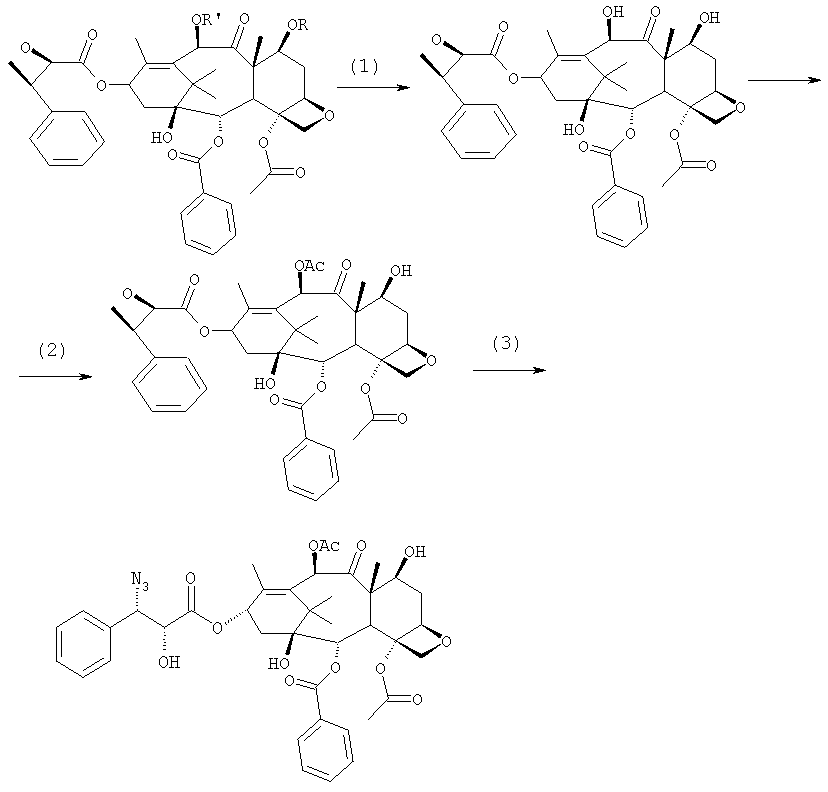Патент на изобретение №2276147
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА
(57) Реферат:
Изобретение относится к новому способу получения паклитаксела, где в качестве исходного используют 10-деацетилбаккатин 111, у которого на первой стадии проводят защиту гидроксилов в положении 7 и 10 одновременно. Способ является многостадийным. Изобретение также относится к новому промежуточному соединению, используемому в этом способе, формулы
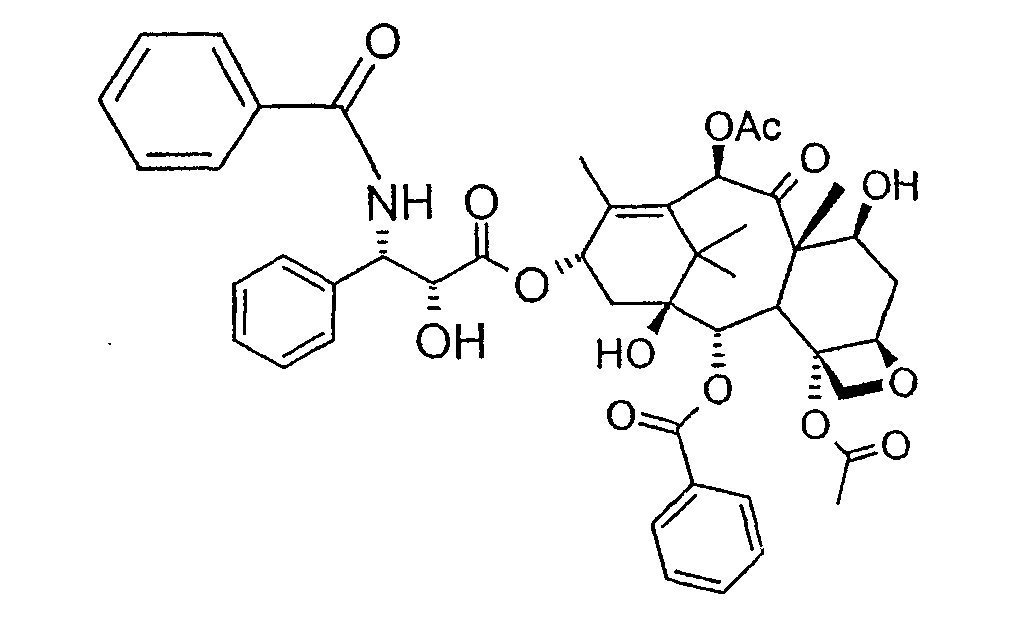
Настоящее изобретение относится к способу получения паклитаксела. Паклитаксел представляет собой природную молекулу, имеющую широкий спектр противоопухолевой активности, со следующей структурной формулой:
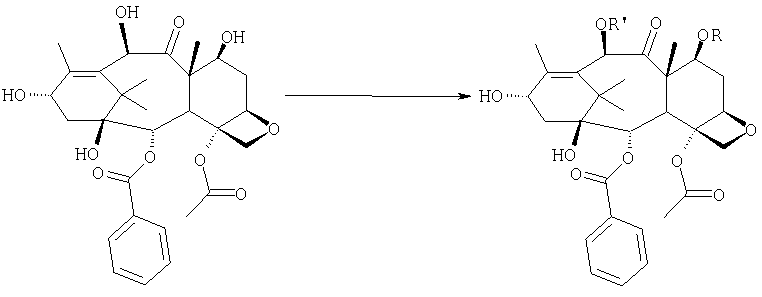
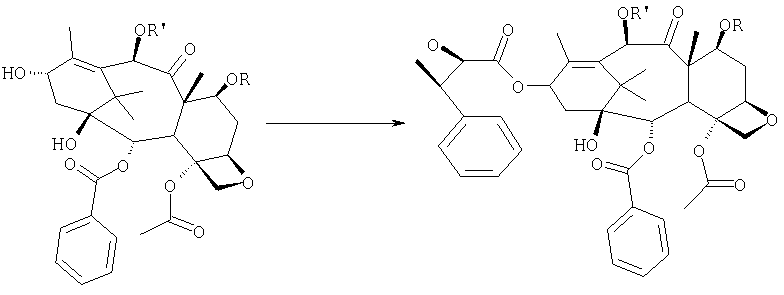
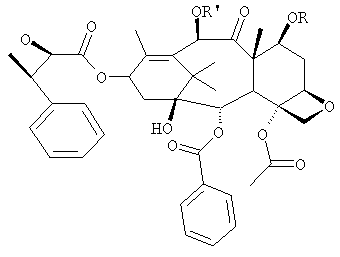
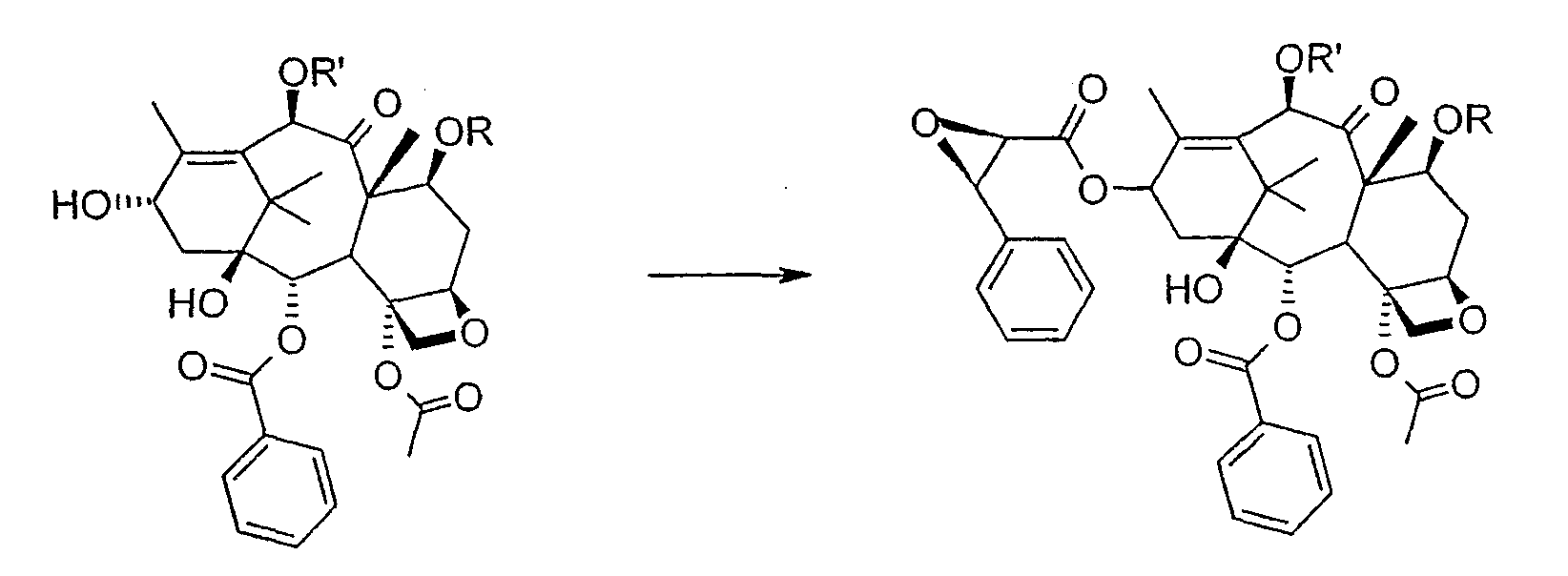
Соединение, впервые выделенное из коры Taxus brevifolia и из других природных источников, можно получить полусинтетически с использованием ряда процедур, описанных как в научной, так и в патентной литературе. US 4924011 описывает полусинтез паклитаксела, использующий 10-деацетилбаккатин III, с защитной триалкилсилильной группой на С-7 гидроксиле и впоследствии ацетилированный на С-10. Полученное в результате промежуточное соединение реагирует с (2R,3S)-N-бензоил-2-О-(1-этоксиэтил)-3-фенилизосерином, и с полученного в результате продукта снимают защиту, получая паклитаксел. WO 93/06094 описывает получение паклитаксела реакцией По US 5476954 паклитаксел получают, используя в качестве исходного вещества 10-деацетилбаккатин III, подвергнутый в положении С-7 эстерификации 2,2,2-трихлорэтоксикарбонильной группой (TROC). По US 5917062 и US 6020507 С-7 гидроксил защищают карбобензоксигруппой (CBZ) или карботретбутоксигруппой (Boc) с последующим селективным ацетилированием С-10 гидроксила. Из литературы ясно, что ключевой аспект полусинтеза паклитаксела заключается в селективной защите гидроксилов дитерпенового фрагмента (скелет 10-деацетилбаккатина III). Положение С-7 является наиболее реакционноспособным и поэтому в него вводят функциональные группы, которые впоследствии можно легко удалить. Обычно такой группой является триэтилсилильная (ТЭС), которая является устойчивой при условиях, используемых для эстерификации других вовлеченных в синтез гидроксилов и предоставляет примерно 85% выход конверсии. Приблизительно 85% выход получают, когда ацетильную группу впоследствии вводят в положение С-10. Неожиданно был обнаружен новый способ синтеза паклитаксела с более высоким конечным выходом, а также другие преимущества по сравнению с известными способами. Способ согласно изобретению включает следующие стадии: a) защиты гидроксилов 10-деацетилбаккатина III (10-ДАБ III) в 7- и 10-положениях
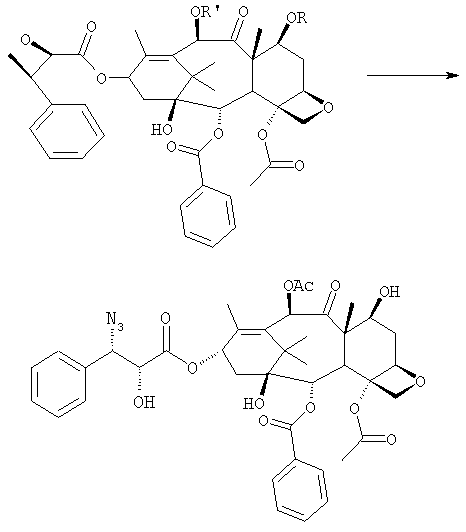
где R=R’= трихлорацетил, или R’= ацетил, а R выбран из трет-бутоксикарбонила и трихлорацетила; b) этерификации гидроксила в 13-положении 3-фенил-2-эпоксипропионовой кислотой
c) удаления защитных групп в 7- и 10-положениях (1), если они обе представляют собой трихлорацетильные группы с последующим селективным ацетилированием в 10-положении (2) и раскрытием эпоксида азидом натрия (3)
или, альтернативно, c’) если R’= ацетил и R= трихлорацетил, раскрытия эпоксида азидом натрия и одновременного снятия защиты в 7-положении
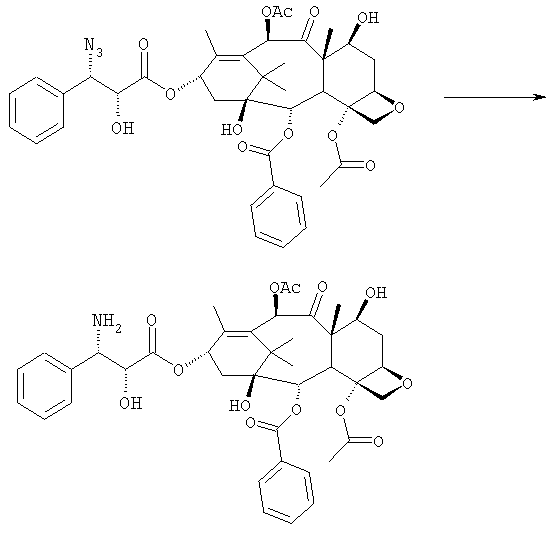
d) восстановления азидогруппы в аминогруппу
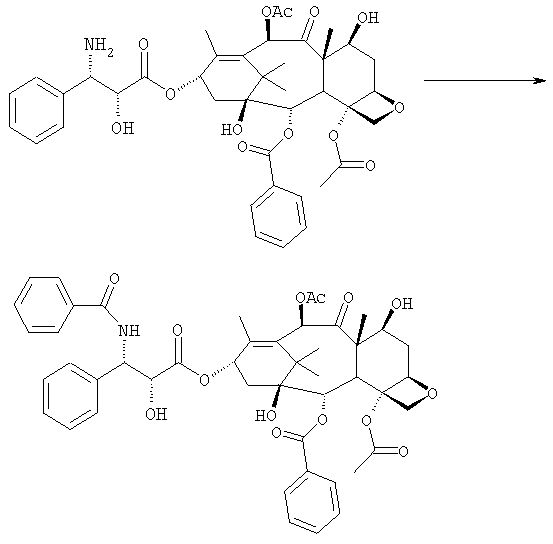
