Патент на изобретение №2155764
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ 8-ОКСО-5,8-ДИГИДРО-6H-ДИБЕНЗО[A,G]ХИНОЛИЗИН-13-ПРОПАНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ
(57) Реферат: Изобретение относится к соединениям общей формулы I 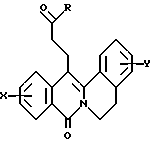 в которой Х представляет собой атом водорода или галогена, (С1-С3)алкильную группу, одну или две (С1-С3)алкокси-группы, или трифторметильную группу, У – атом водорода или галогена, (С1-С3)алкильная или (С1-С3)алкоксигруппа, R представляет собой гидрокси-группу, метоксигруппу, или группу общей формулы NR2R3, в которой R2 и R3 каждый независимо от другого представляет собой атом водорода, (С1-С4)алкильную группу, 2-метоксиэтильную группу, 3-метоксиэтильную группу, 3-аминопропильную группу, группу 2-(диметиламино)этил, группу 3-(диметиламино)пропил или группу 2-пиперидин-2-илэтил, или R2 и R3 образуют вместе с тем атомом азота, с которым они соединены, морфолиновое, пирролидиновое или пиперазиновое кольцо, которое может иметь в положении 4 заместитель в виде метильной группы или группы (1,1-диметилэтокси)карбонил, в виде свободного основания или соли, образованной присоединением кислоты. Соединения формулы I обладают активностью связывания  (бензодиазепиновых)рецепторов, ассоциированных с GABAA рецепторами, и могут найти применения в терапии в качестве лекарств для лечения заболеваний, связанных с нарушением GABAA – эргического переноса GABAA рецепторов, субъединатами (бензодиазепиновых)рецепторов, ассоциированных с GABAA рецепторами, и могут найти применения в терапии в качестве лекарств для лечения заболеваний, связанных с нарушением GABAA – эргического переноса GABAA рецепторов, субъединатами  1, 2, 3 и 5. Предложен также способ получения указанных соединений и фармацевтический состав на их основе. 3 с.п. ф-лы, 1 табл. 1, 2, 3 и 5. Предложен также способ получения указанных соединений и фармацевтический состав на их основе. 3 с.п. ф-лы, 1 табл.
Предметом настоящего изобретения являются производные 8-оксо-5,8-дигидро-6H-дибензо[a, g] хинолизин-13-пропановой кислоты, их получение и применение в терапии. Соединение в соответствии с настоящим изобретением отвечает общей формуле (I) 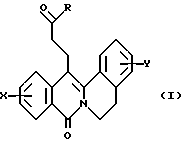 в которой X представляет собой атом водорода или галогена или (C1-C3)алкильную группу, одну или две (C1-C3)алкокси-группы или трифторметильную группу, Y представляет собой атом водорода или галогена, (C-C3)алкильную группу или (C1-C3)алкокси-группу, R представляет собой гидрокси-группу, метокси-группу или группу общей формулы NR2R3, где R2 и R3, независимо один от другого, каждый представляет собой атом водорода, (C1-C4)алкильную группу, 2-метоксиэтильную группу, 2-аминоэтильную группу, 3-аминопропильную группу, группу 2-(диметиламино)этил, группу 3-(диметиламино)пропил или группу 2-пиперидин-1-илэтил, или же R2 и R3 вместе с атомом азота, с которым они соединены, образуют морфолиновое, пирролидиновое или пиперазиновое кольцо, которое может иметь заместитель в положении 4 в виде метильной группы или (1,1-диметилэтокси)карбонильной группы. Эти соединения могут в известных случаях существовать виде оснований или солей, образованных присоединением кислот. В соответствии с изобретением соединения общей формулы (I) можно получить по способу, показанному на схеме 1 (см. в конце описания). Ангидрид общей формулы (II), в котором X такой, как определено ранее, приводят во взаимодействие с имином формулы (III), в которой Y такой, как определено ранее, в ароматическом растворителе, например толуоле, при температуре от 70 до 100oC. Получают кислоту общей формулы (IV), которую этерифицируют действием тионилхлорида в метаноле при температуре от 20 до 50oC, затем полученный таким образом сложный эфир общей формулы (V) превращают в спирт общей формулы (VI), например с помощью смешанного гидрида, такого как борогидрид натрия, в эфирном растворителе, таком как тетрагидрофуран. Затем спирт окисляют в альдегид общей формулы (VII), например по методу Сверна, полученный альдегид обрабатывают метиловым эфиром (диметоксифосфинил)уксусной кислоты (МЭДФУК) в эфирном растворителе, таком как тетрагидрофуран, при температуре от 20 до 75oC, выделяют полученный таким образом сложный эфир общей формулы (VIII) и окисляют его в сложный эфир общей формулы (IX) с помощью хинона, например 2,3-дихлор-5,6-дицианциклогекса-2,5-диен-1,4-диона, например в ароматическом растворителе, таком как толуол, при температуре от 70 до 110oC, и, наконец, сложный эфир общей формулы (IX) подвергают гидрогенизации в присутствии палладированного угля, с получением сложного эфира общей формулы (Ia), которая соответствует