Патент на изобретение №2276149
|
||||||||||||||||||||||||||
(54) 1,3-ДИОКСОЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ
(57) Реферат:
Изобретение относится к новым 1,3-диоксоланам формулы 1:
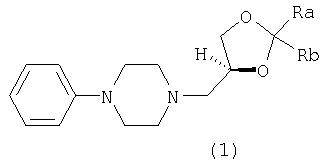
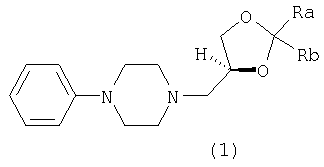
Настоящее изобретение относится к (S)-3-(4-фенил-1-пиперазинил)-1,2-пропандиоловым циклическим ацеталям, которые могут использоваться в качестве противокашлевых средств и в качестве промежуточных продуктов при получении леводропропизина и его солей. Изобретение относится также к способу получения указанных ацеталей. Более конкретно, настоящее изобретение относится к (S)-2,2-замещенным-1,3-диоксоланам формулы (1):
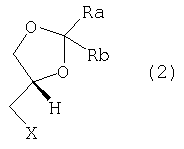
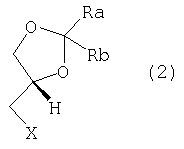
где каждый из Ra и Rb, которые могут быть одинаковыми или разными, представляет собой водород, C1-С6-алкил, фенил или Ra и Rb вместе с атомом С, к которому они присоединены, образуют необязательно замещенное 4-7-членное карбоциклическое кольцо. Предпочтительными соединениями формулы (1) являются соединения, в которых Ra и Rb представляют собой алкильные группы, содержащие менее 6 атомов С. Предпочтительно Ra и Rb являются одинаковыми; более предпочтительно Ra и Rb представляют собой метил или этил или вместе с атомом С, к которому они присоединены, образуют кольцо, содержащее от 5 до 6 атомов углерода. Изобретение относится также к энантиомерно чистым моноосновным солям (S)-2,2-замещенных-1,3-диоксоланов формулы (1) с фармацевтически приемлемыми кислотами. Особенно предпочтительными фармацевтически приемлемыми кислотами являются уксусная, пропионовая, янтарная, фумаровая, малеиновая, L-яблочная, D- и L-винная, D- и L-миндальная, L- и D-камфорсульфоновая кислоты. Особенно предпочтительными соединениями по данному изобретению являются S(-)-1,2-циклопентилиден-3-(4-фенилпиперазин-1-ил)пропан-1, 2-диол; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1, 2-диол; S(-)-1,2-(2-пропилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-(3-пентилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол малеат; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол L-тартрат; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол фумарат; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол D-10-камфорсульфонат. Соединения по данному изобретению формулы (1) получают взаимодействием фенилпиперазина с (R)-1,2-глицерилдиоксоланом формулы (2):
где Х выбран из группы, включающей Cl, Br, I и подходящий сложный сульфоновый эфир (R-SO3-), где R представляет собой C1-С3-алкил, трифторметил, фенил, п-толил или п-метоксифенил. Диоксоланы формулы (2) являются известными соединениями и/или могут быть получены известными методами. Более конкретно, сложные сульфоновые эфиры формулы (2) (X=R-SO3-) получают стандартными методами с использованием ангидрида или хлорангидрида алкил- и/или арилсульфоновой кислоты формулы (3):
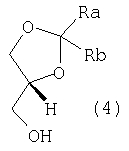
для этерификации (R)-2,2-замещенного-1,3-диоксолан-4-метанола формулы (4):
где Ra и Rb принимают значения, определенные выше. 1,3-Диоксолан-4-метанолы формулы (4) также являются известными соединениями, и их получение достаточно подробно описано в литературе. Например, они могут быть получены ферментативным разделением рацематов формулы (4) в соответствии со способом, описанным в US 5190867 (02.03.1993), или, предпочтительно, окислительным разложением 1,2;5,6-бис-диоксоланов D-маннита в соответствии со способом, описанным Borsa et al., в ЕР 147847 (07.03.1990) и использованным для получения (+)-1,2-изопропилиден-sn-глицерина (формула (4), где Ra и Rb представляют собой метил, и его тозилата (формула (3), где Ra и Rb представляют собой метил и R представляет собой п-толил). Удобные способы получения 1,2;5,6-бис-диоксоланов D-маннита и соответствующих D-глицеральдегидацеталей можно найти также в публикациях J. Org. Chem. 56, 4056 (1991) и Synthesis 587 (1992), где подробно описано получение 2,3-О-(3-пентилиден)-D-глицеральдегида с 55% выходами из D-маннита. 