Патент на изобретение №2281942
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СИНТЕЗ 4АЛЬФА-АРИЛЭПИКАТЕХИНОВ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
(57) Реферат:
Изобретение относится к новому способу получения 4-
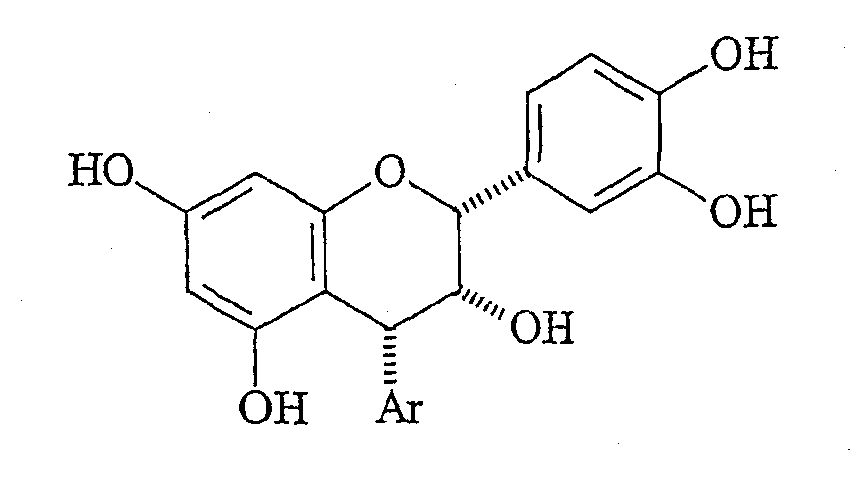
ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ По данной заявке испрашивается приоритет по заявке США, серийный № 09/655,360, поданной 5 сентября, 2000. ОБЛАСТЬ ИЗОБРЕТЕНИЯ Настоящее изобретение, в основном, относится к полифенольным продуктам, в частности к проантоцианидинам. Кроме того, изобретение относится к синтетических способам получения полифенольных природных продуктов и других родственных соединений. ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ Проантоцианидины (не гидролизуемые таннины) составляют группу полифенольных природных продуктов, представляющих в настоящее время интерес по причине их значимости для здоровья человека, связанной с многочисленными биологическими воздействиями и широкой распространенностью в пищевых продуктах. Проантоцианидины являются димерными или олигомерными флаваноидами, содержащими одну или несколько гидроксильных групп на ароматических циклах и, часто, дополнительную гидроксильную группу в положении 3. В природе обнаружено одиннадцать различных гидроксилированных структур в отношении циклов А и В. Характерные представители проантоцианидинов включают:
Стереохимия заместителей на полифенольном мономерном звене проантоцианидина может быть описана в терминах их относительной стереохимии, “альфа/бета” или “цис/транс”. Термин “альфа” ( Выделение чистых проантоцианидинов из природных источников становится все более затрудненным с ростом степени олигомеризации. Разложение путем тиолиза позволяет идентификацию основных мономерных звеньев, но задача выявления положения и стереохимии межфлавановых связей является нетривиальной. Оба этих фактора приводят к нескольким определенным олигомерам выше тетрамера, описание которого известно из уровня техники. Проантоцианидины и их исходные мономеры распространены в природе в форме ряда производных, например, гликозидов или сложных эфиров с гидроксилированными ароматическими карбоновыми кислотами, такими как галловая или гексагидроксидифеновая кислота. Из числа проантоцианидинов два подкласса, процианидины (5,7,3′,4′-гидроксилирование) и продельфинидины (5,7,3′,4′,5′-гидроксилирование), широко распространены в продуктах питания человека, например в какао. Процианидины какао состоят преимущественно из основных блоков эпикатехина (C-3 эпимера катехина). Идентифицированы олигомеры, достигающие размера декамера. Начиная с пентамера, эти олигомеры обладают активностью ингибиторов роста в отношении различных раковых клеточных линий. (Romanczk, L. J. Jr.; Hammerstone, J.F., Jr.; Buck, M. M. Патент США 5.554.645, сент. 10, 1996.) Флаван-3-олы получают путем биосинтеза из (2S)-фенилаланина через флаван-3,4-диолы. Эти последние промежуточные продукты легко образуют высоко стабилизированный ион карбения (или хинонметид) в положении C-4, который атакует A цикл флаван-3-ола, что по существу является процессом алкилирования по Фриделю-Крафтсу, образуя межфлавановую связь. Этот процесс может быть повторен еще один или несколько раз, что приводит к олигомерам цепочечного типа, которые вместе с димерами известны как негидролизуемые таннины, конденсированные таннины или проантоцианидины. Как понятно любому специалисту в данной области, структурная сложность указанных соединений быстро возрастает с длиной цепи как следствие различных гдроксильных замещений и C-3 стереохимии мономерного звена, и различных регио- и стереохимий межфлавановых связей, а также дополнительных структурных модификаций. Кроме того, может происходить разветвление цепей за счет алкилирования мономерного звена по 6- и 8-положениям. Для окончательной проверки структур, приписанных соединениям, выделенным из какао, следует произвести сравнения с синтетически полученными димерами и олигомерами эпикатехина указанных структур. Синтетические мономеры, димеры и олигомеры полезны для обнаружения взаимосвязей структура-активность на различных моделях противораковой активности in vitro и, в конечном счете, in vivo. Синтетическая задача, возникающая в связи с цианидинами, связана со сложностями в регулировании межфлавановой регио- и стереохимии, а также с чувствительностью незащищенных соединений к кислотам, основаниям и окисляющим агентам. Конденсацию между флаван-3-олами и 4-замещенными, электрофильными флаванами традиционно выполняют без использования фенольных защитных групп в слабокислой среде, или, в последнее время, с AgBF4 в качестве 4-заместителя для бензилтио. Продукты являются смесями регио- и, иногда, стереоизомеров, а также высших олигомеров, несмотря на применение избытка основного нуклеофильного блока. Указанные продукты