общей формуле (I), где R представляет собой метокси-группу. Этот сложный эфир затем может быть превращен, как показано на схеме 2 (см. в конце описания): если хотят получить соединение общей формулы (I), где R представляет собой гидрокси-группу, сложный эфир общей формулы (Ia) подвергают гидролизу в щелочной среде с получением кислоты общей формулы (Ib) и, если хотят получить соединение общей формулы (I), где R представляет собой группу общей формулы NR2R3, кислоту общей формулы (Ib) обрабатывают амином общей формулы HNR2R3, проходя через промежуточный имидазолид, полученный in situ с помощью N,N-карбонилдиимидазола. В случае соединения общей формулы (I), где X и Y отличаются от алкокси-группы, можно применить другой способ, показанный на схеме 3 (см. в конце описания). Дибензо[a, g] хинолизин общей формулы (X) подвергают обработке оксихлоридом фосфора в N,N-диметилформамиде при температуре от 20 до 130oC, с получением альдегида общей формулы (XI), затем этот альдегид обрабатывают метиловым эфиром (диметоксифосфинил)уксусной кислоты в эфирном растворителе, таком как тетрагидрофуран при температуре от 20 до 65oC, с получением сложного эфира общей формулы (IX), который окончательно обрабатывают, как описано по этому поводу в схеме 1. Исходный ангидрид общей формулы (II) доступен в продаже, когда X представляет собой водород, и, в других случаях, его можно получить методом, таким как метод, описанный в Arch. Pharm. (1991) 324 509-518. Замещенные имины общей формулы (III) могут быть получены способами, аналогичными описанным в Synthesis (1974) 4 288-289 и Heterocycles (1982) 19(4) 653-656. Хинолизины общей формулы (X) могут быть получены методами, аналогичными описанным в Synthesis (1980) 10 845-847 и в Tetrahedron Lett. (1992) 33(38) 5653-5654. Приводимые далее примеры иллюстрируют получение некоторых соединений согласно изобретению. Элементные микроанализы, ИК- и ЯМР-спектры подтверждают структуры полученных соединений. Цифры, указанные в скобках в заголовках примеров, соответствуют цифрам в первой колонке таблицы, приводимой далее. В названиях соединений тире “-” составляет часть слова, а тире “_” служит только для переноса в конце строки; его следует опустить при отсутствии переноса и нельзя заменять ни обычным тире, ни пробелом. Пример 1 (Соединение N 1) Метиловый эфир 8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин-13-пропановой кислоты. 1.1. 8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин- 13-карбоксальдегид. Охлаждают до 0oC, в атмосфере аргона, 150 мл сухого N,N-диметилформамида, добавляют по каплям 5 мл (53,6 ммоль) оксихлорида фосфора, смесь перемешивают в течение 30 мин при комнатной температуре, добавляют 5 г (20,2 ммоль) 5,6-дигидро-8H-дибензо[a,g]хинолизин-8-она, смесь постепенно нагревают до 110oC и выдерживают при этой температуре в течение 6 ч. Охлаждают ее до комнатной температуры, выпаривают растворитель при пониженном давлении, добавляют к остатку лед и 30%-ный раствор гидроксида натрия, экстрагируют смесь дихлорметаном, сушат органическую фазу над сульфатом натрия, фильтруют ее и выпаривают растворитель при пониженном давлении. Остаток очищают хроматографически на колонке из силикагеля, элюируя смесью циклогексан/дихлорметан, меняя состав от 100/0 до 0/100, затем смесью дихлорметан/этилацетат состава от 100/0 до 80/20. После перекристаллизации из циклогексана выделяют 3,6 г (13,08 ммоль) альдегида в виде твердого белого вещества. Температура плавления: 209-210oC. 1.2. Метиловый эфир (E)-3-(8-окса-5,8-дигидро-6H-дибензо[a, g]хинолизин-13-ил)проп-2-еновой кислоты. В атмосфере аргона промывают 0,5 г (12,5 ммоль) 60%-ной суспензии гидрида натрия пентаном, затем приготовляют из него суспензию в 150 мл сухого тетрагидрофурана, охлаждают смесь на ледяной бане, добавляют по каплям 2 г (11 ммоль) метилового эфира (диметоксифосфинил)уксусной кислоты, смесь перемешивают в течение 15 мин при 0oC, добавляют 2,78 г (10,1 ммоль) 8-оксо-5,8-дигидро-6H-дибензо[a, g] хинолизин-13-карбоксальдегида, смесь постепенно нагревают с обратным холодильником до кипения и кипятят в течение 6 ч. Охлаждают смесь до комнатной температуры, добавляют несколько мл метанола, выпаривают растворители при пониженном давлении, добавляют к остатку лед, 200 мл дихлорметана и 1 М раствор соляной кислоты, отделяют органическую фазу, сушат ее над сульфатом натрия, фильтруют и выпаривают растворитель при пониженном давлении. Остаток очищают хроматографически на колонке из силикагеля, элюируя смесью дихлорметан/этилацетат, меняя состав от 100/0 до 80/20, и перекристаллизовывают продукт из циклогексана. Выделяют 2,78 г (8,4 ммоль) , -ненасыщенного сложного эфира в виде белых кристаллов.