4-Галогенметилдиоксоланы формулы (2), где Х представляет собой Cl, Br или I, в свою очередь, могут быть получены, исходя из соответствующих эфиров формулы (2) (Х= RSO3-, где R принимает значения, определенные выше), взаимодействием с подходящим галогенидом щелочного или щелочноземельного металла (Na, K или Са) в инертном растворителе, выбранном из группы, включающей ацетон, метилэтилкетон, тетрагидрофуран, диоксан, диметилсульфоксид, ацетонитрил, С1-С4-спирт и их смеси. Альтернативно, диоксоланы формулы (2) (Х=Cl, Br) могут быть получены введением диоксолановых групп в соответствующие 3-галоген-1,2-пропандиолы, как описано в ЕР 0930311 (21.07.1999). Особенно предпочтительным является 3-хлорпропандиол. Предпочтительными регентами введения ацетальных групп являются формальдегид, ацетальдегид и бензальдегид, ацетон, диэтилкетон, бензофенон, циклогексанон и ацетали или их простые енольные эфиры, такие как 2,2-диметоксипропан, 2,2-диметоксиэтан и 2-метоксипропен. Альтернативно, диоксоланы формулы (2) (Х=Cl, Br) могут быть получены введением ацетальной группы в хиральные эпихлоргидрины или эпибромгидрины или соответствующие хиральные 3-галогенпропандиолы с помощью циклоалканона в соответствии со способами, описанными для получения (±)2-хлорметил-1,4-диоксаспиро[4,5]декана в FR 1552153 или описанными в более общем виде в публикациях Blicke F.F. et al., J.A.C.S, 74, 1735 (1972) и ibidem, 76, 1226 (1954). Указанные хиральные эпихлоргидрины или эпибромгидрины и соответствующие 3-галогенпропандиолы, в свою очередь, представляют собой доступные промежуточные продукты, которые могут быть получены, например, кинетическим разделением соответствующих рацематов [в соответствии с публикацией Furrow et al., J. Org. Chem., 63, 6776, 1998 или, альтернативно, в соответствии с публикацией T. Takeichi et al., Tetrahedron, 36, 3391 (1980)] или ферментативным разделением (см. публикации Kasai N. et al., JP 02257895 (1990); C.A.: 114, 41064q, 1991). Фенилпиперазины алкилируются 1,2-глицерилдиоксоланом формулы (2) в стандартных условиях реакции превращения вторичного амина в третичный при использовании на каждый моль алкилирующего агента формулы (2), по меньшей мере, одного моля или небольшого молярного избытка фенилпиперазина в присутствии, по меньшей мере, одного моля противооснования. Противооснование выбрано из группы, включающей тонко измельченные неорганические основания, такие как карбонаты или гидрокарбонаты щелочных или щелочноземельных металлов (Na, K, Mg, Ca), оксиды Са или Mg, третичные амины, такие как триэтиламин, диметил- или диэтиланилин, ароматические амины, такие как пиридин, пиколин или этилметилпиридин и, если это желательно, непосредственно фенилпиперазин, который затем может повторно циклизоваться с получением цикла. Реакцию алкилирования можно проводить при нагревании, необязательно в присутствии инертных растворителей, таких как толуол и/или ксилол, которые при кипении растворителя будут преимущественно уменьшать продолжительность реакции. После завершения реакции алкилирования любые нерастворимые вещества отфильтровывают или удаляют центрифугированием, затем органические фазы повторно промывают водой для простого удаления примесей и побочных продуктов и растворитель отгоняют и получают с высокими выходами остаток, состоящий из по существу чистого 1,3-диоксолана по данному изобретению формулы (1), который выделяют прямой кристаллизацией или получением соли желаемой фармацевтически приемлемой кислоты. Соединения (1) и их соли удивительно легко кристаллизуются из обычных растворителей: следовательно, способ по данному изобретению снижает до минимума любой риск загрязнения продукта следовыми количествами глицидолов и/или эпигалогенгидринов в качестве возможных примесей. Моноосновные соли соединений формулы (1) получают обычными стандартными способами, такими как получение солей с эквимолекулярными количествами желаемой кислоты в подходящем растворителе с последующей кристаллизацией полученной соли. Кроме того, соединения по данному изобретению формулы (1) и их моноосновные соли являются по существу безвкусными и не обладают остаточным горьким привкусом леводропропизина. Соединения (1) являются эффективными пролекарствами леводропропизина, в водных растворах которых 1,3-диоксоланы формулы (1) или их соли преобразуются в леводропропизин в результате гидролиза в теплой среде при