бензоилирования с получением конечного продукта Исходным продуктом является 10-деацетилбаккатин III (10-ДАБ III), который экстрагируют из листьев Taxus baccata. На первой стадии гидроксилы 10-ДАБ III в 7 и 10-положениях количественно подвергают реакции этерификации. Когда R=R’= трихлорацетил, 10-ДАБ III реагирует с трихлорацетилхлоридом в метиленхлориде в присутствии триэтиламина и каталитических количеств [4-диметиламинопиридина (ДМАП). Когда R На следующей стадии (b) гидроксил в положении 13 подвергают эстерификации 3-фенил-2-эпоксипропионовой кислотой, предпочтительно ее аммонийной солью в толуоле в присутствии дициклогексилкарбодиимида, ДМАП и п-толуолсульфокислоты, посредством этого получая сложный эфир (2R,3R)-3-фенил-2,3-эпоксипропионовой кислоты с баккатином. Когда обе защитные группы R и R’ представляют собой трихлорацетил, их можно удалить, используя условия и реагенты, описанные в работах Zheng et al., Tetrahedron Lett., 1995, 36, 2001 и Datta et al., J. Org. Chem., 1995, 60, 761. Предпочтительно, две трихлорацетильные группы удаляют двумя эквивалентами гидроксида аммония. Соединение со снятыми защитными группами селективно ацетилируют в положении 10 уксусным ангидридом в присутствии солей церия, скандия, иттербия, предпочтительно CeCl3·7H2O. Полученное в результате соединение реагирует с NaN3 в водном метаноле в присутствии метилформиата в условиях, сообщенных в литературе (Yamaguchi T., Tetrahedron Letters 39, 5575-78, 1998), давая соответствующий азид. Альтернативно, когда R= трихлорацетил и R’= ацетил (d), оксиран реагирует с NaN3, давая соответствующий азид со снятием защиты в 7-положении, соответствующий соединению, полученному на стадии (c’). На следующей далее стадии (d) азид восстанавливают до амина. Восстановление можно провести водородом на катализаторе или PPh3. Полученный на последней стадии (е) продукт бензоилируют в положении аминогруппы, получая паклитаксел. Бензоилирование можно осуществить бензойным ангидридом одновременно с восстановлением либо впоследствии на выделенном восстановленном продукте, используя стехиометрические количества бензоилхлорида в присутствии карбоната калия. Следующие ниже примеры иллюстрируют изобретение более детально. Пример I – синтез 7-трихлорацетилбаккатина III В 25 мл круглодонной колбе растворяют 0,603 г (1,03 моль, 1,0 экв.) баккатина III при перемешивании магнитной мешалкой в 9,7 мл сухого пиридина при 25°С под атмосферой азота. В прозрачный бледно-желтый раствор по каплям добавляют 138 мкл (1,23 ммоль, 1,23 экв.) трихлорацетилхлорида. Через 30 мин после завершения добавления образуется белый осадок. При тех же условиях, как описано выше, в реакционную суспензию по каплям добавляют дополнительные 120 мкл (1,07 ммоль, 1 экв.) трихлорацетилхлорида. После 20 мин раствор имеет желто-коричневую окраску. Тонкослойная хроматография (ТСХ) (SiO2, н-гексан/EtOAc, 2:3) показывает почти полную конверсию исходного баккатина III. Реакционную смесь разбавляют CH2Cl2. Полученный в результате раствор несколько раз промывают насыщенным раствором CuSO4 до полного удаления пиридина (раствор больше не имеет голубую окраску). Органическую фазу концентрируют под вакуумом, сушат над MgSO4, фильтруют и растворитель выпаривают, получая 0,612 г желтовато-белого порошка, соответствующего 7-трихлорацетилбаккатину III, имеющему следующие спектроскопические характеристики. 1Н-ЯМР (400 МГц, CDCl3): Пример II – синтез (2’R,3’R)-7-трихлорацетилбаккатин III-13-(3′-фенил-2,,3′-эпоксипропионата) В 30 мл безводного толуола при 0°С растворяют 0,164 г (1,00 ммоль, 1 экв.) свежеприготовленной 3-фенил-2-эпоксипропионовой кислоты. Затем добавляют 0,5 г (1 ммоль, 0,68 экв.) 7-(трихлорацетил)-баккатина III [7-(ТСА)-баккатина III] при 0°С под атмосферой азота. Наконец последовательно добавляют дициклогексилкарбодиимид (ДЦК, 0,21 г, 1,00 ммоль, 1,0 экв.), 4-диметиламинопиридин (ДМАП, 0,084 г, 0,68 ммоль, 0,66 экв.) и п-толуолсульфокислоту (п-ТСК, 0,17 г, 0,10 ммоль, 0,1 экв.). Затем раствор нагревают до 70°С при перемешивании магнитной мешалкой и под током азота. Развитие реакции контролируют ТСХ (SiO2, н-гексан/EtOAc, 3:2). Первое пятно, имеющее