Температура плавления: 201-202,5oC.
1.3. Метиловый эфир 8-оксо-5,8-дигидро-6H-дибензо[a, g]хинолизин-13-пропановой кислоты.
К раствору 2,8 г (8,4 ммоль) метилового эфира (E)-3-(8-оксо-5,8-дигидро-6H-дибензо[a, g] хинолизин-13-ил)проп-2-еновой кислоты в 150 мл уксусной кислоты добавляют 0,23 г 5%-ного палладированного угля, и суспензию подвергают гидрогенизации в аппарате Парра при давлении около 0,32 MPa при комнатной температуре в течение 30 мин, затем при 40-45oC в течение 2 ч 30 мин.
Катализатор отделяют фильтрованием, концентрируют фильтрат при пониженном давлении, к остатку добавляют 250 мл дихлорметана, ледяную воду и насыщенный раствор гидрокарбоната натрия, органическую фазу отделяют, промывают ее водой и сушат над сульфатом натрия, фильтруют и выпаривают растворитель при пониженном давлении. Остаток очищают хроматографически на колонке из силикагеля, используя как элюент смесь дихлорметана/этилацетат от 100/0 до 70/30, затем смесь дихлорметан/метанол от 95/5 до 90/10, и получают 2,8 г (8,4 ммоль) маслообразного продукта. После перекристаллизации из циклогексана выделяют 2,5 г (7,5 ммоль) сложного эфира в виде белых кристаллов.
Температура плавления: 157-158,5oC.
Пример 2 (Соединение N 2) -ненасыщенного сложного эфира в виде белых кристаллов.
Температура плавления: 201-202,5oC.
1.3. Метиловый эфир 8-оксо-5,8-дигидро-6H-дибензо[a, g]хинолизин-13-пропановой кислоты.
К раствору 2,8 г (8,4 ммоль) метилового эфира (E)-3-(8-оксо-5,8-дигидро-6H-дибензо[a, g] хинолизин-13-ил)проп-2-еновой кислоты в 150 мл уксусной кислоты добавляют 0,23 г 5%-ного палладированного угля, и суспензию подвергают гидрогенизации в аппарате Парра при давлении около 0,32 MPa при комнатной температуре в течение 30 мин, затем при 40-45oC в течение 2 ч 30 мин.
Катализатор отделяют фильтрованием, концентрируют фильтрат при пониженном давлении, к остатку добавляют 250 мл дихлорметана, ледяную воду и насыщенный раствор гидрокарбоната натрия, органическую фазу отделяют, промывают ее водой и сушат над сульфатом натрия, фильтруют и выпаривают растворитель при пониженном давлении. Остаток очищают хроматографически на колонке из силикагеля, используя как элюент смесь дихлорметана/этилацетат от 100/0 до 70/30, затем смесь дихлорметан/метанол от 95/5 до 90/10, и получают 2,8 г (8,4 ммоль) маслообразного продукта. После перекристаллизации из циклогексана выделяют 2,5 г (7,5 ммоль) сложного эфира в виде белых кристаллов.
Температура плавления: 157-158,5oC.