обычно разделяют гель-хроматографией на Sephadex LH-20 способом, требующим значительной затраты времени на выполнение каждой отдельной задачи по разделению ввиду неприемлемости для данного адсорбента, позволяющего быстрый анализ оборудования, такого как ВЭЖХ колонки или пластины для тонкослойной хроматографии. Вдобавок, оптически чистые, незащищенные 4-замещенные катехины и эпикатехины труднодоступны, их получают восстановлением дорогостоящего природного продукта, (+)-таксифолина (4-кетона) или, на месте, расщеплением или тиолитическим расщеплением природных проантоцианидиновых олигомерных фракций, коммерческие источники которых трудноидентифицируемы, либо не существуют. Поэтому неудивительно, что в синтезах, известных из уровня техники, используют в качестве основных блоков защищенные олигомерные процианидины. В качестве дополнительного стимула, защита фенольных, но не спиртовых гидроксилов, дает возможность региоселективного получения производных, таких как 3-сложные эфиры и -гликозиды, как было сделано в случае катехина при использовании ацетильных защитных групп. Описан интересный подход, согласно которому 8-бромпроизводное 3-О-бензил-5,7,3′,4′-тетра-О-метилкатехина подвергают галоген-литиевому обмену и взаимодействию с O-метилированным 4-кетоном, что, таким образом, создает уверенность в полноте региоконтроля. Однако метильные О-блокирующие группы не могут быть удалены с целью получения свободного димера. Последняя опубликованная работа выполнена с применением вышеуказанного способа электрофильного замещения с включением фенольных защитных групп на одном или обоих реагентах. Предыдущая работа авторов заявки касалась синтеза указанных олигомеров эпикатехина с применением TiCl4-опосредованного алкилирования 5,7,3′,4′-тетра-О-бензил-(-)-эпикатехина с помощью 5,7,3′,4′-тетра-О-бензил-4-(2-гидроксиэтокси)эпикатехина. Помимо высших олигомеров, чей выход быстро снижается с увеличением молекулярной массы, был получен один димерный продукт, имеющий бета-стереохимию межфлавановой связи (производное процианидина B2). До совсем недавнего времени аналитические методы, используемые для определения регио- и стереохимии межфлавановой связи в этих соединениях, были необоснованными из-за независимости конформации от структуры димерного проантоцианидина. Применению рентгеновской кристаллографии мешала низкая способность к кристаллизации проантоцианидинов и их производных. Установление стереохимии на основании 1H-ЯМР констант взаимодействия и кругового дихроизма игнорирует тот основной факт, что C кольца являются конформационно гибкими. С традиционной точки зрения положения о специфических конформациях гибких молекул, независимо от их происхождения (интуитивного или вычислительного), не могут считаться разумным подходом к выяснению структуры. Успешное развитие методик синтеза, имевшее место после выделения многочисленных проантоцианидинов из природных источников, дает теперь возможность получать строгое доказательство ранее предполагаемой 4 Для квалифицированного специалиста в данной области очевидна важность наличия достоверного образца противоположного стереоизомера ввиду отсутствия в настоящее время для сравнения используют эпикатехин-4 Вероятным является предположение, что в ходе образования 4 КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Способ по настоящему изобретению позволяет получать незащищенные производные эпикатехина, замещенные в положении 4 Ключевой стадией настоящего способа является превращение, при котором C-4-карбокатион, уже имеющий ароматическое звено на требуемом месте, подвергается атаке гидридным нуклеофилом с Настоящее изобретение касается способа получения 4 (A) получения соответственно защищенного эпикатехина путем защиты C-3 гидроксильной группы 5,7,3′,4′-тетра-О-бензилэпикатехина с помощью защитной группы и получения соединения формулы:
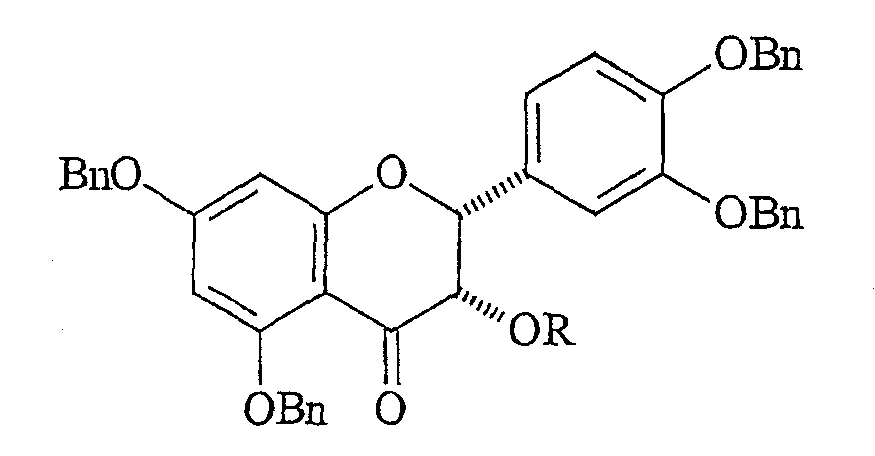
где Bn означает бензильную группу и R означает гидроксильную защитную группу. (B) окисления 4-положения защищенного эпикатехина и получения защищенного флаван-4-она формулы:
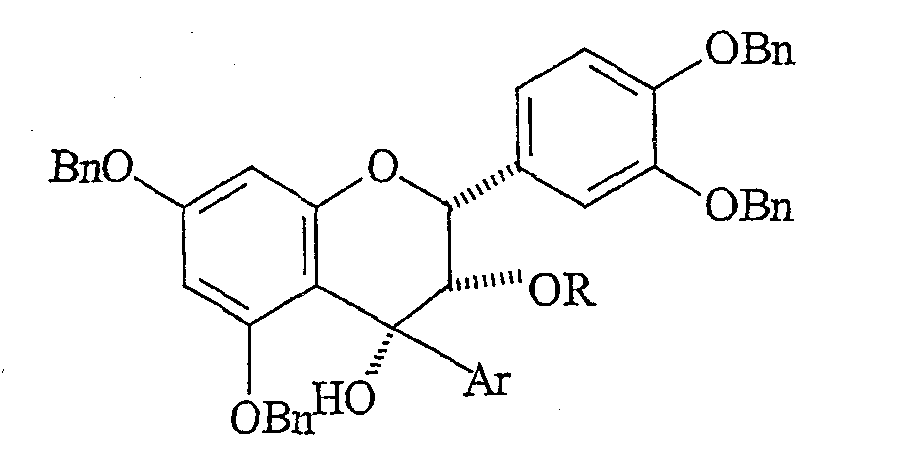
(C) контактирования флаван-4-она с нуклеофильным арильным металлоорганическим реагентом и получения соединения формулы:
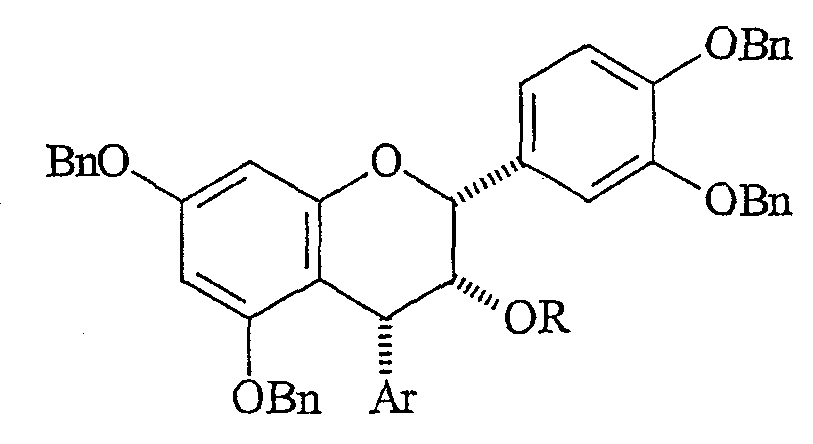
(D) стереоселективного деоксигенирования C-4-положения и получения соединения формулы:
(E) необязательного снятия защиты C-3 гидроксильной группы с помощью подходящего ацилирующего агента и последующего удаления бензильных групп; либо (A) необязательного снятия защиты C-3 гидроксильной группы и удаления бензильных групп и получения свободного 4
Также представлен способ получения других производных 4 По одному из аспектов настоящего изобретения представлен способ получения димеров эпикатехин-катехина, где нуклеофильный арильный металлоорганический реагент получают из защищенного 8-бромэпикатехина или защищенного 8-бромкатехина, либо их производного. ПРЕДПОЧТИТЕЛЬНЫЕ ВАРИАНТЫ ВЫПОЛНЕНИЯ ИЗОБРЕТЕНИЯ Как использовано здесь, термин эпикатехин (эпимер катехина) означает соединение формулы:
5,7,3′,4′-Тетра-О-бензилэпикатехин означает эпикатехин, где протон каждой фенольной гидроксильной группы замещен на бензильную группу. Для специалиста в области органического синтеза очевидно, что может существовать множество способов получения тетра-О-бензилэпикатехина. Один из особенно удобных способов получения такого соединения описан Tackmantel et al. J. Am. Chem. Soc. 1999, 121, 12073-12081. Как использовано здесь, “арил” означает ароматическое углеводородное соединение или гетероцикл. Арильная группа может означать фенил или замещенный фенил, где заместители выбирают из группы, включающей: галоген, арил, C1-C6-алкил, C1-C6-галогеналкил, C1-C6-алкокси, C3-C8-циклоалкил и C3-C8-циклоалкокси при условии, что они совместимы со структурой нуклеофильного металлоорганического реагента. Арильная группа может быть введена в циклическую систему других ароматических или насыщенных углеводородных циклов или гетероциклов. Важным изменением в настоящем способе является защита гидроксильной группы в C-3 положении 5,7,3′,4′-тетра-О-бензилэпикатехина. По одному из вариантов выполнения защита включает алкилирование бензилгалогенидом, таким как бензилбромид, что позволяет получать полностью защищенный эпикатехин. Обычно взаимодействие проводят в присутствии основания, предпочтительно сильного основания, такого как гидрид щелочного металла, диалкиламид, бис(триалкилсилил)амид или гидроксид, более предпочтительно в присутствии гидрида щелочного металла, такого как гидрид натрия. Взаимодействие обычно осуществляют в полярном органическом растворителе. Специалист в данной области способен выбрать растворитель, который совместим с конкретным основанием. Предпочтительным растворителем может быть ацетонитрил, тетрагидрофуран (ТГФ), N,N-диметилформамид, сульфоксид, такой как диметилсульфоксид, или N-метилпирролидинон. Более предпочтительным растворителем является амид, такой как N,N-диметилформамид. Характерная температура реакции находится в пределах приблизительно от 0°C до температуры кипения растворителя, предпочтительно от 15°C до 40°C, более желательно, составляет порядка 23°C. После добавления всех реагентов реакционную смесь обычно перемешивают в течение 15 минут – 24 часов, предпочтительно от 30 минут до 1 часа. По другому варианту выполнения защитная группа, используемая для защиты гидроксильной группы в C-3 положении 5,7,3′,4′-тетра-O-бензилэпикатехина, является ортогональной по отношению к бензильной группе. Обычно взаимодействие включает O-силилирование C-3 гидроксильной группы с получением простого силилового эфира. Силилирующим агентом может быть силилхлорид. Когда алкильными заместителями на кремнии являются третбутилдиметильные группы, силилирующим агентом служит трет-бутилдиметилсилилхлорид, и взаимодействие проводят в присутствии слабого основания, такого как имидазол или триэтиламин с 4-(диметиламино)пиридином. Взаимодействие осуществляют в инертном полярном органическом растворителе, предпочтительно, N,N-диметилформамиде, дихлорметане или ТГФ. Обычно реакция O-силилирования происходит при температуре приблизительно от 0°C до 40°C, предпочтительно, от 15°C до 30°C, более предпочтительно, около 23°C, за период приблизительно от 1 до 24 часов, предпочтительно, за период 6 – 12 часов, более предпочтительно, порядка 12 часов. Специалисту в данной области понятно, что подходят также другие способы силилирования гидроксильных групп. Силилирующим агентом может быть силилтрифторметансульфонат, в этом случае предпочтительным основанием является пиридин или 2,6-лутидин и растворителем служит дихлорметан или хлороформ. Специалисту в данной области известно, что C-3 гидроксил 5,7,3′,4′-тетра-О-бензил-8-бромэпикатехина или 5,7,3′,4′-тетра-О-бензил-8-бромкатехина может быть защищен подобным образом. Другой важной стадией настоящего способа является окисление защищенного эпикатехина. Окисление может быть выполнено с применением любого подходящего оксиданта. Обычно окисление проводят в две стадии. Первая стадия включает превращение C-4 метилена во вторичный спирт, что приводит к получению защищенного 4-гидроксиэпикатехина, предположительно имеющего 4 Второй стадией является дальнейшее окисление, включающее конверсию защищенного 4-гидроксиэпикатехина до защищенного флаван-4-она. Взаимодействие осуществляют в присутствии окисляющего агента, предпочтительно, окисляющего агента, полученного комбинированием N-метилморфолин-N-оксида и тетрапропиламмонийперрутената, и в присутствии нереакционно-способного осушителя, предпочтительно, молекулярных сит. Такой способ окисления описан в Ley et al., J. Chem. Soc., Chem. Commun. 