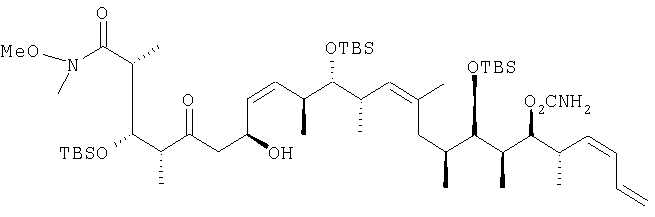
Патент на изобретение №2283309
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ДИСКОДЕРМОЛИДА И ЕГО АНАЛОГОВ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
(57) Реферат:
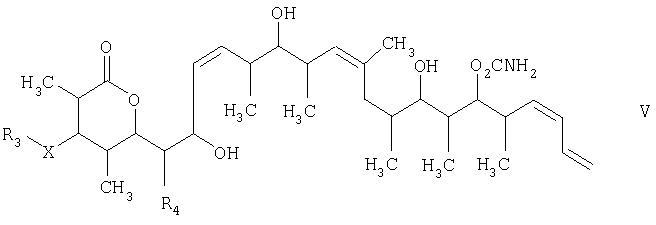
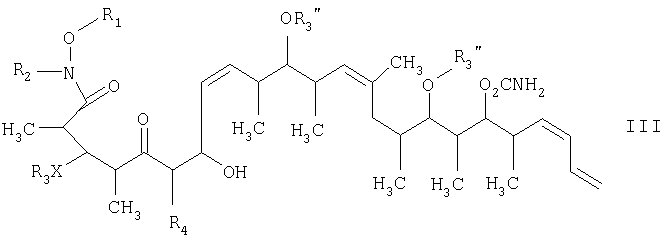
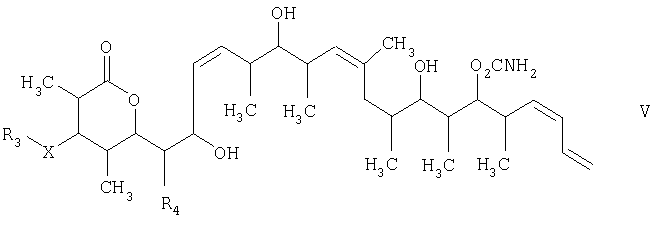
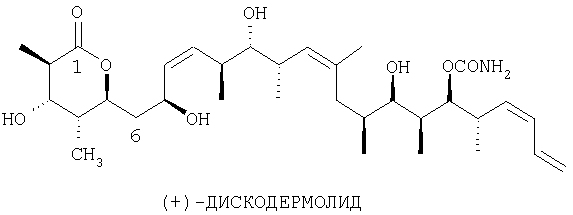
Изобретение относится к способу получения дискодермолида и его аналогов формулы V
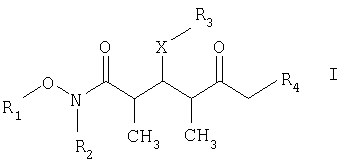
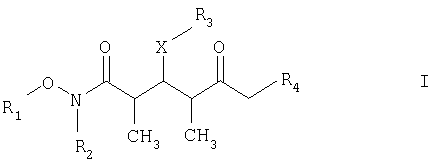
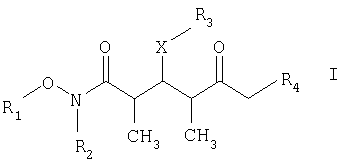
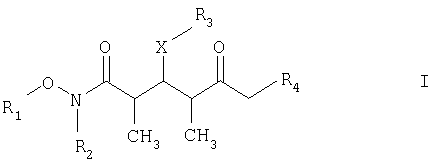
который включает на первой стадии реакцию сочетания кетонового соединения формулы I
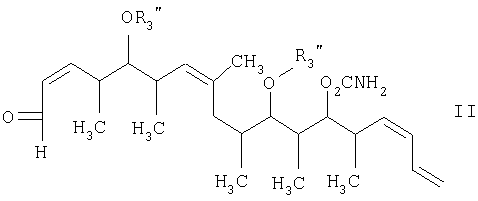
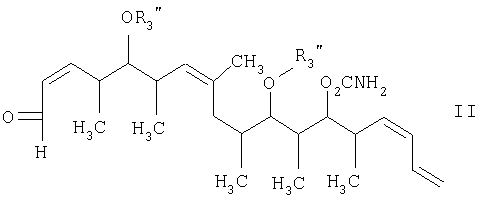
с альдегидным соединением формулы II
в присутствии диалкилборгалогенида или трифлата, аминового основания и полярного органического растворителя с получением
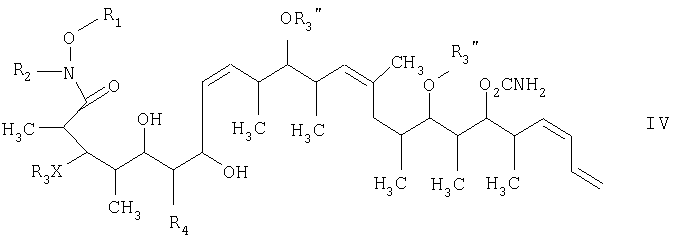
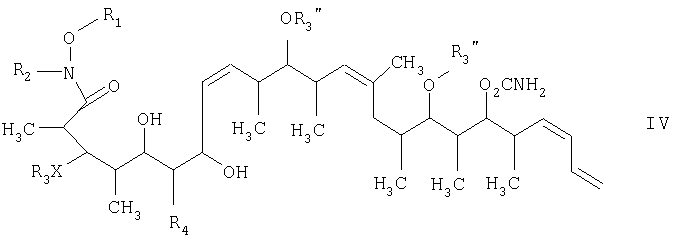
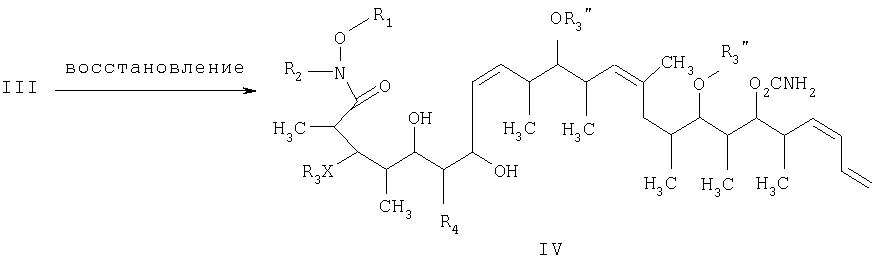