Пример 2 (Соединение N 2)8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин-13-пропановой кислоты. В колбу емкостью 100 мл вводят 2,45 г (7,35 ммоль) метилового эфира 8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин-13-пропановой кислоты, растворенной в 40 мл этанола, добавляют 2 мл 30%-ного водного раствора гидроксида натрия и смесь кипятят с обратным холодильником в течение 3 ч. Растворитель выпаривают при пониженном давлении, остаток обрабатывают водой и 30%-ной соляной кислотой, собирают фильтрованием выпавшую твердую фазу, промывают ее водой и сушат. Получают 2,2 г (6,89 ммоль) кислоты. Температура плавления: 269-271oC (разложение). Пример 3 (Соединение N 12) N,N-диметил-8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин-13-пропанамид. В колбе емкостью 250 мл в атмосфере аргона готовят суспензию 1,2 г (3,76 ммоль) 8-оксо-5,8-дигидро-6H-дибензо[a, g]хинолизин-13-пропановой кислоты в 100 мл дихлорметана, добавляют 1,2 г (7,4 ммоль) N,N’-карбонилдиимидазола и перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь насыщают газообразным диметиламином в течение 1 мин и оставляют при перемешивании в течение 12 ч. Растворитель выпаривают при пониженном давлении, к остатку добавляют 200 мл дихлорметана, промывают раствор 1 н. соляной кислотой, затем водой, затем 1 н. раствором гидроксида натрия, затем снова водой, сушат над сульфатом натрия, фильтруют, упаривают растворитель при пониженном давлении и очищают остаток хроматографически на колонке из силикагеля, используя как элюент смесь дихлорметан/метанол от 100/0 до 90/10. После перекристаллизации из смеси ацетонитрил/дихлорметан получают 0,93 г (2,68 ммоль) твердого белого вещества. Температура плавления: 192,5-193,5oC. Пример 4 (Соединение N 38) 10-метокси-8-оксо-5,8-дигидро-6H-дибензо[a, g] хинолизин-13-пропановая кислота. 4.1. 10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H-дибензо[a,g]хинолизин-13-карбоновая кислота. В колбу емкостью 500 мл вводят 8,1 г (42,15 ммоль) 7-метоксигомофталевого ангидрида в растворе в 350 мл толуола, добавляют 5,8 г (44,2 ммоль) 3,4-дигидроизохинолина, и смесь кипятят с обратным холодильником в течение 3 ч. Дают остыть до комнатной температуры и оставляют на ночь. Собирают образовавшиеся кристаллы, промывают их диэтиловым эфиром и сушат в вакууме. Получают 12,75 г (39,43 ммоль) кислоты в виде смеси транс/цис приблизительно 90/10. Температура плавления: 227-232oC. 4.2. Метиловый эфир транс-10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H- дибензо[a,g]хинолизин-13-карбоновой кислоты. В трехгорлой колбе емкостью 500 мл готовят суспензию 14 г (43,3 ммоль) 10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H-дибензо[a, g] хинолизин-13-карбоновой кислоты в 100 мл метанола, добавляют по каплям 3,2 мл (44,11 ммоль) тионилхлорида и кипятят смесь с обратным холодильником в течение 4 ч. Оставляют ее при комнатной температуре на ночь. Образовавшиеся кристаллы собирают, промывают диэтиловым эфиром и сушат в вакууме. Получают 11,5 г (34,08 ммоль) чистого транс-эфира. Температура плавления: 154,5-157,5oC. 4.3. Транс-13-(гидроксиметил)-10-метокси-5,6,13,13a-тетрагидро-8H-дибензо[a,g]хинолизин-8-он. В колбе емкостью 250 мл приготовляют суспензию, исходя из 9,85 г (29,2 ммоль) метилового эфира транс-10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H-дибензо[a, g] хинолизин-13-карбоновой кислоты и 75 мл тетрагидрофурана, добавляют последовательно 4,68 г (146 ммоль) метанола и 5,52 г (146 ммоль) борогидрида натрия и перемешивают смесь при комнатной температуре в течение 4 ч. Гидролизуют смесь, добавляя 100 мл воды, экстрагируют трижды дихлорметаном (150 мл), сушат органическую фазу над сульфатом натрия, упаривают растворитель при пониженном давлении и очищают остаток хроматографически на колонке силикагеля, используя как элюент смесь дихлорметан/этилацетат от 100/0 до 50/50. После выпаривания растворителей извлекают кристаллы этиловым эфиром и окончательно получают 8,3 г (26,83 ммоль) транс-спирта. Температура плавления: 180-181,5oC. 4.4. 