Rf=0,28, соответствует сложному эпоксиэфиру 7-(ТСА)-баккатина III. Второе пятно, имеющее Rf=0,11, соответствует 7-(ТСА)-баккатину III. После 3 часов смесь охлаждают и суспендированное твердое вещество отфильтровывают. Осажденную дициклогексилмочевину (ДЦМ) промывают CH2Cl2. Объединенные органические фракции концентрируют до сухого состояния. Полученное в результате неочищенное вещество (0,919 г) подвергают флэш-хроматографии (SiO2, н-гексан/EtOAc, 3:2). Получают 0,100 г (0,14 ммоль, 20%) непрореагировавшего 7-(ТСА)-баккатина III и 0,435 г (0,49 ммоль, 73%) (2’R,3’R)-7-трихлорацетилбаккатин III-13-(3′-фенил-2 1Н-ЯМР (400 МГц, CDCl3): Пример III – синтез (2’R,3’R)-баккатин III-13-(3′-азидо-2′-гидрокси-3′-фенилпропионата) В 25 мл одногорлой круглодонной колбе, оборудованной магнитной мешалкой, в 10,0 мл CH3OH при 25°С суспендируют 0,397 г (0,45 ммоль, 1 экв.) (2’R,3’R)-7-трихлорацетилбаккатин III-13-(3′-фенил-2,,3′-эпоксипропионата). Последовательно добавляют 1,26 мл Н2О, 1,26 мл НСООСН3 и 0,735 г (11,3 ммоль, 25,0 экв.) азида натрия. Температуру повышают до 50°С и ход реакции контролируют ТСХ (SiO2, CHCl3/EtOAc/MeOH, 12,0:2,0:0,3). Наблюдают исчезновение исходного продукта и одновременное образование двух продуктов, имеющих Rf=0,22 и 0,29, соответственно. Продукт, имеющий Rf=0,29, впоследствии идентифицируют как конечный продукт, тогда как продукт с Rf=0,22 представляет собой (2’R,3’R)-баккатин III-13-(3′-фенил-2′,3′-эпоксипропионат), образовавшийся в качестве промежуточного продукта реакции. Продукт, имеющий Rf=0,29, растет со временем в ущерб продукту, имеющему Rf=0,22. Реакционный раствор после 46 часов имеет желто-коричневую окраску с белым осадком (не прореагировавший NaN3). Реакцию гасят через 46 часов, добавляя воду, наблюдают два дополнительных пятна с Rf=0,38 и 0,13 (неизвлеченные продукты разложения). Осажденное молочно-белое вещество отфильтровывают, промывают водой и затем AcOEt. Получают двухфазную смесь, причем обе фазы являются прозрачными. Две фазы разделяют. Водную фазу три раза экстрагируют AcOEt и объединенные органические фазы концентрируют и сушат над MgSO4. Смесь фильтруют и растворитель выпаривают, получая 0,335 г желтовато-белого порошка. Полученное в результате неочищенное вещество очищают флэш-хроматографией (SiO2, CHCl3/EtOAc/MeOH, 12: 2:0,3), получая 0,279 г (0,36 ммоль; 80%; Rf = 0,22) (2’R,3’R)-баккатин III-13-(3′-азидо-2′-гидрокси-3′-фенилпропионата). Соединение имеет следующие спектроскопические характеристики: 1Н-ЯМР (400 МГц, CDCl3): Пример IV – синтез N-дебензоилпаклитаксела В 25 мл двугорлой круглодонной колбе в 5,2 мл свежеперегнанного CH2Cl2 растворяют 0,102 г (0,13 ммоль, 1,0 экв.) (2’R,3’R)-7-гидроксибаккатин III-13-(3′-азидо-2′-гидрокси-3′-фенилпропионата) и в полученный в результате бледно- желтый раствор добавляют воду (0,05 мл), затем 0,071 г (0,26 ммоль, 2,0 экв.) PPh3. Смесь реагирует при комнатной температуре при перемешивании магнитной мешалкой. После 16 часов ход реакции проверяют ТСХ (SiO2, CHCl3/CH3OH 9:1). Исходный продукт (Rf=0,61) исчезает и наблюдается пятно с Rf=0,19. Реакцию гасят, разбавляя смесь (раствор бледно-желтого цвета с белым осадком) CHCl3. Затем смесь промывают дистиллированной Н2О и потом насыщенным раствором хлорида натрия (соляным раствором). Органическую фазу ярко-желтого цвета сушат над MgSO4, затем фильтруют и растворитель выпаривают. Получают 0,177 г масла цвета желтой охры. Неочищенное вещество подвергают флэш-хроматографии (SiO2, CHCl3/CH3OH 9:1), получая 0,074 мг (0,10 ммоль; 76%) N-дебензоилпаклитаксела (порошок бледно-желтого цвета). 