1987, 1625. Обычно такое взаимодействие выполняют в органическом растворителе, предпочтительно, хлорированном алкане, таком как дихлорметан, при температуре от 0°C до температуры кипения растворителя, предпочтительно, при интервале температур от 15°C до 30°C, более предпочтительно, при температуре 23°C, и за период времени 1-24 часа, предпочтительно, за период 6-12 часов, более предпочтительно, за 12 часов. Дальнейшее превращение по настоящему способу включает взаимодействие защищенного флаван-4-она с нуклеофильным арильным металлоорганическим реагентом, приводящее к получению защищенного 4-арил-4-гидроксиэпикатехина. Обычно взаимодействие осуществляют, получая сначала нуклеофильный арильный металлоорганический реагент на месте, путем смешивания арилгалогенида, предпочтительно арилбромида, с алкиллитием, предпочтительно трет-бутиллитием, в безводном простом эфире в качестве растворителя, предпочтительно тетрагидрофуране, в инертной атмосфере при температуре приблизительно от -100°C до 0°C, в течение приблизительно 1-24 часов, и затем контактированием полученного нуклеофильного арильного металлоорганического реагента с защищенным флаван-4-оном, также в безводном простом эфире в качестве растворителя, предпочтительно тетрагидрофуране, в инертной атмосфере при температуре от -100°C до 0°C, в течение приблизительно 1-24 часов. Специалисту в данной области понятно, что по настоящему изобретению могут быть использованы другие металлоорганические реагенты, такие как реактивы Гриньяра. Для специалиста в данной области также очевидно, что некоторые арильные металлоорганические реагенты могут быть получены из материалов, отличных от галогенидов. В частности, нуклеофильные арильные металлоорганические реагенты могут быть получены способами, известными в данной области как ортометаллирование и переметалирование. Один из конкретных вариантов выполнения настоящего изобретения представляет взаимодействие 2,4,6-триметоксифениллития с защищенным флаван-4-оном. Другой отдельный вариант выполнения настоящего изобретения предполагает обработку защищенного 8-бромэпикатехина или защищенного 8-бромкатехина трет-бутиллитием для получения нуклеофильного металлоорганического реагента по изобретению. Типичным защищенным 8-бромэпикатехином является 5,7,3′,4′-тетра-O-бензил-8-бром-3-O-(трет-бутилдиметилсилил)эпикатехин, который обрабатывают алкиллитием, предпочтительно, трет-бутиллитием, в безводном простом эфире в качестве растворителя, предпочтительно тетрагидрофуране, в инертной атмосфере при температуре приблизительно от -100°C до 0°C, в течение приблизительно 1-24 часов, и затем осуществляют контактирование полученного ариллития с защищенным флаван-4-оном, также в безводном простом эфире в качестве растворителя, предпочтительно тетрагидрофуране, в инертной атмосфере при температуре от -100°C до 0°C, в течение приблизительно 1-24 часов. Способ по настоящему изобретению предусматривает деоксигенирование защищенного 4-арил-4-гидроксиэпикатехина по С-4, приводящее к защищенному 4 Далее изобретение предусматривает снятие защиты с С-3 гидроксильной группы 4-арил-5,7,3′,4′-тетра-O-бензилэпикатехина, или, когда 4-арильный заместитель сам по себе является катехином или эпикатехином, с обеих С-3 гидроксильных групп. При варианте выполнения, когда С-3 гидроксильная группа защищена силильной группой, реакция снятия защиты проводят с фторидом, обычно с водной фтористоводородной кислотой в ацетонитриле. Специалисту в данной области понятно, что после снятия защиты могут быть получены дальнейшие производные С-3 гидроксильной группы (групп). Обычно получение производных включает ацилирование С-3 гидроксильной группы ацилирующим агентом, таким как активированная кислота или хлорангидрид кислоты. Предпочтительным ацилирующим агентом является агент, полученный активацией три-O-бензилгалловой кислоты до хлорангидрида кислоты. Специалисту в данной области понятно, что кислоты могут быть активированы различными способами в целях получения подходящих ацилирующих агентов. Дополнительные примеры подходящих кислот включают в себя кофеиновую, кумариновую, синаповую, феруловую кислоты, а также такие гидроксизащищенные кислоты, как коричную, галловую и гидроксибензойную кислоты. Еще одна важная стадия способа включает удаление бензильных групп. Бензильные группы могут быть удалены с защищенных ароматических гидроксильных групп (т.