молярном избытке разбавленной минеральной кислоты, такой как соляная кислота, или растворимых в воде карбоновых кислот, таких как уксусная, малоновая или лимонная кислоты. Соединения формулы (1) и водные растворы их моноосновных солей являются стабильными при физиологических значениях рН. Соединения по данному изобретению и их соли обладают противокашлевой активностью, что доказано результатами сравнительных биологических испытаний при внутривенном способе введения и в сравнении с самим леводропропизином. Испытания проводились на самцах морской свинки Dunkin-Hartley (4-6 животных в каждой группе), их обрабатывали аэрозолем 0,0045% (ч./об.) водного раствора капсацина (capsacin) [Lavezzo A., Pulm. Pharmacol., 5, 143-147, 1992; Gallico L. et al., Br. J. Pharmacol., 112, 795-800, 1994] в течение 5 минут после внутривенного введения 0,5 мл раствора (-)-1,2-циклогексилидин-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола фумарата (DF 1689A, 10 мг/кг) или, для сравнения, 0,5 мл раствора леводропропизина (рН 4,5, 10 мг/кг), или равного объема солевого раствора (контроль). Кашель подсчитывался в течение 4 минут обработки аэрозолем капсицина, и таким образом были получены доказательства значительного ингибирования (47,3%) ответа на стимулирование кашля по сравнению с 43,6% ингибированием, вычисленным для контрольной группы. Фактически, получены следующие количественные показатели кашля: 9,17±1,01 для контрольных животных, 5,17±0,54 для животных, обработанных леводропропизином, 4,84±0,54 для животных, обработанных соединением по изобретению DF 1689A. Противокашлевое действие дополнительно характеризуется продолжительным эффектом: фактически, показатели ингибирования кашля, стимулированного аэрозольным введением раствора капсацина, равные 34,6 и 27,3%, были вычислены через 15 и 30 минут после внутривенного введения. Соединения формулы (1) и их соли могут соответствующим образом быть введены в виде аэрозоля; действительно, самцы морской свинки Dunkin-Hartley, подвергшиеся обработке аэрозолем 1% (мас./об.) водных растворов соединения DF 1689A и леводропропизина в течение 10 минут, показали значительное уменьшение количества приступов кашля, индуцированных этими лекарственными средствами, по сравнению с контролем (аэрозоль солевого раствора), при примерно 35% ингибиторном действии для обоих лекарственных средств. Для применения в качестве противокашлевых средств соединения (1) будут вводиться в фармацевтические композиции стандартными методами в сочетании со стандартными наполнителями для введения пероральным, парентеральным способами или с помощью аэрозоля, например, в виде капсул, таблеток, не разлагающихся в желудке, сиропов, препаратов с контролируемым выделением действующего вещества. Величина суточной дозы будет зависеть от различных факторов, таких как частота и сила кашля и общее состояние пациента (возраст, пол и масса тела). Суточная доза для взрослого субъекта массой 60 кг будет колебаться в интервале от 10 мг до 1500 мг соединений формулы (1) в сутки, при этом необязательно может быть разбита на несколько введений. Соединения по данному изобретению могут также вводиться детям, даже в течение длительного периода, в приемлемых дозах благодаря их низкой токсичности. И наконец, соединения по данному изобретению могут использоваться в качестве промежуточных продуктов при получении леводропропизина и его солей. Приведенные далее примеры дополнительно иллюстрируют данное изобретение. Пример 1 К раствору 13,8 г влажного 1,2;5,6-ди-О-(пентилиден)-D-маннита (полученного в соответствии со способом, описанным в публикации Synthesis, 587, 1992) в тетрагидрофуране (ТГФ), охлажденном до 20-25о С, в течение 10 минут добавляют суспензию периодата калия (7,95 г) и бикарбоната калия (0,32 г) в воде (50 мл). Смесь энергично перемешивают в течение трех часов, охлаждают до 5оС и фильтруют. Осажденный йодат калия промывают этилацетатом и две объединенные фазы оставляют нагреваться до комнатной температуры. Водную фазу обрабатывают NaCl и повторно экстрагируют этилацетатом. К объединенным органическим фазам при энергичном перемешивании добавляют раствор 3,2 г NaBH4 и 1,88 г бромида тетрабутиламмония в 120 мл воды. Смесь взаимодействует