1Н-ЯМР (400 МГц, CDCl3): Пример V – синтез паклитаксела В 10 мл круглодонной колбе в 1,25 мл AcOEt растворяют 0,031 г (0,041 ммоль, 1,0 экв.) N-дебензоилпаклитаксела. В прозрачный желтый раствор добавляют 1,25 мл водного насыщенного раствора NaHCO3. В полученную в результате двухфазную смесь при интенсивном перемешивании магнитной мешалкой по каплям добавляют 7,1 мл (0,064 ммоль, 1,5 экв.) бензоилхлорида. Ход реакции контролируют ТСХ (SiO2, CHCl3/СН3OH, 9:1). После исчезновения исходного продукта наблюдается пятно, имеющее Rf=0,50. Реакционную смесь разбавляют AcOEt. Органическую фазу отделяют от водной фазы, которую экстрагируют AcOEt (три экстракции). Объединенные органические фазы сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество (0,037 г) растворяют в смеси 1:1 СН2Cl2/этиловый эфир, затем добавляют н-пентан (0,030 г, 0,035 ммоль, 86%) для осаждения паклитаксела, имеющего спектроскопические характеристики, сообщенные в литературе. Пример VI – синтез (2’R,3’R)-7,10-бис-трихлорацетил-10-деацетилбаккатин III-13-(3′-фенил-2,3′-эпоксипропионата) В 100 мл круглодонной колбе при 0°С в 30 мл безводного толуола растворяют 0,178 г (1,09 ммоль, 1,0 экв.) свежеприготовленной 3-фенил-2-эпоксипропионовой кислоты. В полученном в результате растворе под атмосферой азота и при 0°С суспендируют 0,663 г (0,79 ммоль, 0,73 экв.) 7,10-бис-трихлорацетил-10-деацетилбаккатина III [7,10-бис-(ТХА)-10-ДАБ III]. Наконец, последовательно добавляют дициклогексилкарбодиимид (ДЦК, 0,224 г, 1,09 ммоль, 1,0 экв.), 4-диметиламинопиридин (ДМАП, 0,088 г, 0,72 ммоль, 0,66 экв.) и п-толуолсульфокислоту (п-ТСК, 0,19 г, 0,11 ммоль, 0,1 экв.). Реакцию проводят в гетерогенной фазе при 700С с перемешиванием магнитной мешалкой и под потоком азота. Ход реакции контролируют ТСХ (SiO2, н-гексан/EtOAc, 3:2). Первое пятно, имеющее Rf= 0,28, соответствует сложному эпоксиэфиру 7,10-бис-(ТХА)-10-ДАБ III. Второе пятно, имеющее Rf= 0,15, соответствует 7,10-бис-(ТХА)-10-ДАБ III. Через 3 часа смесь охлаждают и суспендированное твердое вещество отфильтровывают. Темно-желтый осадок промывают CH2Cl2: оставшееся белое твердое вещество представляет собой ДЦМ. Объединенные органические фракции концентрируют и полученное в результате твердое вещество подвергают флэш-хроматографии (SiO2, н-гексан/EtOAc, 3:2). Получают 0,63 г (2’R,3’R)-7,10-бис-трихлорацетил-10-деацетилбаккатин III-13-(3′-фенил-2,3′-эпоксипропионата). 1Н-ЯМР (400 МГц, CDCl3): Пример VII – синтез (2’R,3’R)-10-деацетил-баккатин III-13-(3′-фенил-2′,3′-эпоксипропионата) В 25 мл круглодонной колбе в 3 мл СН3ОН суспендируют 0,174 г (0,18 ммоль, 1,0 экв.) (2’R,3’R)-7,10-бис-(ТХА)-10-ДАБ III-13-(3′-фенил-2′,3′-эпоксипропионата). Полученную в результате суспензию охлаждают до 0°С и к ней по каплям при интенсивном перемешивании магнитной мешалкой добавляют 0,24 мл (0,36 ммоль, 2,0 экв.) 1,57 М водного раствора NH3. Реакцию проводят в течение 15 мин при 0°С, в течение которых суспензия становится зеленовато-желтой. После этого смесь нагревают до комнатной температуры, и она реагирует в течение дополнительных 5 мин до полного растворения осадка, приводя к получению прозрачного зеленовато-желтого раствора. Полное исчезновение исходных соединений контролируют ТСХ (SiO2, н-гексан/EtOAc, 3:2), которая дает одно пятно на основе. Реакционную смесь разбавляют Н2О, получая молочно-белый раствор, из которого экстрагируют органическую фазу (3 экстракции) AcOEt (при добавлении органического растворителя образуется эмульсия, которую разрушают растворением в ней NaCl). Объединенные органические фазы сушат над MgSO4, фильтруют и растворитель выпаривают. Получают 0,194 г белого порошка (2’R,3’R)-10-деацетилбаккатин III-13-(3′-фенил-2′,3′-эпоксипропионата). 