е. фенолов) и защищенных алифатических гидроксильных групп путем гидрогенолиза. Обычно реакцию гидрогенолиза проводят в атмосфере газообразного водорода либо при стандартных температуре и давлении, либо, предпочтительно, при повышенном давлении порядка 1-5 бар, более предпочтительно, порядка 3,5-5 бар, в подходящем органическом растворителе, таком как метанол, этанол, этилацетат или их смесь. Обычно используют металлический катализатор, способствующий удалению бензильных групп. Предпочтительными катализаторами являются Pd, Pt или Ni, адсорбированные на твердом носителе, более предпочтительным катализатором является гидроксид палладия, абсорбированный на угле. Однако специалисту в данной области понятно, что множество других катализаторов может быть использовано для облегчения удаления бензильных групп. По одному из вариантов выполнения удаление бензильных групп дает свободный 4-арилэпикатехин, который может быть выделен и очищен. Альтернативно, сырой продукт гидрогенолиза может быть непосредственно превращен в его ацетатное производное путем взаимодействия сырого продукта с уксусным ангидридом и основанием. В более предпочтительном варианте выполнения, где 4-арильным заместителем является эпикатехин, гидрогенолиз дает свободный эпикатехин-4 По другому варианту выполнения бензильные группы удаляют после ацилирования С-3 гидроксильных групп, что приводит к получению свободного 4-арилэпикатехина, дериватизированного по С-3. Настоящее изобретение также предусматривает удаление бензильных групп, вводимых в качестве компонента ацилирующего агента. В предпочтительном варианте выполнения, где, по меньшей мере, одна С-3 гидроксильная группа ацилирована активированным производным три-О-бензилгалловой кислоты, бензильные группы на гидроксильных группах галлата могут быть удалены за одну стадию, вместе с удалением бензильных групп из ядра эпикатехина или катехина. Настоящее изобретение дополнительно раскрыто следующими примерами, не ограничивающими объем притязаний. ПРИМЕРЫ Общие методики. Катализатор Pearlman’а (20% гидроксид палладия на угле), поставляется Aldrich и содержит <50% воды. 1Н- и 13C- ЯМР спектры получают при номинальных частотах 300 и 75 МГц соответственно. 1Н-ЯМР спектры соотносят с внутренним стандартом ТМС, 13C-ЯМР спектры соотносят с ТМС, если это указано, в противном случае, с сигналом CDCl3 ( Пример 1: Получение 3,5,7,3′,4′-пента-O-бензилэпикатехина. К суспензии 180 мг (4,5 ммоль) гидрида натрия (60% в масле) в 10 мл сухого N,N-диметилформамида (Tackmantel et al. J. Am. Chem. Soc. 1999, 121, 12073-12081) добавляют при комнатной температуре раствор 2,60 г (4,00 ммоль) 5,7,3′,4′-тетра-O-бензилэпикатехина в 10 мл сухого N,N-диметилформамида. Спустя 1 час добавляют 0,56 мл (4,7 ммоль) бензилбромида. Смесь перемешивают в течение ночи, выливают в ледяную воду и экстрагируют 3 раза 50 мл дихлорметана. Объединенные органические фазы промывают водой и насыщенным раствором соли, сушат над сульфатом магния и упаривают. Остаток очищают хроматографией на колонке (дихлорметан/этилацетат/гексан 1:1:6), получая 2,20 г (74%) продукта в виде бесцветного, аморфного твердого вещества: [ Пример 2: Получение 3,5,7,3′,4′-пента-О-бензил-4-гидроксиэпикатехина. К раствору 2,20 г (3,38 ммоль) 3,5,7,3′,4′-пента-O-бензилэпикатехина в 20 мл тетрагидрофурана и 0,16 мл (8,9 ммоль) воды добавляют при комнатной температуре 2,00 г (7,4 ммоль) 2,3-дихлор-5,6-дициано-п-бензохинона. Смесь перемешивают в течение ночи, затем добавляют 0,91 г (7,4 ммоль) 4-(диметиламино)пиридина, перемешивание продолжают в течение 5 мин и добавляют 20 г силикагеля. После упаривания остаток фильтруют через силикагель (этилацетат/гексан 1:4, затем дихлорметан/этилацетат/гексан 1:1:4), получая 1,05 г (47%) продукта в виде белого пенистого вещества: [ Пример 3: Получение (2R,3S)-3,5,7,3′,4′-пентакис(бензилокси)флаван-4-она. К раствору 1,00 г (1,32 ммоль) 3,5,7,3′,4′-пента-О-бензил-4-гидроксиэпикатехина в 8 мл сухого дихлорметана добавляют при комнатной температуре 300 мг молекулярных сит 4Е, 180 мг (1,54 ммоль) N-метилморфолин-N-оксида и 58 мг (165 мкмоль) для тетрапропиламмонийперрутената. Реакционную смесь перемешивают в течение ночи и упаривают, остаток очищают хроматографией на колонке (этилацетат/дихлорметан/гексан 1:1:10), получая 0,66 г (66%) кетона в виде белого пенистого вещества: Пример 4: Получение 3,5,7,3′,4′-пента-О-бензил-4-гидрокси-4-(2,4,6-триметоксифенил)эпикатехина. К раствору 32 мг (130 мкмоль) 1-бром-2,4,6-триметоксибензола в 1 мл сухого тетрагидрофурана добавляют при -78°C 85 мкл (145 мкмоль) трет-бутиллития (1,7 M в пентане). Спустя 1 час при -78°C добавляют раствор 50 мг (66 мкмоль) (2R,3S)-3,5,7,3′,4′-пентакис(бензилокси)флаван-4-она в 1 мл сухого тетрагидрофурана. Спустя еще 3 часа при -78°C добавляют 2 мл водн. раствора хлорида аммония и продукт экстрагируют трижды 10 мл дихлорметана. Объединенные органические фазы сушат над сульфатом магния и упаривают, остаток очищают хроматографией на колонке (этилацетат/гексан 1:4), получая 25 мг (45%) продукта: [ Пример 5: Получение 3,5,7,3′,4′-пента-О-бензил-4 (a) Восстановление смесью триэтилсилан/трифторуксусная кислота: К раствору 22 мг (24 мкмоль) 3,5,7,3′,4′-пента-O-бензил-4-гидрокси-4-(2,4,6-триметоксифенил)эпикатехина в 1 мл дихлорметана добавляют при комнатной температуре 