на второй стадии восстановление кетонового соединения, полученного на первой стадии, его обработкой боргидридным реагентом в полярном органическом растворителе и протонном растворителе с получением 1,3-диола формулы IV
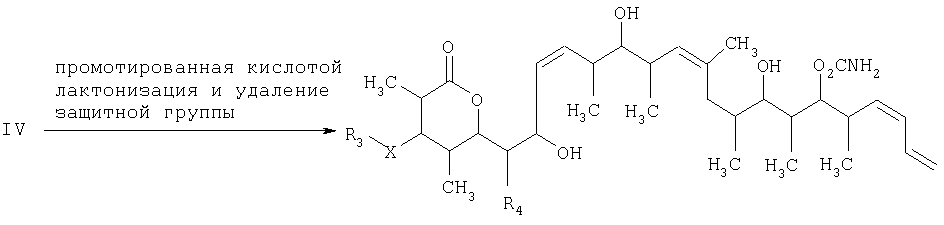
и на третьей стадии лактонизацию и удаление кислотнолабильной гидроксильной защитной группы 1,3-диола, полученного на второй стадии, его обработкой галогенидом водорода, растворенным в полярном растворителе или смеси растворителей, с получением целевого соединения формулы V, где R1 обозначает С1-С6алкил, R2 обозначает С1-С6алкил; R3 обозначает водородный атом или кислотнолабильную гидроксильную защитную группу; R3” обозначает кислотнолабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; а Х обозначает О, при условии, что когда Х обозначает О и R3 обозначает кислотнолабильную гидроксильную защитную группу соединения формулы I, остаток -Х-R3 соединения формулы V представляет собой -ОН. Изобретение также относится к новым промежуточным соединениям формулы I, III и IV и способу получения соединения формулы I. Технический результат – новый способ получения ценного соединения – дискодермолида и его аналогов, позволяющий получать технически приемлемые количества. 5 н. и 11 з.п. ф-лы.
