэлектродный промежуток 3 мм. Регистрацию спектров проводили при помощи дифракционного спектрографа ДФС-13 и многоканального анализатора атомно-эмиссионных спектров МАЭС. В таблице приведены данные подготовки проб и результаты обработки спектров, полученных в результате эксперимента.
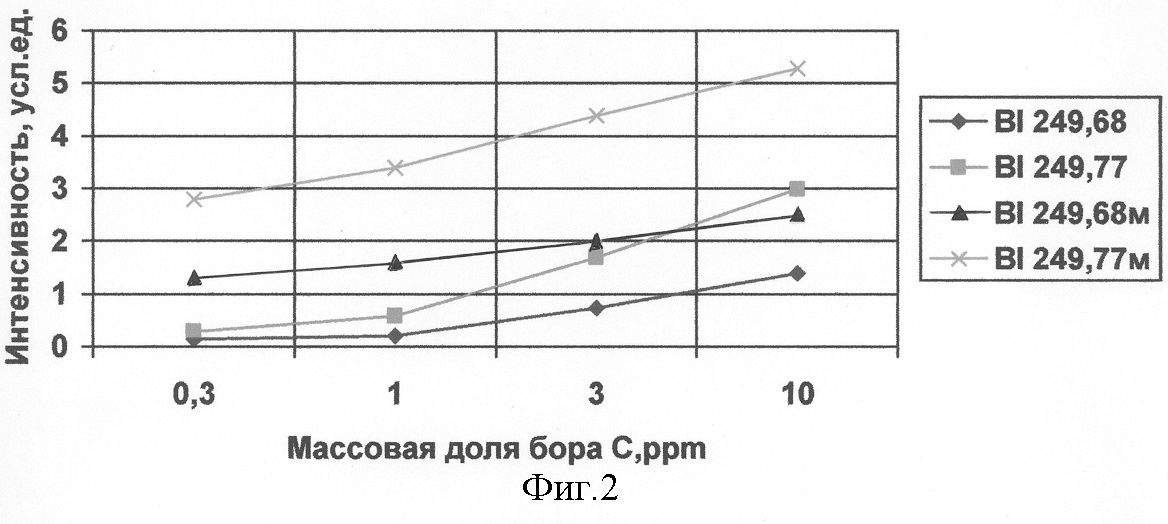
Для наглядности зависимость интенсивности спектральной линии бора от величины добавки мочевины представлена графически на фиг.1. Из таблицы и фиг.1 видно, что максимум интенсивности спектральной линии BI 249.77 нм соответствует 5% добавке мочевины к смеси ГФУ с оксидом алюминия. Экстремальный вид зависимости на фиг.1 свидетельствует также о том, что возможное наличие бора в мочевине в виде примеси не является главной причиной эффекта увеличения интенсивности спектральных линий бора по мере добавления мочевины. В этом случае вид рассматриваемой зависимости имел бы тенденцию к возрастанию или насыщению. Для оценки пределов обнаружения бора при анализе ГФУ с использованием добавки мочевины были приготовлены две серии стандартных образцов на основе ГФУ. Одна серия не содержала мочевины, а в другую добавили около 5% мочевины на стадии смешивания с порошком Al2O3. На фиг.2 показан вид градуировочных графиков, построенных по результатам обработки данных проведенного эксперимента. Градуировочные графики для серии стандартных образцов с добавкой 5% мочевины отмечены индексом “м”. Практическое использование проб для определения бора в гексафториде урана на основе гидролизованного гексафторида урана с добавкой 5% мочевины к смеси равных количеств солевого остатка, полученного после упаривания гидролизованного ГФУ и оксида алюминия, подтвердило повышение чувствительности метода атомно-эмиссионной спектроскопии и снижение предела обнаружения бора до 1·10-5%. Источники информации: 1. Авт. св. SU №945723, G 01 N 1/28. Способ подготовки пробы для определения примесных элементов в многокомпонентных урановых сплавах. / Н.Егоров и И.И.Новикова. 2. Бор. Методика спектрального определения в гексафториде урана. Регистрационный код: рс РФ.31.2003.00049.
Формула изобретения
Способ подготовки пробы для определения бора в гексафториде урана методом атомно-эмиссионного спектрального анализа, включающий смешивание соединения урана и активной добавки, отличающийся тем, что в качестве универсальной активной добавки используют мочевину (карбамид C(NH2)2О) при следующих соотношениях компонентов, мас.%:
при этом мочевину добавляют на стадии смешивания пробы с порошком оксида алюминия.
РИСУНКИ
PD4A – Изменение наименования обладателя патента СССР или патента Российской Федерации на изобретение
(73) Новое наименование патентообладателя:
Адрес для переписки:
Извещение опубликовано: 27.02.2009 БИ: 06/2009
|
||||||||||||||||||||||||||