1Н-ЯМР (400 МГц, CDCl3): Пример VIII – синтез (2’R,3’R)-баккатин III-13-(3′-фенил-2′,3′-эпоксипропионата) К раствору (2’R,3’R)-10-деацетилбаккатин III-13-(3′-фенил-2′,3′-эпоксипропионата) (138 мг) в 3 мл сухого тетрагидрофурана добавляют 7,3 мг CeCl3·7H2O и 0,073 мл уксусного ангидрида. Реакционную смесь перемешивают при комнатной температуре в течение 5 часов, в течение которых реакционная смесь становится гомогенной. Добавляют 1 г льда, продолжая перемешивание в течение 1 часа. Органический растворитель выпаривают под вакуумом и остаток разбавляют 5 мл Н2О. Образовавшийся осадок отфильтровывают и сушат откачкой вакуумным насосом в течение 18 часов. Полученный в результате продукт (белый порошок, 130 мг) имеет следующие характеристики: 1Н-ЯМР (400 МГц, CDCl3): Пример IX – синтез (2’R,3’R)-баккатин III-13-(3′-азидо-2′-гидрокси-3′-фенилпропионата) В 25 мл одногорлой круглодонной колбе, оборудованной магнитной мешалкой, при 25°С в 5 мл СН3ОН суспендируют 0,17 г (0,45 ммоль, 1 экв.) (2’R,3’R)-баккатин III-13-(3′-фенил-2′,3′-эпоксипропионата). Последовательно добавляют 0,63 мл Н2О, 0,23 мл НСООСН3 и 0,36 г (5,5 ммоль, 12,5 экв.) азида натрия. Смесь нагревают до 50°С и ход реакции контролируют ТСХ (SiO2, CHCl3/EtOAc/MeOH, 12,0:2,0:0,3). Реакционная смесь после 46 час имеет желто-коричневую окраску с белым осадком (непрореагировавший NaN3). Добавляют Н2О (10 мл) и осажденное молочно-белое твердое вещество отфильтровывают, промывают водой и затем AcOEt. Разделяют на две фазы, водную три раза экстрагируют AcOEt и объединенные органические фазы концентрируют и сушат над MgSO4, фильтруют и растворитель выпаривают, получая 0,20 г желтовато-белого порошка. Полученное в результате неочищенное вещество чистят флэш-хроматографией (SiO2, CHCl3/EtOAc/MeOH, 12:2:0,3), получая 0,140 г (2’R,3’R)-баккатин III-13-(3′-азидо-2′-гидрокси-3′-фенилпропионата). Соединение имеет такие же спектроскопические характеристики, как соединение, полученное в примере III. ПРИМЕР А: 10-деацетил-7,10-бистрихлорацетилбаккатин III 10-DAB III (10 г, 18,38 ммоль) суспендируют в СН2Cl2 (120 мл), добавляют ДМАР (220 мг, 1,4 ммоль, 0,1 экв.) и смесь охлаждают до 0°С на ледяной бане. Затем добавляют Et3N (10,26 мл, 73,6 ммоль, 4 экв.) и, сразу после этого, добавляют Cl3COCl (4,12 мл, 36,8 ммоль, 2 экв.) в струе азота за 5 мин, поддерживая температуру ниже 10°С. После завершения добавления смесь оставляют при перемешивании на ледяной бане в течение 15 мин, затем баню удаляют и смесь перемешивают при комнатной температуре в течение 1 часа. Через 1 ч реакцию контролируют ТСХ (AcOEt 2/н-гексан 3, Rf 10-ДАБ 111=0,05, Rf 7,10-бистрихлорацетил-10-ДАБ 111=0,26) и добавляют Cl3CCOCl (1 мл, 0,5 экв.). Перемешивание поддерживают при комнатной температуре в течение 10 мин, затем смесь выливают в химический стакан, содержащий 160 г измельченного льда, перемешивая при комнатной температуре до уравновешивания (приблизительно 1 ч). После этого водную фазу отделяют и экстрагируют СН2Cl2 (3×40 мл). Объединенные органические фазы промывают 1N HCl (20 мл), затем насыщенным раствором NaHCO3 (20 мл), сушат над Na2SO4, получая бледно-желтое твердое веществ (16,5 г), у которого после кристаллизации: [ ПРИМЕР В: Получение баккатина-III (7):
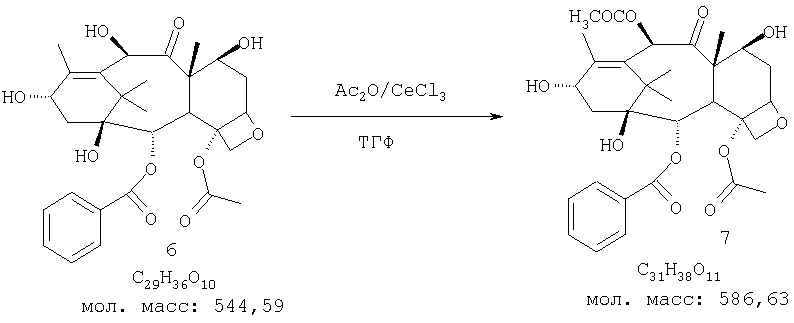
К суспензии 10-дезацетилбаккатина-III (6,382 г, 70 ммоль) в свежеперегнанном сухом ТГФ (350 мл) и уксусном ангидриде (26,5 мл, 280 ммоль) добавляют CeCl3·7Н2O (2,62 г, 7,02 ммоль). Реакционную смесь перемешивают при комнатной температуре. После перемешивания в течение 2 ч реакционная смесь становится однородной, но согласно ТСХ реакция была завершена. Таким образом, перемешивание продолжают в течение ночи при комнатной температуре. В этот момент не остается исходного вещества, в то время как некоторое количество продукта выкристаллизовывается. Смесь гасят ледяной водой (100 мл) и перемешивают в течение 1 ч. Реакционную смесь концентрируют на роторном испарителе для удаления ТГФ и затем выливают в ледяную воду (200 мл). Осажденное твердое вещество фильтруют, промывают водой (2×250 мл) и сушат в вакуумной печи над P2O5 в течение ночи. Выход соединения 7 составляет 41,0 г (100%), 1H-ЯМР-6.