Изобретение относится к способу получения дискодермолида и его аналогов, к новым соединениям, используемым в данном способе, и к новым соединениям, получаемым по этому способу. Предпосылки создания изобретения
(+)-Дискодермолид представляет собой новый поликетидный природный продукт, который был выделен из экстрактов морской губки Discodermolide dissoluta исследователями Harbor Branch Oceanographic Institution (HBOI) (Gunasekera SP, Gunasekera M, Longley RE, Schulte GK. Discodermolide: a new bioactive polyhydroxylated lactone from the marine sponge Discodermolide dissoluta. [В опубликованном номере J.Org.Chem. 1991; 56:1346 содержится опечатка]. J.Org.Chem. 1990; 55:4912-15). У дискодермолида отсутствует явное структурное сходство с паклитакселем, но тем не менее он обладает общей с паклитакселем (активное вещество в лекарственном средстве таксол) способностью стабилизировать микротрубки. В испытаниях по изучению механизма действия дискодермолид оказывается более эффективным, чем паклитаксель. В самом деле, из небольшой группы соединений, которые известны тем, что инициируют полимеризацию очищенного тубулина, дискодермолид является самым эффективным. Однако микротрубки, основной структурный компонент клеток, не являются простыми равновесными полимерами тубулина. Они существуют как динамические ансамбли гетеродимеров Перспективное расширение производства дискодермолида или структурно родственных аналогов затруднено из-за отсутствия надежного природного источника этого соединения или возможного пути синтеза. Встречающегося в природе дискодермолида недостаточно, и добывание организма-продуцента обуславливает проблемы, связанные с поставками. Таким образом, существует постоянно возрастающая потребность в усовершенствованных синтезах, осуществление которых давало бы возможность получения технически приемлемых количеств дискодермолида и структурно родственных аналогов. Краткое изложение сущности изобретения Настоящее изобретение относится к более практичному синтезу дискодермолида и его аналогов. По другому варианту настоящее изобретение относится к новым соединениям, которые могут быть использованы при получении дискодермолида и его аналогов. По еще одному варианту настоящее изобретение относится к новым соединениям, которые получают согласно способу по настоящему изобретению. Подробное описание изобретения В основе настоящего изобретения лежит разработка более практичного синтеза дискодермолида и его аналогов. Более конкретно было установлено, что дискодермолид и его аналоги могут быть получены трехстадийной реакцией следующим образом: СТАДИЯ 1
СТАДИЯ 2
СТАДИЯ 3
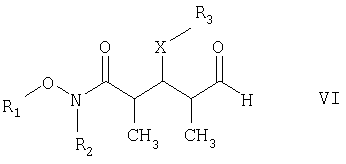
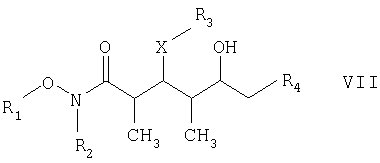
где R1 обозначает C1-С6алкил, бензил или кислотнолабильную гидроксильную защитную группу; R2 обозначает C1-С6алкил или бензил; R3 обозначает водородный атом, C1-С6алкил, бензил, С(O)-С1-С12алкил, C(O)Ph, C(O)O-C1-С12алкил, C(O)OPh, С(O)NH-С1-С12алкил, C(O)NHPh или кислотнолабильную гидроксильную защитную группу; R3” обозначает кислотнолабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; а Х обозначает О, NH, NCH3, S или СН2 при условии, что когда Х обозначает О, а R3 обозначает кислотно-лабильную гидроксильную защитную группу соединения формулы I, остаток -Х-R3 соединения формулы V представляет собой -ОН. Что касается отдельных стадий, то стадия 1 включает реакцию сочетания кетонового соединения формулы I с альдегидным соединением формулы II посредством