10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H-дибензо[a,g]хинолизин-13-карбоксальдегид. В трехгорлую колбу объемом 500 мл в атмосфере аргона вводят 20,2 г (159 ммоль) оксалилхлорида и 100 мл дихлорметана, охлаждают все до -70oC, добавляют по каплям 16,36 г (209 ммоль) диметилсульфоксида, растворенного в 50 мл дихлорметана, и все перемешивают при -70oC в течение 15 мин. Добавляют на протяжении 30 мин 8,2 г (26,68 ммоль) транс-13-(гидроксиметил)-10-метокси-5,6,13,13а-тетрагидро-8H- дибензо[a, g] хинолизин-8-он в растворе в 150 мл дихлорметана, и смесь перемешивают при -60oC в течение 30 мин. Добавляют 21,2 г (209 ммоль) триэтиламина, растворенного в 50 мл дихлорметана, и оставляют смесь нагреваться до комнатной температуры. Выливают ее в 500 мл насыщенного водного раствора хлорида натрия и экстрагируют трижды дихлорметаном (200 мл). Органическую фазу сушат над сульфатом натрия, фильтруют и упаривают растворители при пониженном давлении. Остаток очищают хроматографически на колонке силикагеля, используя как элюент смесь дихлорметан/циклогексан 2/1, затем 1/0, затем смесью дихлорметан/этилацетат от 90/10 до 50/50. После кристаллизации из смеси этилацетата и петролейного эфира получают 8,02 г (26,09 ммоль) альдегида в смеси транс/цис 80/20. Температура плавления: 146-149oC. 4.5. Метиловый эфир (E)-3-(10-метокси-8-оксо-5,8,13,13a- тетрагидро-6H-дибензо[a,g]хинолизин-13-ил)проп-2-еновой кислоты. В трехгорлой колбе емкостью 500 мл промывают пентаном 1,3 г (32,5 ммоль) гидрида натрия в виде 60%-ной суспензии в масле, приготовляют из него суспензию в 200 мл тетрагидрофурана, охлаждают суспензию до 0oC, добавляют по каплям 5,2 г (28,55 ммоль) метилового эфира (диметоксифосфинил)уксусной кислоты и продолжают перемешивать при приблизительно 10oC в течение 20 мин. Добавляют 8 г (26 ммоль) 10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H- дибензо[a, g] хинолизин-13-карбоксальдегид и смесь кипятят с обратным холодильником в течение 4 ч 30 мин. Дают смеси остыть до комнатной температуры, добавляют несколько капель метанола, выпаривают растворители при пониженном давлении, добавляют к остатку 250 мл воды и экстрагируют трижды дихлорметаном (150 мл). Сушат органическую фазу над сульфатом натрия, фильтруют, выпаривают растворитель при пониженном давлении и остаток очищают хроматографически на колонке из силикагеля, элюируя смесью этилацетат/циклогексан от 1/2 до 3/1, и получают 6,55 г (18,02 ммоль) сложного эфира в виде масла, которое используют как есть на следующем этапе. 4.6. Метиловый эфир (E)-3-(10-метокси-8-оксо-5,8-дигидро-6H-дибензо[a,g] хинолизин-13-ил)проп-2-еновой кислоты. В колбе объемом 500 мл растворяют 5,5 г (15,13 ммоль) метилового эфира (E)-3-(10-метокси-8-оксо-5,8,13,13a-тетрагидро-6H- дибензо[a, g]хинолизин-13-ил)проп-2-еновой кислоты в 150 мл толуола, добавляют 5,15 г (22,7 ммоль) 2,3-дихлор-5,6-дицианциклогекса-2,5- диен-1,4-диона и кипятят смесь с обратным холодильником в течение 2 ч 30 мин. Дают остыть до комнатной температуры, выпаривают растворитель при пониженном давлении и остаток очищают хроматографически на колонке силикагеля, элюируя смесью дихлорметан/этилацетат состава от 100/0 до 80/20. Извлекают полученное масло с помощью смеси диэтилового эфира и петролейного эфира, и выделяют 3,31 г (9,2 ммоль) соединения в виде желто-оранжевых кристаллов. Температура плавления: 149-152oC. 4.7. Метиловый эфир 10-метокси-8-оксо-5,8-дигидро-6H- дибензо[a,g]хинолизин-13-пропановой кислоты. К раствору 3,5 г (9,68 ммоль) метилового эфира (E)-3-(10-метокси-8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин-13-ил)проп-2-еновой кислоты в 100 мл уксусной кислоты добавляют 0,5 г 5%-ного палладированного угля и суспензию гидрогенизируют в аппарате Парра при давлении около 0,32 МПа при температуре от 50 до 60oC в течение 3 ч. Дают остыть до комнатной температуры, отделяют катализатор фильтрованием, фильтрат концентрируют при пониженном давлении, добавляют к остатку 250 мл дихлорметана, промывают раствор насыщенным водным раствором гидрокарбаната натрия, сушат над сульфатом натрия, фильтруют и упаривают растворители при пониженном давлении. Остаток очищают хроматографически на колонке силикагеля, элюируя смесью дихлорметана/этилацетат состава от 100/0 до 50/50, и получают 3,4 г (9,36 ммоль) сложного эфира в виде масла, которое используют как есть на следующем этапе. 4.8. 