Патент на изобретение №2290402
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ 2,5,7,8-ТЕТРАМЕТИЛ-2-(2′-КАРБОКСИЭТИЛ)-6-АЦЕТОКСИХРОМАНА – ПРЕДШЕСТВЕННИКА АЛЬФА-СЕНС
(57) Реферат:
Изобретение относится к области синтеза биологически активных веществ, а именно к способу получения 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана (
Предлагаемое изобретение относится к области синтеза биологически активных веществ, конкретно 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана, который является предшественником главного водорастворимого метаболита
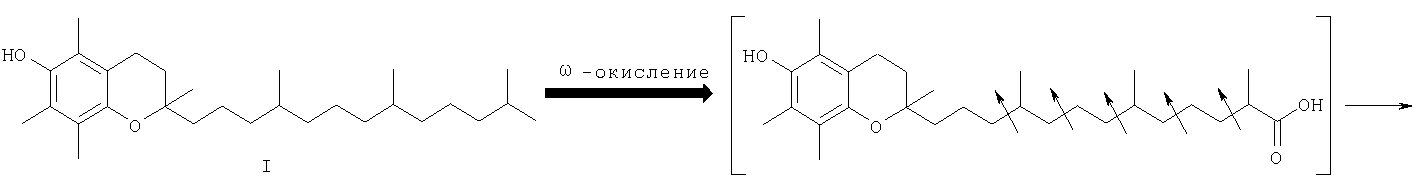
Одним из основных факторов, определяющим биологию Схема 1
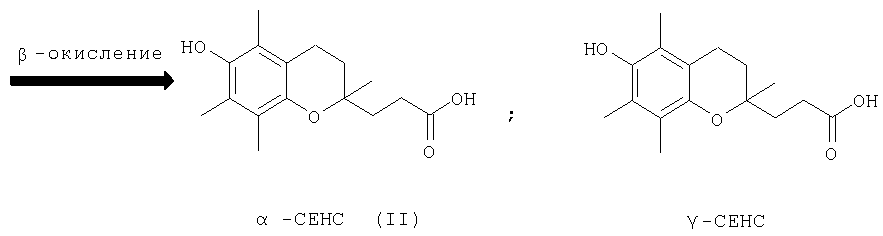
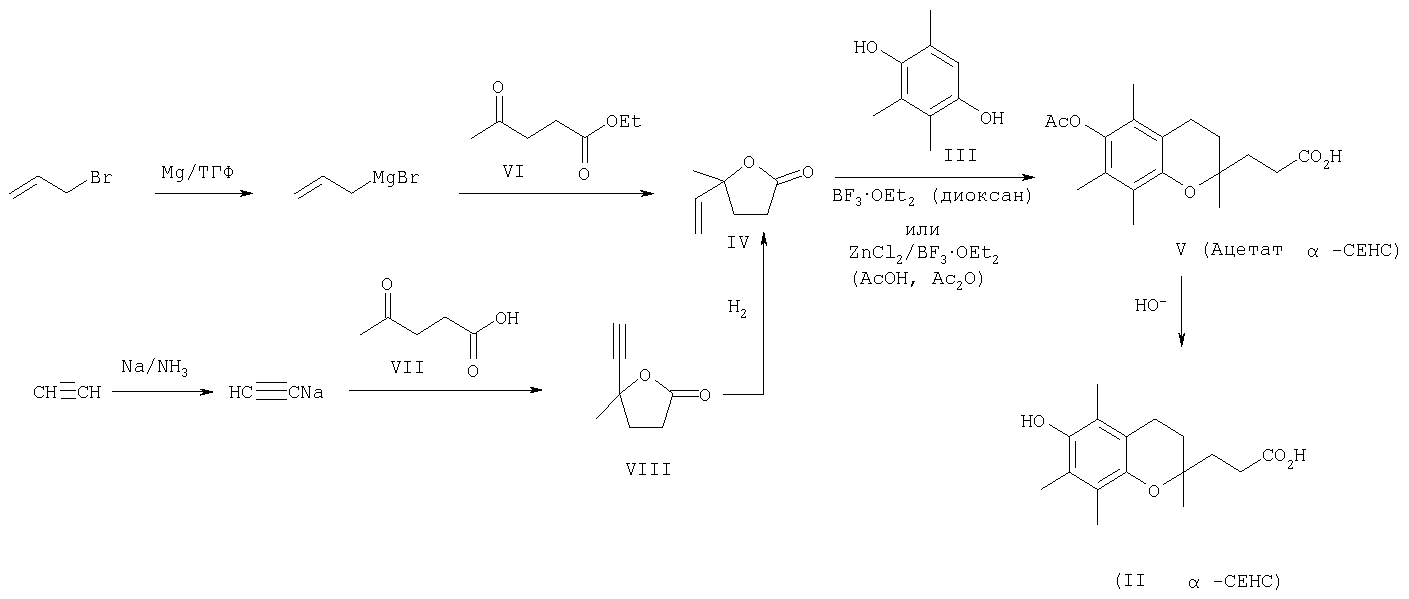
Наличие гидроксильной группы в 6-ом положении хроманового фрагмента токоферола и его аналогов необходимо для проявления их биологической активности. Вместе с тем, некоторые сложные эфиры хроманолов, в том числе ацетаты, проявляют более высокую биологическую активность, чем свободные хроманолы. Например, витаминная активность Изучение метаболизма Препараты, полученные на основе метаболита (II) Высокая эффективность метаболитов Исследования антиоксидантной активности кислоты (II) in vitro в различных модельных окислительных реакциях показали ее высокие антиоксидантные свойства, сравнимые с такими известными препаратами, как Несмотря на обнаруженные уникальные свойства кислоты (II) и открывающиеся перспективы ее использования в биологии и медицине, методы синтеза этого соединения очень ограничены. Известен способ получения кислоты (II) и ее предшественника – 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана (V), основанный на реакции конденсации триметилгидрохинона (ТМГХ) (III) с Схема 2
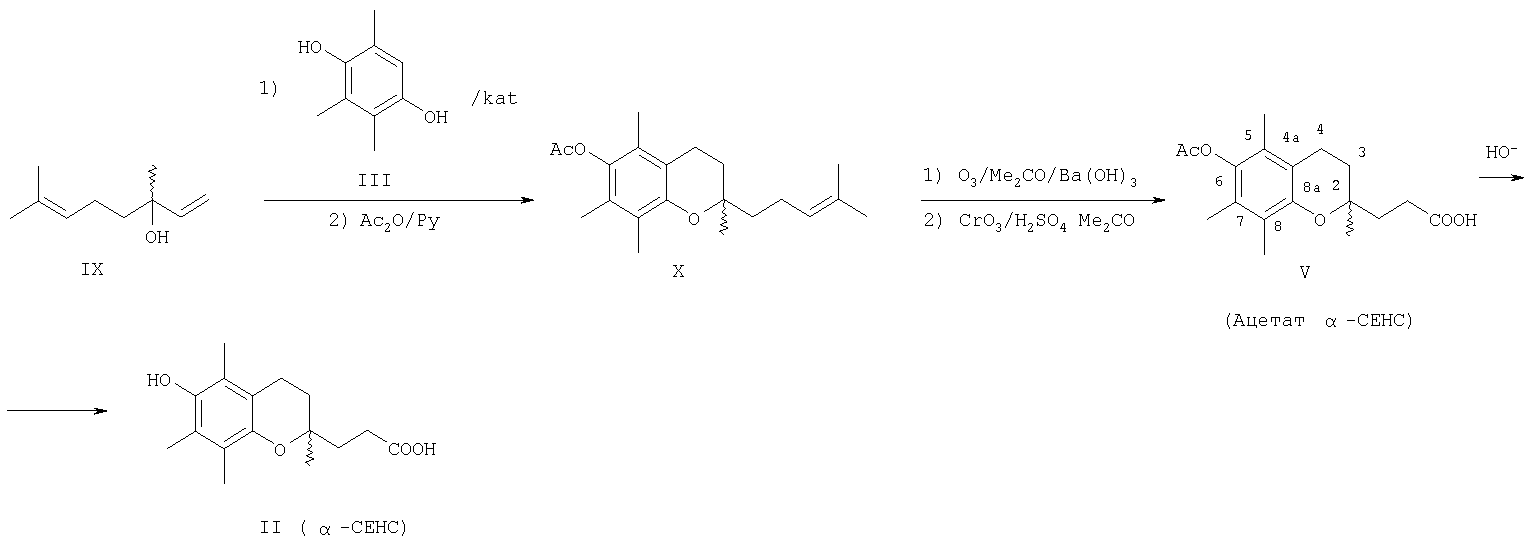
Промежуточный лактон (IV) получали конденсацией винилмагнийбромида с этиловым эфиром левулиновой кислоты (VI) или взаимодействием левулиновой кислоты (VII) с ацетилинидом натрия в жидком аммиаке с последующим частичным гидрированием этинильной группы в промежуточном Как видно, известные способы синтеза ацетатного производного кислоты (II) – 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана (V), приведенные на схеме 2, предполагают предварительное получение в несколько стадий Предлагается новый способ получения ацетатного производного кислоты (II) – 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана (V), заключающийся в проведении реакции конденсации ТМГХ (III) с линалоолом (IX) в кипящем н-октане под действием катализаторов: n-TsOH или (+)-камфор-10-сульфоновой кислоты (CSA) с