реакции альдоля с получением На стадии 2 осуществляют восстановление Что касается стадии 3, то она включает лактонизацию и удаление кислотнолабильных гидроксильных защитных групп соединения формулы IV с получением соединения формулы V. Реакцию лактонизации и удаления защитных групп проводят в присутствии: 1) протонной кислоты, предпочтительно водного раствора протонной кислоты, предпочтительнее водного раствора галогенида водорода, такого, как водный хлорид водорода; и 2) полярного органического растворителя, предпочтительно смеси полярных органических растворителей, более предпочтительно смеси алифатического спирта и простого эфира, таких, как метанол и тетрагидрофуран, при температуре в пределах -20 и 40°С, предпочтительно в пределах 20 и 25°С, в течение периода от 8 ч до 7 дней, предпочтительно в пределах 16 и 72 ч, более предпочтительно в пределах 24 и 48 ч. По другому варианту объектом настоящего изобретения являются новые кетоновые соединения формулы I:
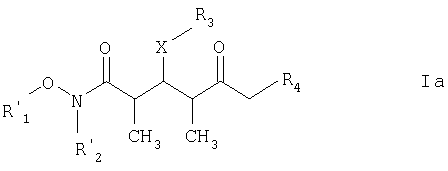
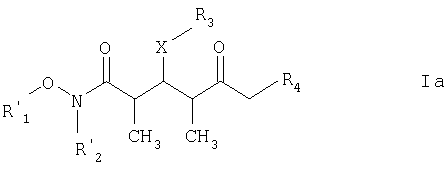
где R1 обозначает C1-С6алкил, бензил или кислотнолабильную гидроксильную защитную группу; R2 обозначает C1-С6алкил или бензил; R3 обозначает водородный атом, C1-С6алкил, бензил, С(O)-С1-С12алкил, C(O)Ph, С(O)O-С1-С12алкил, C(O)OPh, C(O)NH-C1-C12алкил, C(O)NHPh или кислотнолабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; а Х обозначает О, NH, NCH3, S или CH2. Предпочтительными соединениями являются соединения формулы Ia:
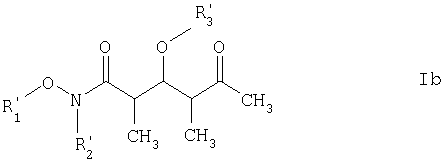
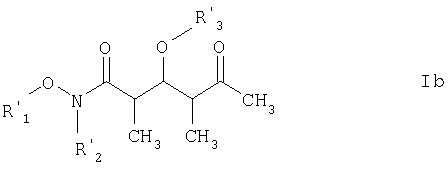
где каждый из R1‘ и R2‘ обозначает C1-С6алкил; Х обозначает О или СН2; а R3 и R4 имеют такие же значения, как указанные выше. Более предпочтительными соединениями являются соединения формулы Ib:
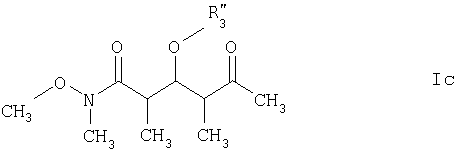
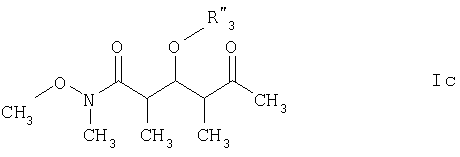
где R3‘ обозначает C1-С6алкил, С(O)-С1-С12алкил, бензил, С(O)O-С1-С12алкил или кислотнолабильную гидроксильную защитную группу; а R1‘ и R2‘ имеют такие же значения, как указанные выше. Еще более предпочтительными соединениями являются соединения формулы Ic:
R3” обозначает кислотнолабильную гидроксильную защитную группу. Среди вышеприведенных определений понятие “C1-С6алкил”, использованное в настоящем описании, относится к прямоцепочечной или разветвленной группе, состоящей только из углеродных и водородных атомов и содержащей от 1 до 6 углеродных атомов, тогда как понятие “С1-С12алкил”, использованное в настоящем описании, относится к прямоцепочечной или разветвленной группе, состоящей только из углеродных и водородных атомов и содержащей от 1 до 12 углеродных атомов. Примеры алкильных групп включают метил, этил, пропил, бутил, пентил, 3-метилпентил и т.д. Понятие “кислотнолабильные гидроксильные защитные группы”, использованное в настоящем описании, относится к любой связанной с кислородным атомом группе, которая может быть удалена под воздействием кислоты. Многочисленные примеры этих групп специалистам в данной области техники известны, их можно найти в работе Greene и Wuts, Protective Groups in Organic Synthesis, издание 2-ое, John Wiley & Sons, New York, 1991. Конкретные примеры включают, хотя ими их список не ограничен, трет-бутилдиметилсилил, триэтилсилил, трет-бутилдифенилсилил, триизопропилсилил, метоксиметил и тетрагидропиранил. По еще одному варианту объектом настоящего изобретения является способ получения новых соединений формулы I. Более конкретно соединения формулы I могут быть получены так, как представлено ниже: СТАДИЯ А