38 мкл (0,24 ммоль) триэтилсилана и затем 22 мкл (0,29 ммоль) трифторуксусной кислоты. Спустя 2 часа добавляют твердый карбонат натрия. Фильтрование, упаривание и очистка тонкослойной хроматографией (этилацетат/гексан 1:3) дают 15 мг (69%) продукта. (b) Восстановление смесью гидрид трибутилолова/трифторуксусная кислота: К раствору 46 мг (24 мкмоль) 3,5,7,3′,4′-пента-O-бензил-4-гидрокси-4-(2,4,6-триметоксифенил)эпикатехина в 1 мл дихлорметана добавляют при комнатной температуре 20 мкл (74 мкмоль) гидрида трибутилолова и затем 75 мкл 1 M раствора трифторуксусная кислота/дихлорметан. Спустя 10 мин добавляют твердый карбонат натрия. Фильтрование, упаривание и очистка тонкослойной хроматографией (этилацетат/гексан 1:2) дают 39 мг (86%) продукта: [ Пример 6: Получение 3,5,7,3′,4′-пента-О-ацетил-4 К раствору 100 мг (110 мкмоль) 3,5,7,3′,4′-пента-О-бензил-4 Пример 7: Получение 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)эпикатехина. Раствор 4,37 г (6,72 ммоль) 5,7,3′,4′-тетра-O-бензилэпикатехина (Tackmantel et al. J. Am. Chem. Soc. 1999, 121, 12073-12081), 0,69 г (10,1 ммоль, 1,5 экв.) имидазола и 1,42 г (9,4 ммоль, 1,4 экв.) трет-бутилдиметилсилилхлорида в 6 мл безводного N,N-диметилформамида перемешивают при комнатной температуре в закрытой колбе в течение 19,5 часов. Непосредственная хроматография на силикагеле с использованием смеси этилацетат/гексан 1:5, с последующим упариванием и сушкой в вакууме, дает 5,02 г (98%) простого силилового эфира в виде желтоватой стеклообразной массы: 1Н ЯМР Пример 8: Получение 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)-4-гидроксиэпикатехина. К раствору 1,52 г (1,98 ммоль) 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)эпикатехина в 10 мл тетрагидрофурана и 0,10 мл (5,6 ммоль) воды добавляют при комнатной температуре 1,34 г (5,9 ммоль) 2,3-дихлор-5,6-дициано-п-бензохинона. Смесь перемешивают в течение ночи, затем добавляют 0,61 г (5,0 ммоль) 4-(диметиламино)пиридина, перемешивание продолжают в течение 5 мин, и добавляют 20 г силикагеля. После упаривания остаток фильтруют через силикагель (этилацетат/гексан 1:4), получая 1,12 г (72%) продукта в виде белого пенистого вещества: [ Пример 9: Получение (2R,3S)-5,7,3′,4′-тетра-О-бензил-3-О-[(трет-бутилдиметилсилил)окси]флаван-4-она. К раствору 0,39 г (0,50 ммоль) 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)-4-гидроксиэпикатехина в 2 мл сухого дихлорметан добавляют при комнатной температуре 100 мг молекулярных сит 4Е, 60 мг (0,55 ммоль) N-метилморфолин-N-оксида и 20 мг (55 мкмоль) тетрапропиламмонийперрутената. Реакционную смесь перемешивают в течение ночи и упаривают, остаток очищают хроматографией на колонке (этилацетат/гексан 1:4), получая 0,38 г (99%) кетона в виде белого пенистого вещества: [a]D -32,5°, [a]546 -39,2° (c 12 гл-1, этилацетат); 1Н ЯМР (CDCl3) Пример 10: Получение 5,7,3′,4′-тетра-О-бензил-8-бром-3-О-(трет-бутилдиметилсилил)эпикатехина. Раствор 180 мг (247 мкмоль) 5,7,3′,4′-тетра-O-бензил-8-бромэпикатехина (Tackmantel et al. J. Am. Chem. Soc. 1999, 121, 12073-12081), 56 мг (0,37 ммоль) трет-бутилдиметилсилилхлорида и 49 мг (0,72 ммоль) имидазола в 1 мл безводного N,N-диметилформамида перемешивают при комнатной температуре в течение ночи. Смесь выливают в ледяную воду трижды экстрагируют 20 мл диэтилового эфира. Объединенные органические фазы трижды промывают 20 мл воды и 20 мл насыщенного раствора соли и сушат над сульфатом магния. Упаривание и хроматография на колонке (силикагель, дихлорметан/этилацетат/гексан 1:1:4) дают 187 мг (88%) продукта в виде пенистого вещества: 1Н ЯМР (CDCl3) Пример 11: Получение 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)-4-гидроксиэпикатехин-4,8-[5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)эпикатехин]а. К раствору 450 мг (533 мкмоль) 5,7,3′,4′-тетра-O-бензил-8-бром-3-O-(трет-бутилдиметилсилил)эпикатехина в 2 мл сухого тетрагидрофурана добавляют в атмосфере азота при -78°C 0,64 мл (1,1 ммоль) трет-бутиллития (1,7 M в пентане). После перемешивания при -78°C в течение 60 мин добавляют раствор 280 мг (359 мкмоль) (2R,3S)-5,7,3′,4′-тетра-О-бензил-3-О-[(трет-бутилдиметилсилил)окси]флаван-4-она в 2 мл сухого тетрагидрофурана. Спустя еще 3 часа при -78°C добавляют 2 мл водного хлорида аммония, смеси дают нагреться до комнатной температуры и трижды экстрагируют 20 мл дихлорметана. Объединенные органические фазы сушат над сульфатом магния и упаривают, остаток хроматографируют на силикагеле (дихлорметан/этилацетат/гексан 1:1:10), получая 410 мг (74%) продукта в виде бесцветного пенистого вещества: [ Пример 12: Получение 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)эпикатехин-4 К раствору 240 мг (155 мкмоль) 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)-4-гидроксиэпикатехин-4,8-[5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)-эпикатехин]а в 1 