последующей обработкой продукта конденсации уксусным ангидридом в пиридине при комнатной температуре. Реакция конденсации приводит к 2,5,7,8-тетраметил-2-(4-метил-3-пентен-1-ил)-6-ацетоксихроману (X) с селективностью 68% и 75% (катализ n-TsOH и CSA соответственно). Озонолиз 2,5,7,8-тетраметил-2-(4-метил-3-пентен-1-ил)-6-ацетоксихромана (X) в ацетоне в присутствии Ва(ОН)2 при комнатной температуре и последующее окисление образующегося альдегида реактивом Джонса в ацетоне при комнатной температуре приводят к целевому ацетатному производному 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-гидроксихроман ( Схема 3
Преимущества предлагаемого способа: 1. В реакции конденсации используется легкодоступный винилкарбинол – линалоол. 2. В качестве катализаторов используются стабильные сульфокислоты (n-TsOH и CSA), работа с которыми в отличие от используемых ранее катализаторов (ZnCl2 и BF3·OEt2) не требует особых условий (отсутствие влаги, инертная атмосфера). 3. Помимо реакции конденсации предлагаемая схема синтеза ацетатного производного ( Изобретение поясняется следующими примерами: ПРИМЕР 1. Получение 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана (V). К кипящей суспензии 0.4 г (2.6 ммоля) ТМГХ III и 0.061 г (0.26 ммоля) катализатора CSA в 3 мл н-октана при кипении медленно прибавили 0.40 г (2.6 ммоля) винилкарбинола IX. Реакционную смесь кипятили 3 ч, затем охладили до комнатной температуры и вылили в насыщенный раствор NaHCO3 (30 мл). Продукт экстрагировали EtOAc, органические слои промыли насыщенным раствором NaCl и сушили над MgSO4. Фильтрат упарили и остаток хроматографировали на колонке с SiO2 (16 г). Элюированием н-гексаном выделили фракцию (Rf 0.9, н-гексан – EtOAc, 1:1) неполярных веществ, затем смесью (10:1) н-гексан – EtOAc элюировали фракцию (Rf 0.7, «-гексан – EtOAc, 1:1), упариванием которой получили 0.57 г маслообразного вещества, которое растворили в 5.6 мл сухого пиридина, при перемешивании добавили 4.3 мл уксусного ангидрида, выдержали 0.5 ч при комнатной температуре, затем вылили в 15 мл ледяной воды, экстрагировали EtOAc, промыли последовательно 3 н. раствором HCl, насыщенным раствором NaHCO3, Н2O, и сушили MgSO4. После упаривания получили 0.63 г продукта, содержащего по данным ГЖХ 75% ацетата X, или 0.47 г (1.4 ммоля), который растворили в 6 мл ацетона, добавили 0.6 г (3.5 ммоля) Ва(ОН)2, 0.15 мл Н2О и при комнатной температуре пропускали озонокислородную смесь со скоростью 30 л·ч-1 в течение 9 мин (1.5 ммоля О3 при производительности озонатора 10 ммоль О3·ч-1), после чего реакционную смесь отфильтровали, фильтрат