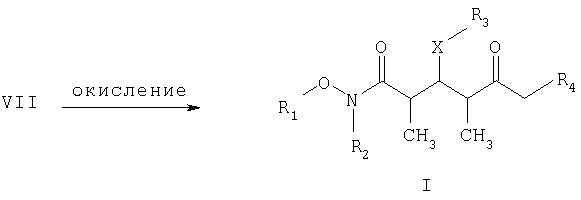
СТАДИЯ Б
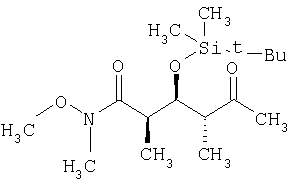
где R1, R2, R3, R4 и Х имеют такие же значения, как указанные выше. Что касается отдельных стадий, то стадия А включает присоединение метильной или этильной группы к альдегидному соединению формулы VI с получением спиртового соединения формулы VII. Это присоединение проводят в присутствии: 1) металлорганического реагента, предпочтительно литийалкил или магнийалкилгалогенида, такого, как магнийметилбромид; и 2) полярного органического растворителя, предпочтительно простого эфира, такого, как диэтиловый эфир, при температуре в пределах -78 и 40°С, предпочтительно -78 и 0°С, более предпочтительно при примерно -40°С, в течение периода в пределах от 5 мин до 24 ч, предпочтительно в пределах 30 мин и 2 ч, более предпочтительно в течение примерно 1 ч. Стадия Б включает окисление спиртового соединения формулы VII с получением целевого кетонового соединения формулы I. Окисление проводят в присутствии: 1) окислителя, предпочтительно сочетания диметилсульфоксида и активирующего агента, более предпочтительно сочетания диметилсульфоксида и комплекса триоксида серы с пиридином; 2) основания, предпочтительно органического основания, более предпочтительно триалкиламина, такого, как триэтиламин; и 3) полярного органического растворителя, предпочтительно хлорированного углеводорода, такого, как дихлорметан. Окисление целесообразно проводить при температуре в пределах -78 и 40°С, предпочтительно в пределах 5 и 20°С, в течение периода в пределах 5 мин и 24 ч, предпочтительно в пределах 1 и 12 ч, более предпочтительно в пределах 4 и 6 ч. Альдегидные соединения формул II и VI либо известны, либо могут быть получены аналогично способам, которые приведены в литературе для других структурно идентичных альдегидов. Тем не менее по еще одному варианту объектом настоящего изобретения являются новые Хотя продукт каждой реакции, описанной выше, можно, если необходимо, очищать по обычным методам, таким, как хроматография или перекристаллизация (если твердое вещество), сырой продукт одной реакции целесообразно без очистки использовать в последующей реакции. Как очевидно для специалистов в данной области техники, соединения формул I и с III по V содержат асимметрические углеродные атомы и, таким образом, необходимо иметь в виду, что отдельные стереоизомеры следует рассматривать как охватываемые объемом настоящего изобретения. Следующие примеры приведены только в иллюстративных целях, поэтому их нельзя рассматривать как ограничивающие каким-либо образом объем настоящего изобретения. Пример 1 (2R,3S,4R)-3-[[(1,1-диметилэтил)диметилсилил]окси]-N-метокси-N,2,4-триметил-5-оксогексанамид
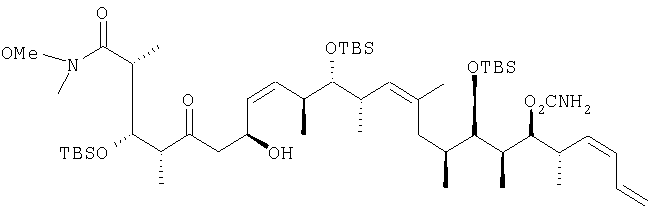
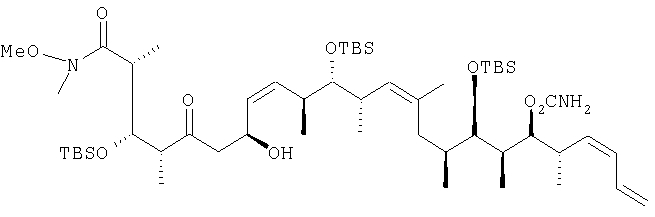
В раствор 13,1 г (39,5 ммоль) диастереоизомерной смеси, полученной на стадии 1.1, в смеси, включающей 150 мл метиленхлорида, 50 мл диметилсульфоксида и 25 мл триэтиламина, при 0°С с помощью капельной воронки по каплям добавляют 19,0 г (120 ммоль) комплекса пиридина стриоксидом серы в 150 мл диметилсульфоксида. Затем полученный раствор перемешивают в течение 1,5 ч при 0°С и по прошествии этого времени реакционную смесь концентрируют с помощью роторного испарителя на охлаждающей бане при <10°С. Затем раствор разбавляют 200 мл диэтилового эфира, после чего последовательно экстрагируют 200 мл 1 М раствора бисульфата натрия и 200 мл рассола. Далее органический слой сушат над сульфатом натрия и полученную сырую смесь очищают экспресс-хроматографией, используя при этом в качестве элюента первоначально гексан, а затем элюентную смесь из 5% этилацетата в гексане, с получением целевого соединения в виде прозрачного масла. 