10-метокси-8-оксо-5,8-дигидро-6H-дибензо[a, g]хинолизин-13- пропановая кислота. В колбу емкостью 100 мл вводят 3,4 г (9,36 ммоль) метилового эфира 10-метокси-8-оксо-5,8-дигидро-6H-дибензо[a, g]хинолизин-13-пропановой кислоты в растворе в 50 мл этанола, добавляют 3 мл 30%-ного водного раствора гидроксида натрия и смесь кипятят с обратным холодильником в течение 3 ч. Выпаривают растворители при пониженном давлении, к остатку добавляют воду и 30%-ную соляную кислоту, собирают выпавшую в осадок твердую фазу, промывают ее водой и сушат. В результате получают 3,0 г (8.58 ммоль) кислоты. Температура плавления: 260-264oC. Пример 5 (Соединение N 40) 10-метокси-N-метил-8-оксо-5,8-дигидро-6H-ди6ензо[a, g]хинолизин-13- пропанамид. В колбе емкостью 250 мл в атмосфере аргона приготовляют суспензию 1 г (2,86 ммоль) 10-метокси-8-оксо-5,8-дигидро-6H- дибензо[a,g]хинолизин-13-пропановой кислоты в 100 мл дихлорметана, добавляют 0,93 г (5,72 ммоль) N, N’-карбонилдиимидазола и перемешивают смесь при комнатной температуре в течение 2 ч. Затем смесь насыщают газообразным метиламином в течение 1 мин и продолжают перемешивание в течение 22 ч. Упаривают растворитель при пониженном давлении, к остатку добавляют 200 мл дихлорметана, раствор промывают водой, затем 1 н. раствором соляной кислоты, затем 1 н. раствором гидроксида натрия, снова водой, сушат над сульфатом натрия, фильтруют, выпаривают растворитель при пониженном давлении и очищают остаток хроматографически на колонке из силикагеля, используя как элюент смесь дихлорметан/метанол состава от 100/0 до 97/3. После перекристаллизации из этилацетата получают в результате 0,43 г (1,19 ммоль) твердого белого вещества. Температура плавления: 182-183oC. Таблица, приведенная в конце описания, иллюстрирует химические структуры и физические свойства некоторых соединений в соответствии с изобретением. Соединения в соответствии с настоящим изобретением были подвергнуты фармакологическим испытаниям, которые показали их полезность в качестве терапевтически активных веществ. Исследование мембранного связывания по отношению к совокупности (бензодиазепиновых) рецепторов, ассоциированных с GABAA рецепторами, содержащими субъединицу 5.
Эти рецепторы могут быть селективно помечены в мембранах гиппокампа крысы, инкубированных в присутствии [3H]флумазенила и 5 мкМ золпидема (для блокирования других подтипов рецепторов).
Соединения в соответствии с настоящим изобретением явились предметом исследования in vitro в отношении своего сродства к рецепторам, меченным [3H]флумазенилом.
Использовали самцов крыс OFA (Iffa Credo) весом от 200 до 250 г. После обезглавливания извлекали гиппокамп и измельчали его с помощью аппарата Ultra-TurraxTM или PolytronTM в течение 20 с при 6/10 максимальной скорости в 80 объемах 50 мМ буферного раствора Трис с pH 7,4, точно установленном с использованием соляной кислоты, содержащего 120 мМ хлорида натрия и 5 мМ хлорида калия.
Связывание с [3H] флумазенилом (1 нМ; удельная активность: 80-87 Ки/ммоль; Du Pont de Nemours/New England Nuclear) определяли инкубацией 200 мкл суспензии мембран в конечном объеме 1 мл буферного раствора, содержащего 5 мкМ золпидема и испытуемое соединение. После инкубации в течение 45 мин при 0oC мембраны выделяют фильтрованием на фильтрах Whatman GF/BTM, которые дважды промывают 5 мл ледяного буферного раствора. Жидкостной сцинтиграфией определяет величину радиоактивности, удержанной фильтром.
Специфическое связывание [3H] флумазенила определяют как величину радиоактивности, удерживаемой на фильтрах, и которая может быть ингибирована ко-инкубацией с 1 мкМ флунитразепама. Для каждой концентрации исследуемого соединения определяют процент ингибирования связывания [3H]флумазенила, затем концентрацию CI50, концентрацию, которая ингибирует 50% специфического связывания.
Соединения в соответствии с настоящим изобретением, наиболее активные в этом испытании, имеют CI50 порядка от 1 до 100 нМ.
Исследование мембранного связывания по отношению к 2 (бензодиазепиновым типа II)рецепторам, ассоциированным с GABAA рецепторами, в большинстве содержащими субъединицы 2 и 3.
Сродство соединений к 2 рецепторам спинного мозга определяли с помощью разновидности метода, описанного S.Z.Langer и S.Arbilla в Fund. Clin. Pharmacol. (1988) 2 159-170, с использованием [3H]флумазенила вместо [3H]диазепама в качестве радиоактивного лиганда.
Ткань спинного мозга гомогенизируют в течение 60 с и 30 объемах ледяного буферного раствора (50 мл Трис/HCl, pH/7,4, NaCl 120 мМ, KCl 5 мМ), затем, после разбавления 1/3 суспензию инкубируют с [3H]флумазенилом (удельная активность: 78 Ки/ммоль; New England Nuclear) в концентрации 1 нМ и с соединениями в соответствии с настоящим изобретением в разных концентрациях в конечном объеме 525 мкл. После 30 мин инкубации при 0oC образцы фильтруют в вакууме на фильтрах Whatman GF/BTM и немедленно промывают ледяным буферным раствором. Специфическое связывание [3H]флумазенила определяют в присутствии 1 мкМ немеченого диазепама. Данные анализируют обычными методами и рассчитывают CI50, концентрацию, которая ингибирует 50% связывания [3H]флумазенила.