упарили в вакууме. Полученный продукт растворили в 15 мл Et2O, сушили MgSO4, упарили, остаток (0.54 г) растворили в 20 мл ацетона, раствор охладили льдом и при интенсивном перемешивании прикапали 2 мл реагента Джонса, приготовленного из 1.33 г CrO3, 3.8 мл H2O и 1.2 мл H2SO4. При этом наблюдалось расслоение реакционной смеси на нижний зеленый слой хромовых солей и верхний слой, содержащий продукты реакции, растворенные в ацетоне. Смесь перемешивали 2 ч при комнатной температуре, экстрагировали EtOAc (15 мл), органический слой промыли насыщенным раствором NaCl и сушили MgSO4. Остаток после упаривания хроматографировали на колонке с SiO2 (6 г). Элюированием смесью (5:1) к-гексан – EtOAc выделили фракцию (Rf 0.7, н-гексан – EtOAc, 1:1), содержащую минорные примеси, образующиеся в реакции конденсации ТМГХ III с винилкарбинолом IX. Затем элюировали метанолом, получили фракцию с Rf 0.5 (CHCl3-МеОН 1:1), упаривание которой дало 0.24 г (29% в расчете на винилкарбинол IX) 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана V, т.пл. 149-150°С. [ср.: J.Weichet, L.Blaha, В.Kakac. Collect. Of Czechoslovak Chem. Commun. 1959, v.24, pp.1689-1694]. ИК-спектр, ПРИМЕР 2. Из 4.0 г (26 ммоля) ТМГХ III, 4.01 г (26 ммоля) IX и 0.45 г (2.6 ммоля) n-TsOH в условиях, приведенных в примере 1, получили 5.3 г продукта, который растворили в 52 мл сухого Ру, при перемешивании добавили 40.2 мл уксусного ангидрида, выдержали 0.5 ч. при комнатной температуре, затем вылили в 120 мл ледяной воды, экстрагировали EtOAc, промыли 3 н. раствором HCl, насыщенным раствором NaHCO3, Н2О, сушили MgSO4, получив 5.9 г маслообразного вещества, содержащего по данным ГЖХ 68%, или 4.01 г (12.1 ммоля) ацетата X, которое растворили в 60 мл ацетона, добавили 5.12 г (29.9 ммоля) Ва(ОН)2, 1.5 мл Н2О и при комнатной температуре пропускали озонокислородную смесь со скоростью 30 л·ч-1 в течение 75 мин (12.5 ммоля О3 при производительности озонатора 10 ммоль О3·ч-1), после чего реакционную смесь отфильтровали, фильтрат упарили при пониженном давлении. Полученный продукт растворили в 100 мл Et2O, сушили MgSO4, упарили. Остаток (5.2 г) растворили в 20 мл ацетона, охладили льдом и при интенсивном перемешивании прикапали 7 мл реагента Джонса, приготовленного из 2.66 г CrO3, 7.6 мл Н2О и 2.4 мл Н2SO4. При этом наблюдалось расслоение реакционной смеси на нижний зеленый слой хромовых солей и верхний слой, содержащий продукты реакции, растворенные в ацетоне. Смесь перемешивали 2 ч при комнатной температуре, экстрагировали EtOAc, органические слои промыли насыщенным раствором NaCl и сушили MgSO4. Фильтрат упарили и остаток хроматографировали на колонке с SiO2 (50 г), как описано в примере 1. Получили фракцию с Rf 0.5 (CHCl3-MeOH, 1:1), упаривание которой дало 2.3 г (28% в расчете на винилкарбинол IX) 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана V, спектры ЯМР 1Н и 13С идентичны спектрам, приведенным в примере 1.