1H-ЯМР (300 МГц, CDCl3) Стадия 1.1: (2R,3S,4S)-3-[[(1,1-диметилэтил)диметилсилил]окси]-5-гидрокси-N-метокси-N,2,4-триметилгексанамид В раствор 6,9 г (21,8 ммоль) (2R,3S,4R)-3-[[(1,1-диметилэтил)диметилсилил]окси]-N-метокси-N,2,4-триметил-5-оксопентанамида в 50 мл диэтилового эфира по каплям при -40°С добавляют 10,2 мл (30,5 ммоль) 3 М раствора метилмагнийбромида в диэтиловом эфире, после чего смесь перемешивают при -20°С в течение 1 ч. Затем реакционную смесь разбавляют 200 мл диэтилового эфира и реакцию гасят добавлением реакционной смеси к 20 г измельченного льда при 0°С. Затем смесь промывают 100 мл 1 М раствора бисульфата натрия и разделяют между двумя 150-миллилитровыми порциями диэтилового эфира. После этого органические слои объединяют и сушат над сульфатом натрия. Далее полученную сырую смесь хроматографируют, используя при этом первоначально элюентную смесь из 20% этилацетата в гексане, а затем элюентную смесь из 40% этилацетата в гексане с получением двух диастереоизомеров в виде светло-желтых масел, которые без дополнительной очистки применяют на следующей стадии. Диастереоизомер 1: 1H-ЯМР (300 МГц, CDCl3) d: 4,08 (dd, J=9,8, 6,8 Гц, 2H), 3,64 (s, 3H), 3,54 (d, J=2,6 Гц, 2H), 3,08 (s, 3H), 3,00 (m, 2H), 1,48 (m, 2H), 1,07 (d, J=6,8 Гц, 3H), 1,05 (d, J=6,4 Гц, 3H), 0,81 (s, 9H), 0,76 (d, J=7,2 Гц, 2H), 0,01 (d, J=3,8 Гц, 6H). Диастереоизомер 2: 1Н-ЯМР (300 МГц, CDCl3) d: 4,19 (dd, J=12,1, 6,0 Гц, 2Н), 3,86 (dd, J=9,0, 1,51 Гц, 2Н), 3,56 (s, 3Н), 3,18 (m, 2H), 3,01 (s, 3H), 1,28 (m, 2H), 1,06 (d, J=6,8 Гц, 3Н), 0,94 (d, 6,4 Гц, 3Н), 0,88 (d, 7,2 Гц, 3Н), 0,78 (s, 9H), 0,01 (d, 1,3 Гц, 6Н). Пример 2 (2R,3S,4R,7S,8Z,10S,11S,12S,13Z,16S,17R,18R,19S,20S,21-E)-19-[(аминокарбонил)оксил]-3,11,17-трис[[(1,1-диметилэтил)диметилсилил]окси]-7-гидрокси-N-метокси-N,2,4,10,12,14,16,18,20-нонаметил-5-оксо-8,13,21,23-тетракозатетраенамид
В перемешиваемый раствор 2,18 г (6,79 ммоль, 10 экв.) (+)- [ 13С ЯМР (100, 6 МГц, CDCl3) d: 212,9, 175,9, 156,9, 136,0, 133,7, 132,1, 131,9, 131,3, 129,8, 129,6, 117,9, 80,6, 78,7, 76,8, 73,6, 64,9, 62,1, 61,3, 54,7, 53,1, 51,7, 49,0, 45,1, 44,9, 37,9, 37,1, 36,2, 35,9, 35,0, 34,4, 30,0, 29,1, 26,26, 26,24, 25,97, 23,0, 18,51, 18,5, 18,43, 18,14, 17,43, 16,44, 13,5, 10,99, 10,1, -3,29, -3,4, 3,5, -3,9, -4,1, -4,4; m/z (ESI+) 1017 (100 (MNa+). Пример 3 (2R,3S,4S,5S,7S,8Z,10S,11S,12S,13Z,16S,17R,18R,19S,20S,21E)-19-[(аминокарбонил)оксил]-3,11,17-трис[[(1,1-диметилэтил)диметилсилил]окси]-5,7-дигидрокси-N-метокси-N,2,4,10,12,14,16,18,20-нонаметил-8,13,21,23-тетракозатетраенамид
В раствор 1,02 г (3,9 ммоль) тетраметиламмонийтриацетоксиборгидрида в 2,2 мл безводного ацетонитрила добавляют 2,2 мл безводной уксусной кислоты и смесь перемешивают при комнатной температуре в течение 30 мин. Затем смесь охлаждают до -29°С и в холодную смесь добавляют раствор 450 мг (0,453 ммоль) соединения примера 2 в 1 мл безводного ацетонитрила. Далее образовавшуюся смесь перемешивают при -29°С в течение 18 ч и по прошествии этого времени реакцию гасят 2 мл 0,5н. водного натрийкалийтартрата. Затем смеси дают медленно нагреться до комнатной температуры, после чего ее разбавляют метиленхлоридом и промывают насыщенным