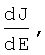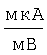Патент на изобретение №2276354
|
|||||||||||||||||||||||||||||||
(54) СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СТРЕПТОМИЦИНА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ
(57) Реферат:
Изобретение относится к области аналитической химии. Технический результат изобретения: повышение селективности, экспрессности, снижение минимально определяемой концентрации стрептомицина. Сущность: стрептомицин переводят из пробы в раствор и проводят вольтамперометрическое определение в перемешиваемом растворе в течение 90÷180 с при потенциалах электролиза – (0,10÷0,20) В на фоне (0,1÷0,2)н. раствора гидрофосфата натрия с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 15÷25 мВ/с, а концентрацию стрептомицина определяют по высоте пика в диапазоне потенциалов от 0,66 до 0,70 В относительно насыщенного хлоридсеребряного электрода методом добавок аттестованных смесей. 1 табл.
Изобретение относится к области аналитической химии, в частности к инверсионному вольтамперометрическому способу определения антибиотика (АБ) стрептомицина, который является производным трисахаридов и представляет 1,3-дигуанидино-2,4,5,6-тетрагидроксициклогексан-2-2-(N-метил-2 глюкозаминозидо)-4-стрептозид. Стрептомицин относится к антибактериальным веществам и обладает высокой клинической эффективностью в лечении ряда тяжелых инфекций. Его применяют в терапии туберкулеза, туляремии и чумы; в ветеринарии и животноводстве в качестве лечебно-профилактических средств. АБ способен переходить в молоко, мясо животных и оказывать токсическое действие на организм человека. Предельно допустимая концентрация (ПДК) стрептомицина нормируется гигиеническими требованиями безопасности и пищевой ценности пищевых продуктов. Санитарно-эпидемиологическими правилами и нормативами СанПиН 2.3.2.1078-01 и составляет 0,5 ед/г, что соответствует 0,5 мг/кг. Бесконтрольное применение антибиотиков и злоупотребление ими становится с каждым годом все более серьезной проблемой. Поэтому важной задачей современной аналитической химии является совершенствование методов определения антибиотиков, разработка экспрессных и высокочувствительных методик их контроля в сложных по составу объектах. Известны химические методы определения, основанные на цветных реакциях, характерных для специфических группировок, входящих в молекулу стрептомицина [Мелентьева Г.А. Фармацевтическая химия. – М.: Медицина, 1976. – 828 с.]. Среди них наибольшее распространение получили титриметрические методы анализа: цериметрическое титрование, перманганатометрический метод [Полюдек-Фабини Р., Бейрих Т. Органический анализ. – Л.: Химия, 1981 – 624 с.] определения антибиотика в лекарственных препаратах. Чувствительность таких методов невелика и составляет 10-4÷10-3 моль/л. -4÷10-3 моль/л) не позволяет использовать эти методы для определения стрептомицина на уровне микро- и наномолярных концентраций. Все большее применение в последнее время за рубежом находят хроматографические методы, особенно жидкостная (ЖХ) и высокожидкостная хроматографии (ВЖХГ). -6÷10-8 моль/л), высокая стоимость приборов, а также длительность анализа с учетом времени пробоподготовки ограничивает их массовое использование. Возможность полярографического определения стрептомицина и его солей описана в монографии [Мискиджьян С.П., Кравченюк Л.П. Полярография лекарственных препаратов. Киев: Вища школа, 1976. – 232 с.] и основана на восстановлении его альдегидной группы в щелочных средах или на фоне фосфатноцитратного буферного раствора с использованием ртутно-капельного электрода при потенциале полуволны Е1/2=1,4÷1,5 В относительно насыщенного каломельного электрода (нас.к.