при комнатной температуре в течение 3 часов, фазы разделяют, водную фазу экстрагируют этилацетатом (2х30 мл), фильтруют, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Полученный маслянистый остаток отгоняют при пониженном давлении с получением 7,12 г 2,3-О-(3-пентилиден)-D-глицерина. [ К раствору 6,45 г полученного спирта в этилацетате (18 мл) добавляют 6 мл триэтиламина, затем при перемешивании с внешним охлаждением раствор 7,76 г п-толуолсульфохлорида в AcOEt (18 мл). Смесь перемешивают в течение 12 часов при комнатной температуре, затем разбавляют водой (10 мл), фазы разделяют, промывают водой (3х10 мл), сушат над сульфатом натрия, фильтруют и выпаривают досуха при пониженном давлении с получением 11,98 г 2,3-О-(3-пентилиден)-D-глицерина тозилата, [ К раствору тозилата в н-бутаноле (70 мл) последовательно добавляют при энергичном перемешивании 4,5 г тонко измельченного карбоната натрия и 6 мл фенилпиперазина. Смесь кипятят с обратным холодильником при перемешивании, затем оставляют для взаимодействия при температуре кипения растворителя еще на 20 часов. Затем бутанол выпаривают при пониженном давлении, остаток обрабатывают водой и повторно экстрагируют этилацетатом. Объединенные органические фазы сушат и фильтруют, затем растворитель выпаривают в вакууме. Полученный остаток кристаллизуют из водного метанола с получением 8,95 г S(-)-1,2-(3-пентилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола, другим названием которого является 4-фенилпиперазин, 1-(2,2-диэтил-1,3-диоксолан-4-илметил). Пример 2 В соответствии с методикой примера 1 при использовании 2,3-О-(2-пропилиден)-D-глицерина тозилата получают S(-)-1,2-(2-пропилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол, другим названием которого является 4-фенилпиперазин, 1-(2,2-диметил-1,3-диоксолан-4-илметил). Пример 3 К раствору 3,1 г 2,3-О-(3-циклогексилиден)-D-глицерина [ 1Н ЯМР: К раствору мезилата в толуоле (16 мл) добавляют 5,45 мл фенилпиперазина, смесь кипятят с обратным холодильником до завершения реакции (примерно 8 часов). Реакционную смесь охлаждают до примерно 50°С, добавляют 10 мл воды и энергично перемешивают в течение, по меньшей мере, 10 минут. Фазы разделяют и органическую фазу повторно промывают водой. Растворитель выпаривают с получением густого масла, которое растворяют в теплом изопропаноле (15 мл). Затем раствор медленно охлаждают для отделения твердого кристаллического S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола. Т. пл. 63-64°С, [ 1Н ЯМР: Пример 4 В соответствии с методикой, описанной в примерах 1 и 2, взаимодействием 1,3-диоксолана, выбранного из группы, включающей 2S-1,4-диоксаспиро[4.4]нонан-2-метанол п-толуолсульфонат; 2S-1,4-диоксаспиро[4.4]нонан-2-хлорметил; 2S-1,4-диоксаспиро[4.5]декан-2-метанол трифторметансульфонат; 2S-1,4-диоксаспиро[4.5]декан-2-хлорметил; 2S-1,4-диоксолан-4-хлорметил-2,2-диметил; 2S-1,4-диоксолан-4-йодметил-2,2-диметил, с фенилпиперазином получают следующие соединения: S(-)-1,2-циклопентилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-(2-пропилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол. Пример 5 0,232 г Фумаровой кислоты при перемешивании добавляют к раствору S-(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола (0,632 г) в абсолютном этаноле (8 мл). Смесь перемешивают до полного растворения веществ, затем растворитель выпаривают в вакууме и твердый остаток кристаллизуют из ацетона с получением твердого кристаллического S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола фумарата (DF1689A, 0,52 г), т. пл. 162-164°С, [ 1Н ЯМР: Пример 6 В соответствии со способом получения соли S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола и L-винной, малеиновой, D(+)-10-камфорсульфоновой, L-миндальной кислот получают следующие соединения: S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол L-тартрат, т. пл. 130-132°С, [ S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол малеат, т. пл. 160-162°С, [ S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан- 1,2-диол, D(+)-10-камфорсульфонат, т. пл. 110-115°С, [ S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол, L-манделат, [ Пример 7 Суспензию 3,5 г S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола в 70 мл водной уксусной кислоты (10% мас./об.) кипятят с обратным холодильником в течение 2 часов, затем в нее барботируют пар для отгонки циклогексанона, который отделяют. Водную фазу нейтрализуют до рН 7 добавлением 10% раствора NaOH, затем охлаждают до 5-10°С с получением 2,05 г (-)3-(4-фенилпиперазин-1-ил)пропандиола, т.