Формула изобретения
1. Способ получения паклитаксела, который включает следующие стадии: а) защиты гидроксилов 10-деацетилбаккатина III (10-ДАБ III) в 7- и 10-положениях
где R=R’ = трихлорацетил, или R’ = ацетил, a R выбран из третбутоксикарбонила и трихлорацетила, b) этерификации гидроксила в 13-положении 3-фенил-2-эпоксипропионовой кислотой
с) удаления защитных групп в положениях 7 и 10 (1), если они обе представляют собой трихлорацетильные группы, с последующим селективным ацетилированием в 10-положении (2) и раскрытием эпоксида азидом натрия (3),
или, альтернативно, с’) если R’= ацетил и R= трихлорацетил, раскрытия эпоксида азидом натрия и одновременного снятия защиты в положении 7
d) восстановления азидогруппы в аминогруппу
е) бензоилирования с получением конечного продукта
2. Способ по п.1, где 10-ДАБ III защищают в 7- и 10-положениях трихлорацетильной группой путем взаимодействия с трихлорацетилхлоридом в метиленхлориде в присутствии триэтиламина и каталитических количеств 4-диметиламинопиридина (ДМАП). 3. Способ по п.1, где 10-ДАБ III сначала ацетилируют в 10-положении реакцией с уксусным ангидридом в присутствии солей церия, скандия или иттербия, а затем защищают гидроксил в 7-положении третбутоксикарбонильной или трихлорацетильной группой. 4. Способ по п.1, где гидроксил в положении 13 этерифицируют аммонийной солью фенил-2-эпоксипропионовой кислоты в толуоле в присутствии дициклогексилкарбодиимида (ДЦК), ДМАП и п-толуолсульфокислоты. 5. Способ по п.1, где защитные группы R=R’= трихлорацетил удаляют гидроксидом аммония. 6. Способ по п.1, где эпоксид раскрывают NaN3 в водном метаноле в присутствии метилформиата. 7. Способ по п.1, где азид восстанавливают до амина водородом на катализаторе или PPh3. 8. Способ по п.1, где бензоилирование на последней стадии проводят бензойным ангидридом одновременно с восстановлением либо впоследствии на выделенном восстановленном продукте бензоилхлоридом в присутствии карбоната калия. 9. Соединения формулы