раствором бикарбоната натрия. Затем водный слой четыре раза экстрагируют метиленхлоридом. После этого объединенные органические слои промывают рассолом, сушат сульфатом натрия и концентрируют под вакуумом. После этого полученный остаток очищают экспресс-хроматографией (продукт Biotage, силикагель, этилацетат/гексан с градиентом от 10 до 30%) с получением целевого соединения в виде белого твердого вещества. [ 13С-ЯМР (100, 6 МГц, CDCl3) d: 156,88, 140,02, 134,19, 133,66, 132,10, 131,88, 131,40, 131,30, 131,11, 130,06, 129,79, 117,91, 115,44, 80,79, 80,69, 78,61, 78,32, 74,31, 70,68, 65,55, 61,66, 45,69, 40,38, 38,36, 37,92, 37,83, 37,29, 36,29, 35,07, 34,91, 34,45, 32,36, 29,68, 26,21, 26,12, 26,03, 25,95, 22,95, 18,52, 18,43, 18,12, 17,41, 17,07, 16,57, 13,44, 12,29, 10,32, 10,14, -3,20, -3,43, -3,96, -4,16, -4,48. m/z (ESI+) 1019 (100 (MNa+). Пример 4 Получение (+)-дискодермолида В раствор 450 мг (0,452 ммоль) соединения примера 3 в 56 мл тетрагидрофурана добавляют 56 мл 4н. водного раствора соляной кислоты. Затем полученный раствор перемешивают при комнатной температуре в течение 24 ч, далее добавляют 10 мл метанола, после чего этот раствор перемешивают в течение дополнительных 24 ч при комнатной температуре. Далее в этот раствор добавляют 50 мл этилацетата с последующим добавлением бикарбоната натрия при 0°С до значения рН 8. Затем органический раствор промывают рассолом. После этого водный слой три раза экстрагируют 30-миллилитровыми порциями этилацетата и объединенные экстракты сушат над сульфатом натрия. В результате фильтрования и концентрирования и последующей экспресс-хроматографии с использованием при этом первоначально элюентной смеси из 50% метиленхлорида в этилацетате, после чего 100% этилацетата получают (+)-дискодермолид. [
Формула изобретения
1. Способ получения соединения формулы V
который включает на первой стадии реакцию сочетания кетонового соединения формулы I
с альдегидным соединением формулы II
в присутствии диалкилборгалогенида или трифлата, аминового основания и полярного органического растворителя с получением
на второй стадии, восстановление кетонового соединения, полученного на первой стадии, его обработкой боргидридным реагентом в полярном органическом растворителе и протонном растворителе с получением 1,3-диола формулы IV
и на третьей стадии лактонизацию и удаление кислотно-лабильной гидроксильной защитной группы 1,3-диола, полученного на второй стадии, его обработкой галогенидом водорода, растворенным в полярном растворителе или смеси растворителей, с получением целевого соединения формулы V, где R1 обозначает С1-С6алкил, R2 обозначает С1-С6алкил; R3 обозначает водородный атом или кислотно-лабильную гидроксильную защитную группу; R3” обозначает кислотно-лабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; а Х обозначает О, при условии, что когда Х обозначает О и R3 обозначает кислотно-лабильную гидроксильную защитную группу соединения формулы I, остаток -Х-R3 соединения формулы V представляет собой -ОН. 2. Способ по п.1, в котором стадию реакции сочетания осуществляют в присутствии 3. Способ по п.1, в котором стадию восстановления осуществляют в присутствии тетраметиламмонийтриацетоксиборгидрида, ацетонитрила и уксусной кислоты при температуре в пределах (-40) и (-10)°С. 4. Способ по п.1, в котором стадию лактонизации и удаления кислотно-лабильных гидроксильных защитных групп осуществляют в присутствии водного хлорида водорода, метанола и тетрагидрофурана при температуре в пределах -20 и 40°С. 5. Соединение формулы I