Формула изобретения
Способ получения 2,5,7,8-тетраметил-2-(2′-карбоксиэтил)-6-ацетоксихромана – предшественника
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 17.06.2007
Извещение опубликовано: 10.02.2009 БИ: 04/2009
NF4A – Восстановление действия патента СССР или патента Российской Федерации на изобретение
Дата, с которой действие патента восстановлено: 20.09.2010
Извещение опубликовано: 20.09.2010 БИ: 26/2010
|
||||||||||||||||||||||||||
 -СЕНС), который заключается в конденсации триметилгидрохинона с линалоолом в кипящем н-октане в присутствии катализатора n-толуолсульфокислоты или (+)-камфор-10-сульфокислоты в течение трех часов при мольном соотношении триметилгидрохинон:линалоол:катализатор 1:1:0.1 с последующим ацетилированием полученного продукта уксусным ангидридом в пиридине при комнатной температуре в течение 0.5 часа, озонированием в ацетоне в присутствии Ba(OH)2, окислением реагентом Джонса в ацетоне и выделением колоночной хроматографией на силикагеле. Данное соединение является ацетатным производным основного метаболита
-СЕНС), который заключается в конденсации триметилгидрохинона с линалоолом в кипящем н-октане в присутствии катализатора n-толуолсульфокислоты или (+)-камфор-10-сульфокислоты в течение трех часов при мольном соотношении триметилгидрохинон:линалоол:катализатор 1:1:0.1 с последующим ацетилированием полученного продукта уксусным ангидридом в пиридине при комнатной температуре в течение 0.5 часа, озонированием в ацетоне в присутствии Ba(OH)2, окислением реагентом Джонса в ацетоне и выделением колоночной хроматографией на силикагеле. Данное соединение является ацетатным производным основного метаболита  -токоферола подвергается
-токоферола подвергается 
 /см-1: 1730, 1700 (С=0), 1210, 1080 (С-O). УФ-спектр (СН3ОН),
/см-1: 1730, 1700 (С=0), 1210, 1080 (С-O). УФ-спектр (СН3ОН),  max/нм(
max/нм( ): 278(1415); 284(1489). Спектр ЯМР 1Н (
): 278(1415); 284(1489). Спектр ЯМР 1Н ( , м.д., J/Гц): 1.21 (с, 3Н, С(2)СН3); 1.79-2.20 (м, 6Н, Н(3), Н(1′), Н(2′)); 1.94, 1.97, 2.06 (с, 9Н, Ar-СН3), 2.28 (с, 3Н, СН3-ОАс); 2.62 (м, 2Н, Н(4)). Спектр ЯМР 13С (
, м.д., J/Гц): 1.21 (с, 3Н, С(2)СН3); 1.79-2.20 (м, 6Н, Н(3), Н(1′), Н(2′)); 1.94, 1.97, 2.06 (с, 9Н, Ar-СН3), 2.28 (с, 3Н, СН3-ОАс); 2.62 (м, 2Н, Н(4)). Спектр ЯМР 13С (