где R и R’ являются таким, как определено в п.1, или водородом, в качестве промежуточных продуктов.
|
||||||||||||||||||||||||||
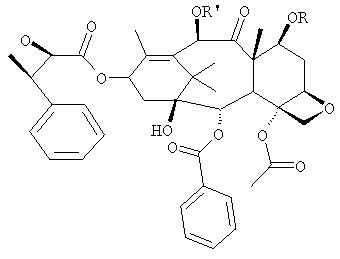
 -лактамного предшественника с 7-О-триэтилсилилбаккатином III, с последующим мягким кислотным гидролизом.
-лактамного предшественника с 7-О-триэтилсилилбаккатином III, с последующим мягким кислотным гидролизом.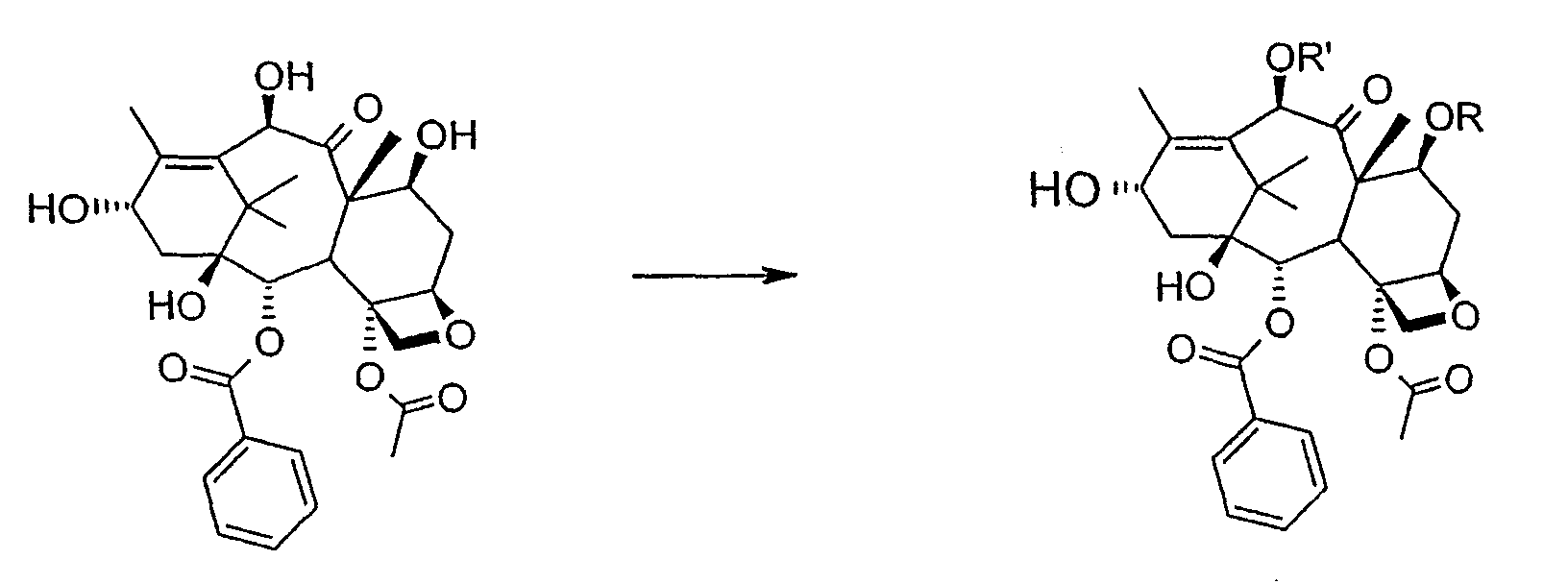
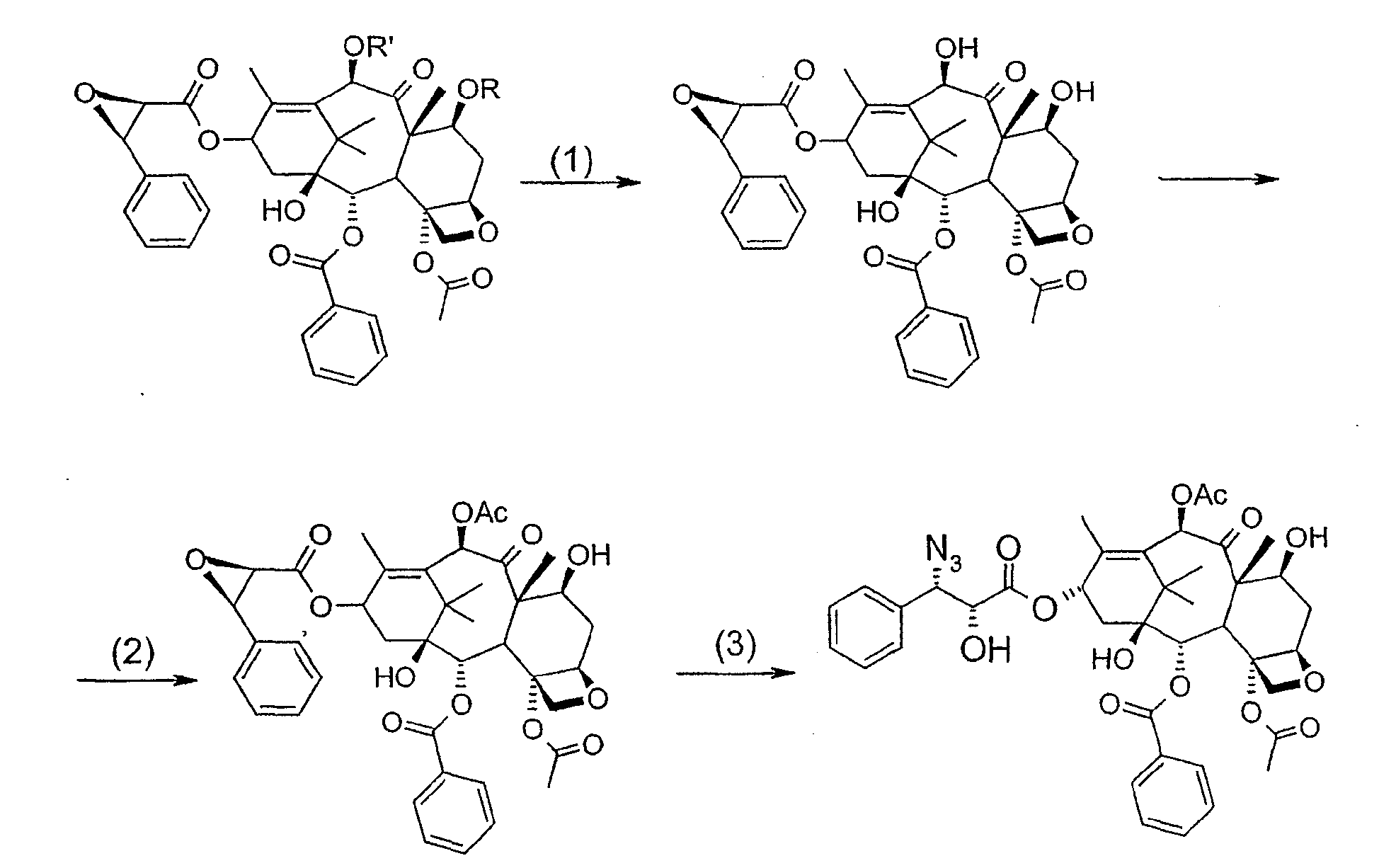
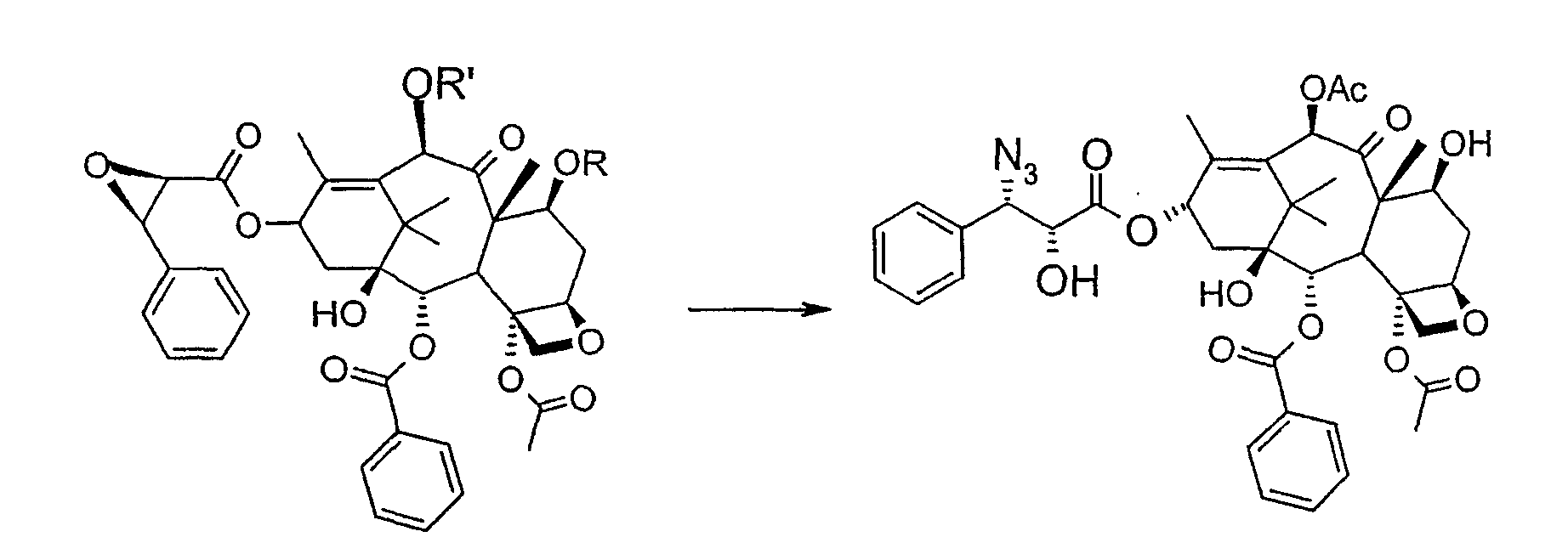
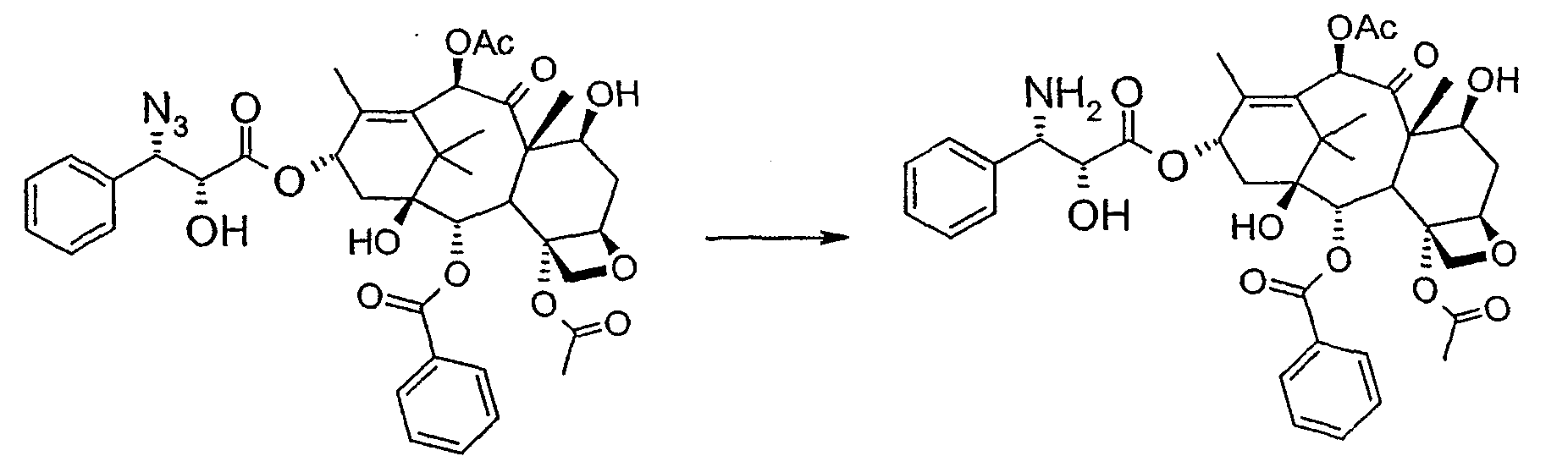
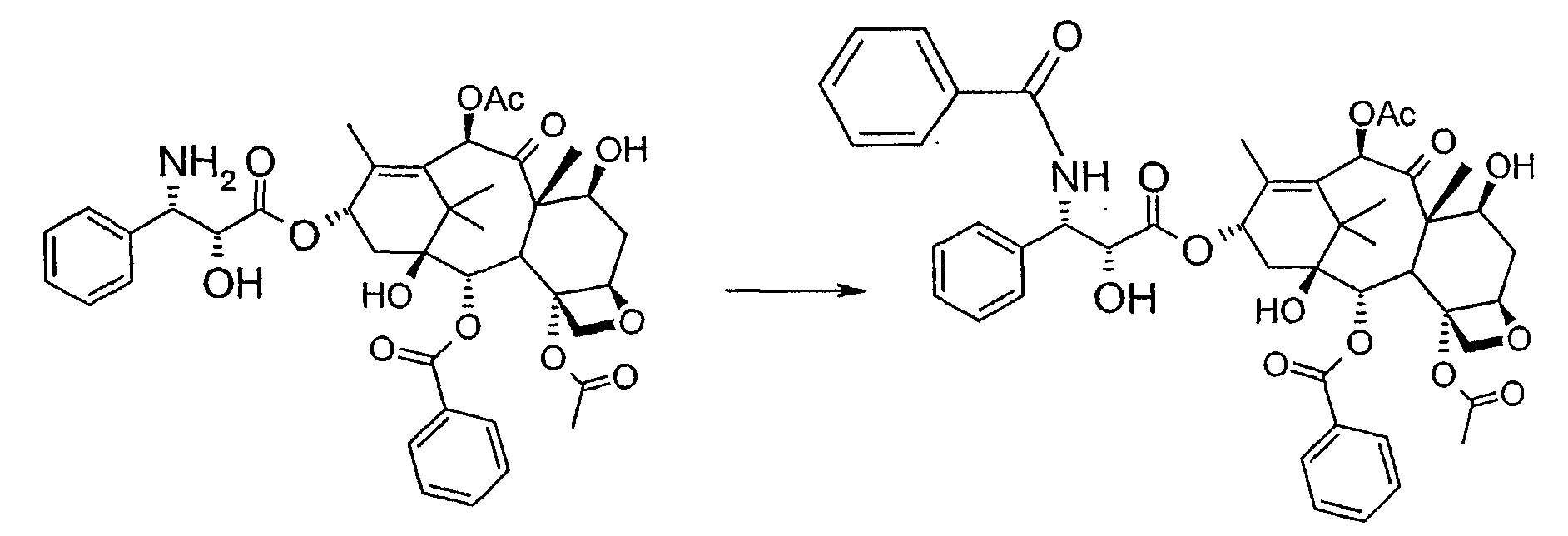
 R’, сначала 10-ДАБ III селективно ацетилируют уксусным ангидридом в присутствии солей церия, скандия, иттербия, предпочтительно CeCl3·7H2O. Полученный в результате баккатин III затем защищают в положении С-7 трет-бутоксикарбонильной или трихлорацетильной группой. Первая может быть введена реакцией баккатина III с трет-бутоксипирокарбонатом в присутствии ДМАП и этилдиизопропиламина или, альтернативно, следуя процедуре, описанной в US 5917062. Трихлорацетильная группа может быть введена в положение 7 реакцией с трихлорацетилхлоридом в пиридине.
R’, сначала 10-ДАБ III селективно ацетилируют уксусным ангидридом в присутствии солей церия, скандия, иттербия, предпочтительно CeCl3·7H2O. Полученный в результате баккатин III затем защищают в положении С-7 трет-бутоксикарбонильной или трихлорацетильной группой. Первая может быть введена реакцией баккатина III с трет-бутоксипирокарбонатом в присутствии ДМАП и этилдиизопропиламина или, альтернативно, следуя процедуре, описанной в US 5917062. Трихлорацетильная группа может быть введена в положение 7 реакцией с трихлорацетилхлоридом в пиридине. м.