пл. 102-103оС, [ Пример 8 Альтернативно, 0,35 моль-эквивалентов одного из 1,3-диоксоланпроизводных, описанных в примерах 1-4, а именно S(-)-1,2-(2-пропилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола; S(-)-1,2-(циклопентилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола; S(-)-1,2-(2-пропилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диола, при перемешивании добавляют частями к 36% раствору соляной кислоты (36 мл) в 45 мл воды; суспензию нагревают до 80°С с получением прозрачного раствора, который выдерживают при этой температуре в течение 30 минут, затем охлаждают до 20-25°С. Водную фазу экстрагируют дихлорметаном (3х15 мл), затем добавляют н-бутанол (0,5 л). Двухфазную смесь кипятят для отгонки азеотропной смеси вода:н-бутанол, выделяя примерно 300 мл дистиллята, затем охлаждают для ускорения кристаллизации (-)-3-(4-фенилпиперазин-1-ил)пропандиола гидрохлорида (85 г). Раствор гидрохлорида в 125 мл воды обесцвечивают активированным углем (2,12 г), нагревая до 50°С и выдерживая при этой температуре в течение 15 минут, фильтруют и затем нейтрализуют добавлением водного раствора гидроксида аммония (30% мас./мас.). Раствор нагревают до 50°С и инициируют кристаллизацию добавлением кристаллов (-)-3-(4-фенилпиперазин-1-ил)-1,2-пропандиола. Суспензию оставляют для самопроизвольного охлаждения, затем выдерживают в течение 2 часов при +2 – +4°С и фильтруют с получением 70-72 г (-)3-(4-фенилпиперазин-1-ил)-1,2-пропандиола. Физико-химические константы: 1) S(-)-1,2-Циклопентилиден-3-(4-фенилпиперазин-1-ил)-пропан-1,2-диол (соединение 1, пункт 4 формулы изобретения) 1H ЯМР 2) S(-)-1,2-(2-Пропилиден)-3-(4-фенилпиперазин-1-ил)-пропан-1,2-диол (соединение 3, пункт 4 формулы изобретения) 1Н ЯМР 3) S (-)-1,2-(3-Пентилиден)-3-(4-фенилпиперазин-1-ил)-пропан-1,2-диол (соединение 4, пункт 4 формулы изобретения) 1H ЯМР
Формула изобретения
1. Соединения формулы (1)
где каждый из Ra и Rb, которые могут быть одинаковыми или разными, представляет собой водород или C1-С6-алкил; или Ra и Rb вместе с атомом С, к которому они присоединены, образуют необязательно замещенное 4-7-членное карбоциклическое кольцо. 2. Соединения по п.1, где Ra и Rb являются одинаковыми. 3. Соединения по п.1 или 2, где Ra и Rb представляют собой метил или этил. 4. Соединения по п.1, выбранные из следующих соединений: S(-)-1,2-циклопентилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-(2-пропилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-(3-пентилиден)-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол малеат; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диол L-тартрат; S(-)-1,2-циклогексилиден-3-(4-фенилпиперазин-1-ил)пропан-1,2-диолфумарат; S(-)-1,2-циклогексилиден-З -(4-фенилпиперазин-1-ил)пропан-1,2-диол D-10-камфорсульфонат. 5. Соединения по пп.1-4 в качестве активного ингредиента лекарственного средства с противокашлевой активностью. 6. Способ получения соединений формулы (1) взаимодействием фенилпиперазина с (R)-1,2-глицерилдиоксоланом формулы (2):
где Х выбран из группы, включающей Cl, Br, I и подходящий сульфоновый эфир (R-SO3-), R представляет собой C1-С3-алкил, трифторметил, фенил, п-толил или п-метоксифенил. 7. Способ по п.6, где взаимодействие проводят в присутствии основания при использовании толуола или ксилола в качестве растворителей. 8. Фармацевтическая композиция, обладающая противокашлевой активностью, содержащая соединение по пп.1-4 в смеси с подходящим носителем.
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
(73) Патентообладатель(и):
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 17.10.2006 № РД0013235
Извещение опубликовано: 27.11.2006 БИ: 33/2006
|
||||||||||||||||||||||||||
 С.П.А. (IT)
С.П.А. (IT)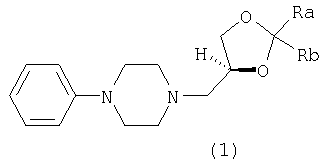
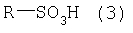
 ]D=±17,2 (EtOH).
]D=±17,2 (EtOH). 4.37 (м,1Н);
4.37 (м,1Н);