мл сухого дихлорметана добавляют при 0°C 50 мкл (186 мкмоль) гидрида три-н-бутилолова с последующим добавлением 154 мкл трифторуксусной кислоты (1 M в дихлорметане). Спустя 1 час добавляют 1 г твердого карбоната натрия и раствор фильтруют и упаривают. Остаток хроматографируют на силикагеле (дихлорметан/этилацетат/гексан 1:1:10), получая 181 мг (76%) продукта в виде бесцветной пенистой массы: [ -5,42, -6,22, Анал. Рассчит. для C98H102O12Si2: C, 77,03; Н, 6,73, Найдено: C, 77,02; Н, 6,63. Пример 13: Получение 5,7,3′,4′-тетра-О-бензилэпикатехин-4 К раствору 130 мг (85 мкмоль) 5,7,3′,4′-тетра-О-бензил-3-О-(трет-бутилдиметилсилил)эпикатехин-4 Пример 14: Получение 5,7,3′,4′-тетра-О-бензилэпикатехин-4 К суспензии 68 мг (154 мкмоль) три-О-бензилгалловой кислоты и 1 мкл N,N-диметилформамида в 1 мл безводного дихлорметана добавляют 15 мкл (0,17 ммоль) оксалилхлорида. После перемешивания при комнатной температуре в течение 2 часов без доступа влаги полученный раствор упаривают и остаток сушат в вакууме. Раствор 40 мг (31 мкмоль) 5,7,3′,4′-тетра-О-бензилэпикатехин-4 Пример 15: Получение эпикатехин-4 Раствор 22 мг (13 мкмоль) 5,7,3′,4′-тетра-О-бензилэпикатехин-4 Пример 16: Получение 3,5,7,3′,4′-пента-О-бензил-8-бром-эпикатехина. К суспензии 29 мг (0,73 ммоль) гидрида натрия (60% в масле) в 2 мл безводного N,N-диметилформамида добавляют при комнатной температуре 450 мг (617 мкмоль) 5,7,3′,4′-тетра-О-бензилэпикатехина (Tackmantel et al. J. Am. Chem. Soc. 1999, 121, 12073-12081) в 3 мл безводного N,N-диметилформамида. После перемешивания в течение 30 мин добавляют 90 мкл (0,73 ммоль) бензилбромида и 20 мг (54 мкмоль) тетрабутиламмонийиодида. Смесь перемешивают в течение ночи, выливают в ледяную воду и трижды экстрагируют 50 мл этилацетата. Объединенные органические фазы трижды промывают 50 мл воды и 50 мл насыщенного раствора соли, сушат над сульфатом магния и упаривают. Хроматография на колонке (этилацетат/гексан 1:2) дает 480 мг (95%) продукта: 1Н ЯМР (CDCl3) Пример 17: Получение 3,5,7,3′,4′-пента-О-бензил-4-гидроксиэпикатехин-4,8-(пента-О-бензилэпикатехин)а. К раствору 200 мг (244 мкмоль) 3,5,7,3′,4′-пента-O-бензил-8-бромэпикатехина в 1 мл сухого тетрагидрофурана добавляют в атмосфере азота при -78°C 0,30 мл (0,51 ммоль) трет-бутиллития (1,7 M в пентане). После перемешивания при -78°C в течение 90 мин добавляют раствор 120 мг (159 мкмоль) (2R,3S)-3,5,7,3′,4′-пентакис(бензилокси)флаван-4-она в 1 мл сухого тетрагидрофурана. После еще 3 часов при -78°C добавляют 2 мл водного хлорида аммония, смеси дают нагреться до комнатной температуры и трижды экстрагируют 20 мл дихлорметана. Объединенные органические фазы сушат над сульфатом магния и упаривают, остаток очищают хроматографией на силикагеле (дихлорметан/этилацетат/гексан 1:1:8), получая 165 мг (69%) продукта в виде бесцветной пенистой массы: [ Пример 18: Получение пента-О-бензилэпикатехин-4 К раствору 70 мг (46,8 мкмоль) 3,5,7,3′,4′-пента-O-бензил-4-гидроксиэпикатехин-4,8-(пента-O-бензилэпикатехин)а в 1 мл сухого дихлорметана добавляют при 0°C 20 мкл (74 мкмоль) гидрида н-бутилолова с последующим добавлением 71 мкл трифторуксусной кислоты (1 M в дихлорметане). Спустя 1 час добавляют 1 г твердого карбоната натрия, раствор фильтруют и упаривают. Остаток хроматографируют на силикагеле (дихлорметан/этилацетат/гексан 1:1:8), получая 55 мг (79%) продукта в виде бесцветной пенистой массы: [ Пример 19: Получение эпикатехин-4 Раствор 40 мг (31 мкмоль) 5,7,3′,4′-тетра-O-бензилэпикатехин-4 Пример 20: Получение пента-О-ацетилэпикатехин-4 К раствору 75 мг (51 мкмоль) пента-О-бензилэпикатехин-4
Формула изобретения
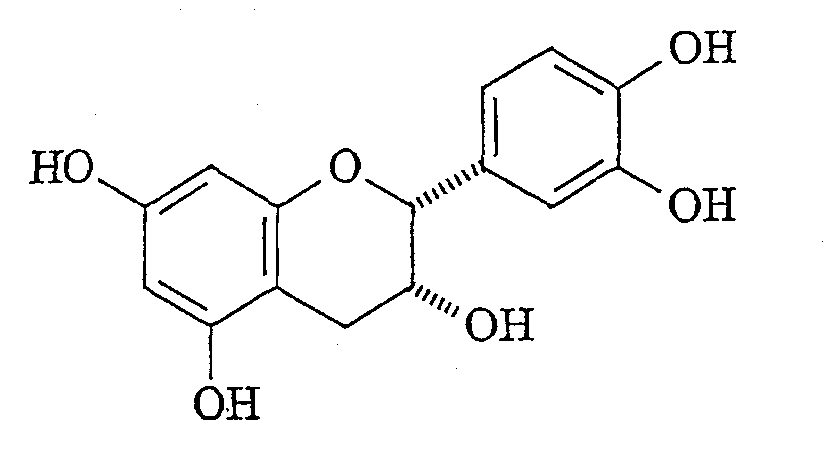
1. Способ получения димера эпикатехин-4 (a) защита бензильной защитной группой С-3 гидроксильной группы 5,7,3′,4′-тетра-O-бензилэпикатехина с получением 3,5,7,3′,4′-пента-O-бензилэпикатехина; (b) окисление 4-положения эпикатехина со стадии (а) с получением 3,5,7,3′,4′-пента-O-бензил-4-гидроксиэпикатехина; (c) окисление С-4 гидроксигруппы эпикатехина со стадии (b) с получением (2R,3S)-3,5,7,3′,4′-пента-O-бензил-4-кетонэпикатехина; (d) добавление трет-бутиллития к 3,5,7,3′,4′-пента-O-бензил-8-бромэпикатехину с получением реагента защищенного эпикатехинлития; (e) добавление эпикатехина со стадии (с) к реагенту эпикатехинлития со стадии (d) с получением димера 3,5,7,3′,4′-пента-O-бензил-4-гидроксиэпикатехин-4( (f) восстановление С-4 гидроксильной группы димера со стадии (е) с получением димера 3,5,7,3′,4′-пента-O-бензилэпикатехин-4 (g) замена бензильных групп димера со стадии (f) на водород с получением димера эпикатехин-4 2. Способ получения защищенного димера эпикатехин-4 (a) защита трет-бутилдиметилсилильной группой С-3 гидроксильной группы 5,7,3′,4′-тетра-O-бензилэпикатехина с получением 5,7,3 ‘,4’-тетра-O-бензил-3-O-(трет-бутилдиметилсилил)эпикатехина; (b) окисление