д. = 1,08 (с, 3Н, Ме), 1,13 (с, 3Н, Ме), 1,86 (с, 3Н, Ме), 1,97 (ддд, 1 Н, J1=14,4 Гц, J2=10,3 Гц, J3=1,9 Гц, С6-Н), 2,13 (д, 3Н, J= 1,2 Гц, Ме), 2,15 (с, 3Н, Ме), 2,30 (с, 3Н, Ме), 2,32-2,28 (м, 2Н, С14-Н2), 2,68 (ддд, 1Н, J1=14,4 Гц, J2=9,3 Гц, J3=7,3 Гц, С6-Н), 4,04 (д, 1Н, J= 7,0 Гц, С3-Н), 4,17 (дд, 1Н, J1=8,4 Гц, J2=1,0 Гц, С20-Н), 4,34 (д, 1Н, J=8,4 Гц, С20-Н), 4,86 (т, 1Н, J=7,5 Гц, С13-Н), 4,98 (дд, 1Н, J1=9,5 Гц, J2=1,7 Гц, С5-Н), 5,65 (д, 1Н, J=7,0 Гц, С2-Н), 5,70 (дд, 1Н, J1=10,4 Гц, J2=7,4 Гц, С7-Н), 6,42 (с, 1Н, С10-Н), 7,52-7,46 (м, 2Н, аром), 7,62 (м, 1Н, аром), 8,10 (м, 2Н, аром); 13С-ЯМР (100 МГц, CDCl3):
м.д. = 1,08 (с, 3Н, Ме), 1,13 (с, 3Н, Ме), 1,86 (с, 3Н, Ме), 1,97 (ддд, 1 Н, J1=14,4 Гц, J2=10,3 Гц, J3=1,9 Гц, С6-Н), 2,13 (д, 3Н, J= 1,2 Гц, Ме), 2,15 (с, 3Н, Ме), 2,30 (с, 3Н, Ме), 2,32-2,28 (м, 2Н, С14-Н2), 2,68 (ддд, 1Н, J1=14,4 Гц, J2=9,3 Гц, J3=7,3 Гц, С6-Н), 4,04 (д, 1Н, J= 7,0 Гц, С3-Н), 4,17 (дд, 1Н, J1=8,4 Гц, J2=1,0 Гц, С20-Н), 4,34 (д, 1Н, J=8,4 Гц, С20-Н), 4,86 (т, 1Н, J=7,5 Гц, С13-Н), 4,98 (дд, 1Н, J1=9,5 Гц, J2=1,7 Гц, С5-Н), 5,65 (д, 1Н, J=7,0 Гц, С2-Н), 5,70 (дд, 1Н, J1=10,4 Гц, J2=7,4 Гц, С7-Н), 6,42 (с, 1Н, С10-Н), 7,52-7,46 (м, 2Н, аром), 7,62 (м, 1Н, аром), 8,10 (м, 2Н, аром); 13С-ЯМР (100 МГц, CDCl3):  ,3′-эпоксипропионата), имеющего следующие спектроскопические характеристики:
,3′-эпоксипропионата), имеющего следующие спектроскопические характеристики: ]D-34° (CH2Cl2 C5,8); ИК(KBr) 3517, 1771, 1728, 1240, 981, 819, 787, 675 см-1; 1H-ЯМР (200 МГц):
]D-34° (CH2Cl2 C5,8); ИК(KBr) 3517, 1771, 1728, 1240, 981, 819, 787, 675 см-1; 1H-ЯМР (200 МГц):