Значения CI50 соединений согласно настоящему изобретению в этом испытании располагаются между 1 и 500 нМ.
Исследование мембранного связывания 1 (бензодиазепиновых типа I) рецепторов, ассоциированных с GABAA рецепторами, содержащими субъединицу 1.
Сродство соединений к 1 рецепторам мозжечка определяли по варианту метода, описанного S. Z. Langer и S.Arbilla в Fund. Clin. Pharmacol. (1988) 2159-170, с использованием [3H]флумазенила вместо [3H]диазепама в качестве радиоактивного лиганда.
Ткань мозжечка гомогенизируют в течение 60 с в 120 объемах ледяного буферного раствора (50 мл Трис/HCl, pH 7,4, NaCl 120 мМ, KCl 5 мМ), затем, после разбавления 1/3, инкубируют суспензию с [3H]флумазенилом (удельная активность: Ки/ммоль, New England Nuclear) в концентрации 1 нМ и с соединениями в соответствии с изобретением в разных концентрациях в конечном объеме 525 мкл. После инкубации в течение 30 мин при 0oC образцы фильтруют в вакууме на фильтрах Whatman GF/BTM и немедленно промывают ледяным буферным раствором. Специфическое связывание [3H]флумазенила определяли в присутствии 1 мкМ немеченного диазепама. Данные анализировали обычными методами и рассчитывали CI50, концентрацию, которая ингибирует 50% связывания [3H]флумазенила.
Значения CI50 соединений согласно данному изобретению в этом испытании располагаются между 1 и 500 нМ.
Результаты испытаний, проведенных с соединениями в соответствии с изобретением, показывают, что in vitro некоторые из них селективно вытесняют [3H] флумазенил из его сайтов мембранного связывания по отношению к совокупности (бензодиазепиновых) рецепторов, ассоциированных с рецепторами GABAA, содержащими субъединицу 5, по сравнению с подтипом рецепторов 1, ассоциированных с рецепторами GABAA, содержащими субъединицу 1, и по сравнению с совокупностью 2 (бензодиазепиновых типа II) рецепторов, ассоциированных с рецепторами GABAA, содержащими в большинстве субъединицы 2 и 3.
Другие соединения обладают сильным сродством к субъединицами 5, 1, 2 и 3 и не являются селективными. Другими словами, эти соединения имеют сродство– сильное к сайтам мембранного связывания [3H]флумазенила по отношению к совокупности (бензодиазепиновых) рецепторов, ассоциированных с GABAA рецепторами, содержащими субъединицу 5,– сильное, среднее или слабое к подтипам 1 (бензодиазепиновых типа I) рецепторов, ассоциированных с GABAA рецепторами, содержащими субъединицу 1,– сильное, среднее или слабое к совокупности 2 (бензодиазепиновых типа II) рецепторов, ассоциированных с GABAA рецепторами, в большинстве содержащими субъединицы 2 и 3.
Селективность, представленная соотношением CI50 1-мозжечок/CI50 -гиппокамп заключена между 1 и 25, и селективность, представленная соотношением CI50 2-костный мозг/CI50 -гиппокамп также заключена между 1 и 25.
Соединения в соответствии с изобретением могут быть использованы при лечении заболеваний, связанных с нарушением GABAA-эргического переноса GABAA рецепторов, ассоциированных с субъединицей 5. Предпочтительное распределение рецепторов, ассоциированных с субъединицей 5 GABAA рецепторного комплекса, в обонятельной луковице, в лимбических структурах, таких как гиппокамп и гипоталамус, и в спинном мозгу наводит на мысль, что соединения в соответствии с изобретением могут быть использованы при лечении расстройств обоняния, познавательной функции, гормональных нарушений, связанных с дисфункцией гипоталамуса, некоторых эмоциональных расстройств и нарушения болевого восприятия. Они могут быть использованы также при лечении спастических реакций и мышечных контрактур.
Соединения в соответствии с настоящим изобретением равным образом могут быть использованы для лечения заболеваний, связанных с нарушением GABA-эргического переноса GABAA рецепторов, связанных с субъединицами 1, 2 и 3, то есть для лечения тревоги, нарушений сна, эпилепсии и нарушений, возникающих при лечении алкоголизма. Наконец, они могут быть использованы как анестезирующие средства, как миорелаксанты или как болеутоляющие средства.