где R1 обозначает С1-С6алкил, R2 обозначает С1-С6алкил; R3 обозначает водородный атом или кислотно-лабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; и Х обозначает О. 6. Соединение по п.5 формулы Ia
где каждый из R1‘ и R2‘ обозначает С1-С6алкил; X’ обозначает О; а R3 и R4 имеют такие же значения, как указано в п.5. 7. Соединение по п.6 формулы Ib
где R3‘ обозначает кислотно-лабильную гидроксильную защитную группу; и R1‘ и R2‘ имеют такие же значения, как указанные в п.6. 8. Соединение по п.7 формулы Ic
где R3” обозначает кислотно-лабильную гидроксильную защитную группу. 9. Соединение по п.8, которое представляет собой (2R,3S,4R)-3-[[(1,1-диметилэтил)диметилсилил]окси]-N-метокси-N,2,4-триметил-5-оксогексанамид, отвечающий формуле
10. Способ получения соединения формулы I по п.5, который включает на первой стадии присоединение метил- или этилсодержащего металлорганического реагента к альдегиду формулы VI
в присутствии полярного органического растворителя с получением спирта формулы VII
и на второй стадии окисление спиртового соединения, полученного на первой стадии, его обработкой окислителем и основанием в полярном органическом растворителе с получением целевого соединения формулы I, в которой R1, R2, R3, R4 и Х имеют такие же значения, как указанные в п.5. 11. Способ по п.10, в котором стадию присоединения осуществляют в присутствии метилмагнийбромида и диэтилового эфира при температуре в пределах -78 и 40°С. 12. Способ по п.10, в котором стадию окисления осуществляют в присутствии комплекса триоксида серы с пиридином, триэтиламина и дихлорметана при температуре в пределах -78 и 40°С. 13. Соединение формулы III
где R1 обозначает С1-С6алкил, R2 обозначает С1-С6алкил; R3 обозначает водородный атом или кислотно-лабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; Х обозначает О, и каждый R3” обозначает кислотно-лабильную гидроксильную защитную группу. 14. Соединение по п.13, которое представляет собой (2R,3S,4R,7S,8Z,10S,11S,12S,13Z,16S,17R,18R,19S,20S,21Е)-19-[(аминокарбонил)окси]-3,11,17-трис[[(1,1-диметилэтил)диметилсилил]окси]-7-гидрокси-N-метокси-N,2,4,10,12,14,16,18,20-нонаметил-5-оксо-8,13,21,23-тетракозатетраенамид, отвечающий формуле
15. Соединение формулы IV
где R1 обозначает С1-С6алкил, R2 обозначает С1-С6алкил; R3 обозначает водородный атом или кислотно-лабильную гидроксильную защитную группу; R4 обозначает водородный атом или метил; X обозначает О, и каждый R3” обозначает кислотно-лабильную гидроксильную защитную группу. 16. Соединение по п.15, которое представляет собой (2R,3S,4S,5S,7S,8Z,10S,11S,12S,13Z,16S,17R,18R,19S,20S,21E)-19-[(аминокарбонил)окси]-3,11,17-трис[[(1,1-диметилэтил)диметилсилил]окси]-5,7-дигидрокси-N-метокси-N,2,4,10,12,14,16,18,20-нонаметил-8,13,21,23-тетракозатетраенамид, отвечающий формуле
|
||||||||||||||||||||||||||
 -гидроксикетона формулы III
-гидроксикетона формулы III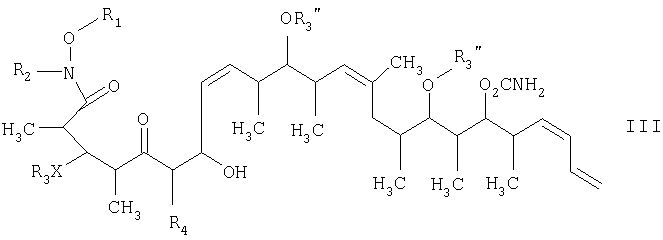
 – и
– и 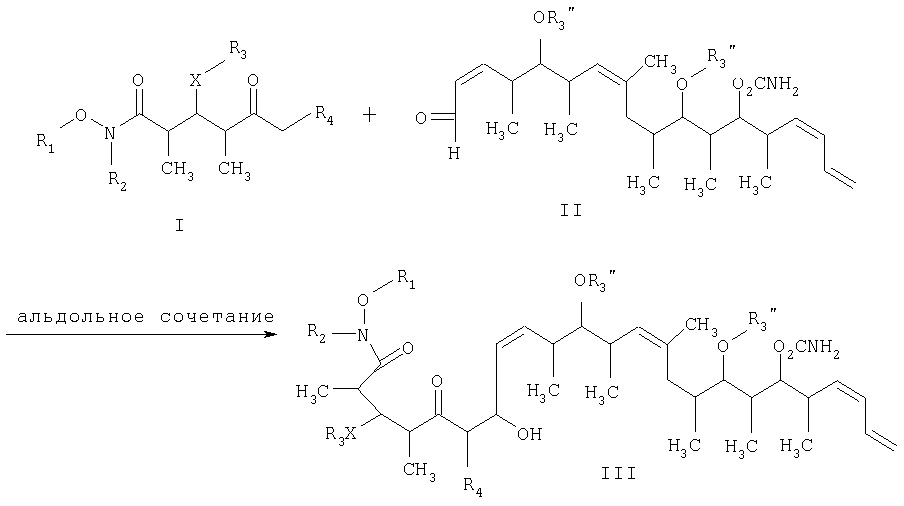
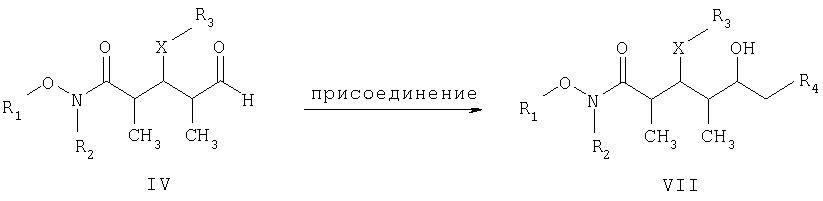
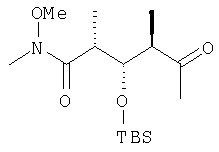
 : 4,23 (dd, J=7,5, 4,2 Гц, 2Н), 3,61 (s, 3H), 3,01 (s, 3H), 2,92 (m, 2H), 2,64 (m, 2Н), 2,08 (s, 3H), 1,03 (d, J=6,8 Гц, 3H), 0,98 (d, J=7,2 Гц, 3H), 0,81 (s, 9H), 0,00 (s, 6H).
: 4,23 (dd, J=7,5, 4,2 Гц, 2Н), 3,61 (s, 3H), 3,01 (s, 3H), 2,92 (m, 2H), 2,64 (m, 2Н), 2,08 (s, 3H), 1,03 (d, J=6,8 Гц, 3H), 0,98 (d, J=7,2 Гц, 3H), 0,81 (s, 9H), 0,00 (s, 6H).