э.). Авторы обращают внимание на изменение полярографических кривых со временем, особенно в щелочных растворах. Метод пригоден для анализа антибиотика в чистых препаратах, однако он не позволяет проводить количественное определение на уровне микроконцентраций. Одним из самых чувствительных электрохимических методов и достаточно простым и дешевым в аппаратурном оформлении является метод вольтамперометрии (ВА) и его высокочувствительный вариант – инверсионная вольтамперометрия (ИВА). Для вольтамперометрического анализа часто допустимы более простые методики предварительного выделения соединений из сложной многокомпонентной смеси чем, например, хроматографические. Пробоподготовка может быть существенно упрощена из-за необязательной дополнительной очистки определяемого вещества перед собственно электрохимическим анализом и возможностью проведения вольтамперометрических измерений в мутных и окрашенных средах. Нижняя граница определяемых опытным путем содержаний CH=1·10-8 моль/л, относительное среднеквадратичное отклонение для концентрации 4·10-8 моль/л равно 5,1%. Показана возможность определения стрептомицина в моче на уровне 1·10-6 моль/л (и более) после разбавления мочи 0,01 М NaOH. Авторы отмечали, что для определения более низких концентраций лекарственного вещества в моче (<1·10-6 моль/л) или в других жидкостях организма, требуются разработки процедуры пробоподготовки вместо шага разбавления. В данных условиях определение стрептомицина в сложных природных и биологических смесях, пищевых продуктах, сточных водах, невозможно на уровне наномолярных концентраций. Поэтому разработка экспрессных, селективных и высокочувствительных методов определения стрептомицина в сложных смесях представляет интерес. Задачей заявляемого изобретения является снижение минимально определяемой опытным путем концентрации, повышение селективности и экспрессности определения стрептомицина методом анодной инверсионной вольтамперометрии. Поставленная задача достигается тем, что стрептомицин переводят из пробы в раствор и проводят электрохимическое концентрирование на стеклоуглеродном электроде (СУ) с последующей регистрацией вольтамперограмм. Новым в способе является то, что проводят накопление стрептомицина в перемешиваемом растворе в течение 90÷180 с при потенциалах электролиза ЕЭ=-(0,10÷0,20) В на фоне (0,1÷0,2)н. раствора гидрофосфата натрия с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала (15÷25) мВ/с и концентрацию стрептомицина определяют по высоте пика в диапазоне потенциалов от 0,66 до 0,70 В относительно насыщенного хлоридсеребряного электрода (нас.х.с.э.) методом добавок аттестованных смесей. В прототипе количественное определение стрептомицина основано на реакции восстановления антибиотика на стационарном ртутном капельном электроде. В предлагаемом способе впервые установлена способность стрептомицина окисляться на стеклоуглеродном электроде (СУ). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов как в водных, так и в неводных средах, а также простотой механического обновления поверхности и требованиями техники безопасности. Ртуть токсична и может вредно влиять на чувствительные биологические материалы. Реакция электроокисления стрептомицина на СУ является электрофильной. Роль электрофильного реагента с электронным дефицитом выполняет анод, а субстрата – органическая молекула. Способность к окислению существенно зависит от материала электрода и состояния его поверхности. Стрептомицин достаточно легко окисляется и на других типах графитовых электродов (на электродах, пропитанных полиэтиленом с парафином в вакууме или эпоксидной смолой), но они меньше удовлетворяли требованиям чисто аналитической практики из-за большого остаточного тока. Для определения антибиотика использовали «игольчатые» по форме индикаторные электроды (СУ) после предварительного электрохимического модифицирования их поверхности. Такая обработка приводит к улучшению воспроизводимости вольтамперометрических измерений. СУ электрод впервые использован для определения стрептомицина. В прототипе описано использование в качестве фона 0,01 М NaOH. При этом на вольтамперограмме регистрировали два катодных пика с близкими потенциалами -1,58 и -1,70 В, что существенно снижает селективность определения. Кроме того, пики регистрируют вблизи разряда фона, вследствие чего ухудшается воспроизводимость. Определение стрептомицина по анодным пикам на фоне 0,01 М NaOH невозможно, что, по-видимому, связано с формой нахождения вещества в растворе. В предлагаемом заявленном изобретении фоне (0,1÷0,2)н. гидрофосфата натрия (Na2HPO4) регистрируется при съемке вольтамперной кривой только один анодный пик при потенциале (0,66÷0,70) В относительно нас.