Для этого они могут быть представлены во всех галеновых формах, в сочетании с подходящими эксипиентами, для энтерального или парэнтерального введения, например в форме таблеток, драже, гелей, капсул, растворов или суспензий для питья или инъекций, суппозориев и т.д., дозированных так, чтобы дневная доза активного вещества составляла от 1 до 1000 мг.
Примеры фармацевтических составовРаствор для инъекций: Действующее начало – 5 мг; Глюкоза – 250 мг; Вода для инъекций – сколько необходимо для получения 5 мл раствора на одну 5 мл ампулу. Желатиновая капсула: Действующее начало – 100 мг; Тальк – 24 мг; Силикагель – 1 мг на одну 125 мг капсулу. Таблетка: Действующее начало – 400 мг; Силикагель – 10 мг; Стеариновая кислота – 20 мг; Кукурузный крахмал – 45 мг на одну 475 мг таблетку. Сироп: Действующее начало – 5 г; Метиловый эфир 4-гидроксибензойной кислоты – 150 мг; Сахароза – 50 г; Дистиллированная вода – сколько необходимо для получения 100 мл раствора на один 100 мл флакон. Формула изобретения
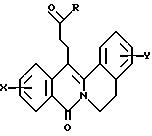 в которой X представляет собой атом водорода или галогена, (C1-C3)алкильную группу, одну или две (C1-C3)алкокси-группы, или трифторметильную группу; Y представляет собой атом водорода или галогена, (C1-C3)алкильную группу или (C1-C3)алкокси-группу; R представляет собой гидрокси-группу, метокси-группу, или группу общей формулы NR2R3, в которой R2 и R3 каждый независимо от другого представляет собой атом водорода, (C1-C4)алкильную группу, 2-метоксоэтильную группу, 2-аминоэтильную группу, 3-аминопропильную группу, группу 2-(диметиламино)этил, группу 3-(диметиламино)пропил или группу 2-пиперидин-1-илэтил, или же R2 и R3 образуют вместе с тем атомом азота, с которым они соединены, морфолиновое, пирролидиновое или пиперазиновое кольцо, которое может иметь в положении 4 заместитель в виде метильной группы или группы (1,1-диметилэтокси)карбонил, в виде свободного основания или соли, образованной присоединением кислоты. 2. Способ получения производных 8-оксо-5,8-дигидро-6H-дибензо[a,g]хинолизин-13-пропановой кислоты по п.1, отличающийся тем, что ангидрид общей формулы II 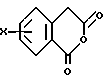 в которой X такой, как определено в п.1, приводят во взаимодействие с имином общей формулы III 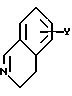 в которой Y такой, как определено в п.1, с получением кислоты общей формулы IV 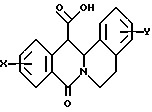 которую этерифицируют тионилхлоридом в метаноле, а затем превращают полученный таким образом сложный эфир общей формулы V 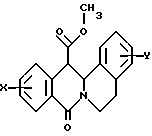 в спирт общей формулы VI 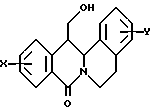 а затем этот спирт окисляют в альдегид, общей формулы VII 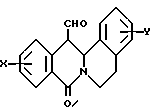 затем этот альдегид обрабатывают метиловым эфиром (диметокси-фосфинил)уксусной кислоты, и выделяют полученный таким образом сложный эфир общей формулы VIII 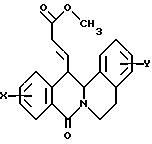 затем окисляют этот сложный эфир в сложный эфир общей формулы IX 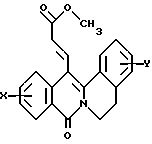 и, наконец, этот эфир подвергают каталитической гидрогенизации с получением сложного эфира общей формулы Ia 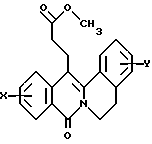 которая соответствует общей формуле I, в которой R представляет собой метоксигруппу, затем, если хотят получить соединение общей формулы I, в которой R представляет собой гидроксигруппу, сложный эфир общей формулы Ia подвергают гидролизу с получением кислоты общей формулы Ib 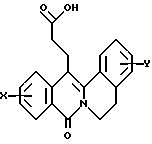 и, если хотят получить соединение общей формулы I, в которой R представляет собой группу общей формулы NR2R3, кислоту общей формулы Ib обрабатывают амином общей формулы HNR2R3. 3. Фармацевтический состав для связывания (бензодиазепиновых) рецепторов, ассоциированных с GABAA рецепторами, отличающийся тем, что он содержит соединение по п.1 в сочетании с эксципиентом.
РИСУНКИ
PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
Номер и год публикации бюллетеня: 12-2001
(73) Новое наименование патентообладателя:
Извещение опубликовано: 27.04.2001
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 29.12.2001
Извещение опубликовано: 20.11.2004 БИ: 32/2004
|
||||||||||||||||||||||||||