С-4 положения эпикатехина со стадии (а) с полученим 5,7,3′,4′-тетра-O-бензил-3 – (7-(трет-бутилдиметилсилил)-4-гидроксиэпикатехина; (c) окисление С-4 гидроксильной группы эпикатехина со стадии (b) с получением 5,7,3 ‘,4’-тетра-O-бензил-3-O-трет-бутилдиметилсилил-4-кетонэпикатехина; (d) добавление трет-бутиллития к 5,7,3′,4′-тетра-O-бензил-8-бром-3-O-(трет-бутилдиметилсилил)эпикатехину с получением реагента эпикатехинлития; (e) добавление эпикатехина со стадии (с) к эпикатехинлитиевому реагенту со стадии (d) с получением димера 5,7,3 ‘,4’-тетра-O-бензил-3-O-(трет-бутилдиметилсилил)-4-гидроксиэпикатехин-4 (f) восстановление С-4 гидроксильной группы димера со стадии (е) с получением димера 5,7,3′,4′ -тетра-O-бензил-3-O-(трет-бутилдиметилсилил)эпикатехин-4 (g) замену трет-бутилдиметилсилильных групп димера со стадии (f) на водород с получением димера 5,7,3′,4′-тетра-O-бензилэпикатехин-4 (h) введение ацильной группы в 3-ОН положение нижнего мономерного звена димера со стадии (g) путем взаимодействия с активированной кислотой или хлорангидридом кислоты с получением димера 5,7,3′,4′ -тетра-O-бензилэпикатехин-4 (i) замену бензильных защитных групп димера со стадии (h) на водород с получением димера эпикатехин-4 3. Способ по п.1, где стадию замены (а) осуществляют с помощью бензилбромида в присутствии гидрида натрия, где стадию окисления (b) осуществляют с помощью 2,3-дихлор-5,6-дициано-п-бензохинона и 4-(диметиламино)пиридина, и где стадию окисления (с) осуществляют с помощью тетрапропиламмонийперрутената и N-метилморфолино-N-оксида, где стадию восстановления (f) осуществляют с помощью гидрида три-н-бутилолова и затем трифторуксусный кислоты; и где стадию замены бензила (g) осуществляют гидрированием в присутствии гидроксида палладия на угле. 4. Способ по п.2, где стадию защиты (а) осуществляют с помощью трет-бутилдиметилсилилхлорида, где стадию окисления (b) осуществляют с помощью 2,3-дихлор-5,6-дициано-п-бензохинона и 4-(диметиламино)пиридина, где стадию окисления (с) осуществляют с помощью N-метилморфолино-N-оксида и тетрапропиламмонийперрутената, где стадию восстановления (f) осуществляют гидридом три-н-бутилолова и трифторуксусной кислотой. 5. Способ по п.3, где стадию замены трет-бутилдиметилсилила (g) осуществляют с помощью фтористоводородной кислоты, и где стадию замены бензила (i) осуществляют гидрированием в присутствии гидроксида палладия на угле. 6. Способ по п.2, где кислота выбрана из группы, включающей: кофеиновую, кумаровую, феруловую и синаповую кислоту. 7. Способ по п.2, где кислота представляет собой гидрокси-защищенную кислоту, выбранную из группы, включающей: коричную кислоту, галловую кислоту и гидроксибензойную кислоту. 8. Соединение формулы
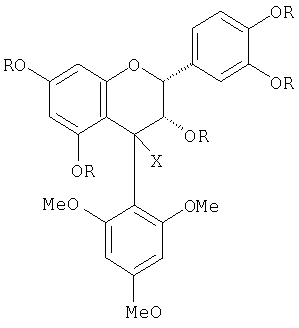
где R означает водород, бензил или ацетил, и Х означает гидрокси или 9. Соединение формулы
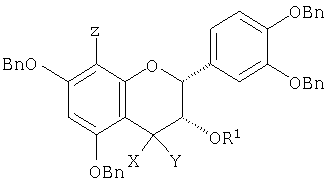
где R1 означает защитную группу силил или защитную группу бензил, и Х и Y независимо означают водород или гидрокси, или Х и Y вместе означают кислород, и Z означает галоген. 10. Соединение формулы
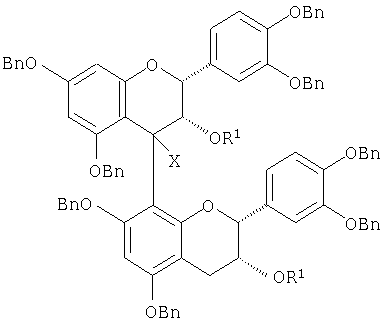
где R1 означает защитную группу на основе силила или защитную группу бензильного типа, и Х означают гидрокси. 11. Соединение, выбранное из (1) 5,7,3′,4′-тетра-O-бензил-3-O-(третбутилдиметилсилил)эпикатехин-4 (2) 3,5,7,3′,4′ -пента-O-бензилэпикатехин-4 (3) 5,7,3′,4′-тетра-O-бензилэпикатехин-4 (4)пента-O-ацетил-эпикатехин-4
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 -арилэпикатехинов, в частности к способу получения димера эпикатехин-4
-арилэпикатехинов, в частности к способу получения димера эпикатехин-4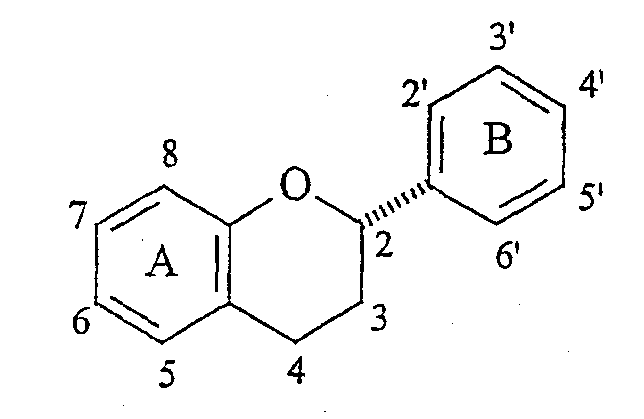
 ) означает, что заместитель расположен над плоскостью цикла. Термин “цис” означает, что два заместителя расположены по одну сторону цикла, тогда как “транс” означает, что два заместителя расположены по противоположным сторонам цикла.
) означает, что заместитель расположен над плоскостью цикла. Термин “цис” означает, что два заместителя расположены по одну сторону цикла, тогда как “транс” означает, что два заместителя расположены по противоположным сторонам цикла.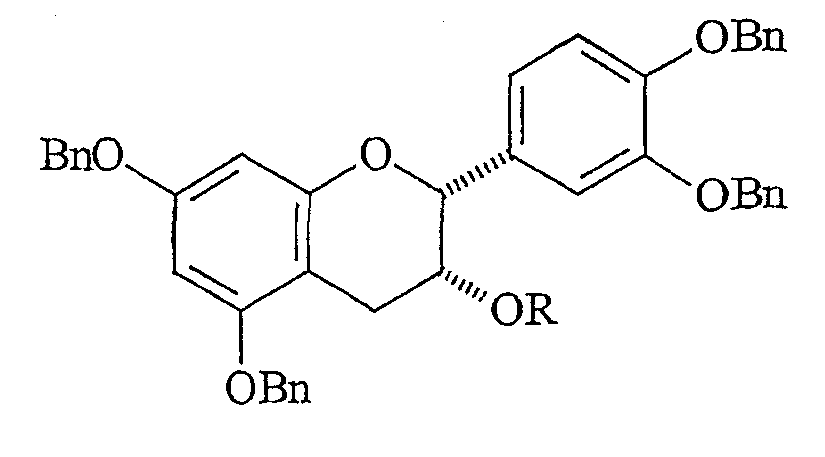
 77,00). Органический элементарный анализ: Micro-Analysis, Inc. (Wilmington, DE). Хроматография на колонке (cc): Merck силикагель 60 (No. 7734-7), размер частиц 63-200 мкм. Тонкослойная хроматография: Merck силикагель 60 F254 (No. 7734-7), толщина слоя 250 мкм; визуализация щелочным раствором перманганата калия.
77,00). Органический элементарный анализ: Micro-Analysis, Inc. (Wilmington, DE). Хроматография на колонке (cc): Merck силикагель 60 (No. 7734-7), размер частиц 63-200 мкм. Тонкослойная хроматография: Merck силикагель 60 F254 (No. 7734-7), толщина слоя 250 мкм; визуализация щелочным раствором перманганата калия.