х.с.э. в широком диапазоне определяемых концентраций от 7,0·10-9 до 1,0·10-7 моль/л с хорошей воспроизводимостью. Относительное стандартное отклонение Sr для диапазона концентраций (1÷4)·10-8 моль/л составляет 0,030÷0,025, т.е. <3,0% против 5,1% в прототипе для концентрации 4·10-8 моль/л. Возможность электрохимического поведения определяемого вещества очень сильно зависит от природы и концентрации фонового электролита. Фон (0,1÷0,2)н. Na2HPO4 подобран экспериментально и соответствует рН Абсолютной новизной является экспериментально подобранный фон Na2HPO4 и установленные значения его концентрации (0,1÷0,2)н. и значения рН (8,0÷8,6). Другим отличительным признаком являются установленные условия электрохимического накопления: потенциал электролиза ЕЭ=-(0,10÷0,20) В. Опытные данные показали зависимость тока пика окисления стрептомицина от ЕЭ (табл.). Величина тока увеличивалась и достигала максимального значения в области потенциалов -(0,10÷0,20) В. При -0,20>ЕЭ>-0,10 В уменьшалась величина тока окисления органического вещества. Использование предварительного электролиза при значениях потенциала -(0,10÷0,20) В позволяет регистрировать вольтамперограммы с четко выраженным максимумом, и это позволяет повысить точность, разрешающую способность способа и экспрессно определять концентрацию менее 1·10-7 моль/л стрептомицина в сложных по составу смесях. Время предварительного электролиза ( Важным для определения стрептомицина является выбор скорости развертки потенциала. Оптимальной является скорость 15÷25 мВ/с. Увеличение скорости более 25 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и может появляться на вольтамперной кривой дополнительный небольшой предпик при потенциале 0,5 В, что приводит к уменьшению разрешающей способности способа и затрудняет обработку аналитического сигнала. Использование скорости <15 мВ/с существенно снижает величину анодного тока и понижается чувствительность определения. При скорости 15÷25 мВ/с происходит практически полная десорбция вещества с поверхности электрода. Нижняя граница определяемых содержаний стрептомицина зависит от режима съемки вольтамперных кривых. В дифференциальном режиме высокая точность определяется тем, что потенциал пика измеряется в точке, в которой круто падающая первая производная пересекает нулевую линию. Использование режима дифференцирования позволяет фиксировать четкие пики и разрешает обнаружение стрептомицина даже на уровне наномолярных концентраций. Для определения стрептомицина он ранее не применялся. При линейном наложении потенциала в интегральном режиме определение концентрации стрептомицина на уровне n·10-8 моль/л практически не возможно из-за большого значения остаточного тока, особенно в многокомпонентных смесях. Поэтому для количественного определения стрептомицина рекомендован дифференциальный режим съемки вольтамперограмм. Таким образом, установленные условия впервые позволили количественно определять стрептомицин на основе реакции электроокисления с использованием нетоксичного стеклоуглеродного электрода (в прототипе использован стационарный ртутный капельный электрод). Для повышения чувствительности определения использовали предварительное адсорбционное электроконцентрирование на поверхности СУ электрода. Предлагаемый вольтамперометрический способ позволил повысить селективность определения и существенно улучшить обработку вольтамперограмм при анализе за счет регистрации одного анодного пика и установленных оптимальных условий проведения электродного процесса. Относительное стандартное отклонение не превышает 3% против 5,1% для концентрации 4·10-8 моль/л. Предел обнаружения, рассчитанный по 3 Определению не мешают вещества, присутствие которых возможно в биологических объектах: аскорбиновая и мочевая кислоты, витамины B1 В2, РР, Вс, ионы Zn2+, Cu2+, Pb2+, Ca2+. Пример 1. Определение стрептомицина на уровне 10-9÷10-8 моль/л В кварцевый стаканчик емкостью 20 мл наливают 10 мл 0,2н. раствора гидрофосфата натрия. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение пяти минут. Не прекращая перемешивания, проводят электролиз раствора при условии: ЕЭ=-0,20 В, Предлагаемый способ впервые применен нами для определения стрептомицина в молоке. Для освобождения антибиотика и устранения мешающего влияния белка использовали кислотный гидролиз и осаждение водорастворимого белка из гидролизата с последующим вольтамперометрическим определением стрептомицина в безбелковом гидролизате путем регистрации анодных пиков на СУ электроде в условиях предлагаемого нами способа (в прототипе молоко и молочные смеси не анализировались). Пример 2. Определение стрептомицина в молоке В коническую колбу вместимостью 100,0 см3 вносят 25,0 см3 молока и 1 мл (6-7 моль/л) HCl порциями по 0,2 см3. Смесь встряхивают в течение 15 минут. Затем к смеси добавляют 3-4 г (NH4)2SO4 порциями в 2-3 приема. Снова встряхивают содержимое колбы в течение 10 минут. Полученную смесь переносят в центрифужные пробирки и центрифугируют 15 минут при скорости 6000 об/мин. Центрифугат отфильтровывают через двойной бумажный фильтр в коническую колбу вместимостью 100,0 см3. Доводят рН фильтрата до 8 раствором (4-5)н. NaOH. Полученный раствор является подготовленной пробой для анализа. Аликвоту пробы объемом 1-3 см3 помещают в электролизер, добавляют фоновый электролит (0,1÷0,2)н. раствор Na2HPO4 до 10 см3 и затем проводят вольтамперометрические измерения при условиях: потенциал электролизера ЕЭ=-0,10В, время электролиза Предложенный способ прост, экспрессен, селективен, высокочувствительный (позволяет определять стрептомицин в молоке на уровне ПДК или 0,5 мг/кг), может быть применен не только для контроля качества сырья пищевых продуктов (молока, молочных смесей, творога, мяса), но и для анализа антибиотика в биологических жидкостях (крови, плазме, лимфе, моче), а также при анализе лекарственных форм в технологических средах и сточных водах производства стрептомицина и родственных ему соединений. Методики с использованием предлагаемого способа легко могут быть автоматизированы, особенно в настоящее время, когда налажен выпуск отечественной электронной аппаратуры с компьютерным управлением и обработкой данных (типа СТА, ТА, ВАМ и др.). Способ может быть использован не только в научных исследованиях, но и в широкомасштабном серийном аналитическом контроле.
Формула изобретения
Способ количественного определения стрептомицина методом адсорбционной инверсионной вольтамперометрии, заключающийся в том, что стрептомицин переводят из пробы в раствор и проводят вольтамперометрическое определение, отличающийся тем, что проводят накопление стрептомицина в перемешиваемом растворе в течение 90÷180 с при потенциалах электролиза -(0,10÷0,20) В на фоне (0,1÷0,2) н раствора гидрофосфата натрия с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 15÷25 мВ/с и концентрацию стрептомицина определяют по высоте пика в диапазоне потенциалов от 0,66 до 0,70 В относительно насыщенного хлоридсеребряного электрода методом добавок аттестованных смесей.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 18.02.2007
Извещение опубликовано: 10.11.2008 БИ: 31/2008
|
|||||||||||||||||||||||||||||||
 8,0÷8,6. При этих значениях рН стрептомицин находится в устойчивой форме и способен окисляться на СУ электроде в водных средах. При концентрации раствора Na2HPO4<0,1н. (т.е. при рН<8,0) регистрация пиков окисления затруднена, а при концентрации Na2HPO4>0,2н. (рН>8,6) пики практически не получены. По-видимому, при рН>8,6 происходит щелочной гидролиз стрептомицина и распад его молекулы с образованием электрохимически неактивного мальтола.
8,0÷8,6. При этих значениях рН стрептомицин находится в устойчивой форме и способен окисляться на СУ электроде в водных средах. При концентрации раствора Na2HPO4<0,1н. (т.е. при рН<8,0) регистрация пиков окисления затруднена, а при концентрации Na2HPO4>0,2н. (рН>8,6) пики практически не получены. По-видимому, при рН>8,6 происходит щелочной гидролиз стрептомицина и распад его молекулы с образованием электрохимически неактивного мальтола. Э) выбирают в зависимости от концентрации определяемого вещества. Максимальное значение величины тока окисления достигается при
Э) выбирают в зависимости от концентрации определяемого вещества. Максимальное значение величины тока окисления достигается при 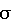 -критерию, равен 5,3·10-10 моль/л при
-критерию, равен 5,3·10-10 моль/л при