|
(21), (22) Заявка: 2004129763/04, 03.03.2003
(24) Дата начала отсчета срока действия патента:
03.03.2003
(30) Конвенционный приоритет:
05.03.2002 ES 200200531
(45) Опубликовано: 20.03.2006
(15) Информация о коррекции:
Версия коррекции № 1 (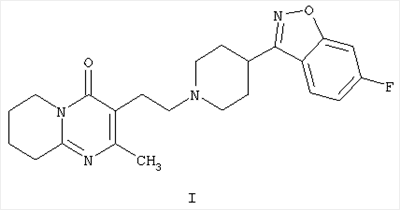
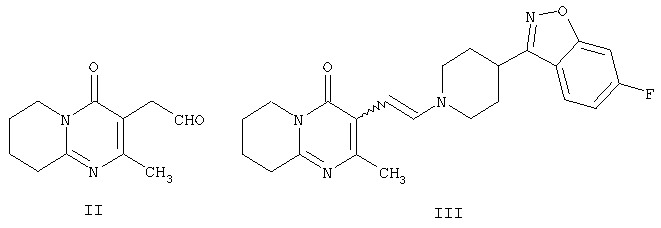
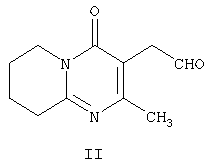
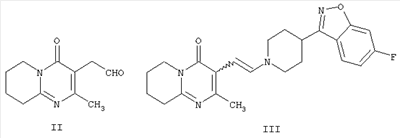
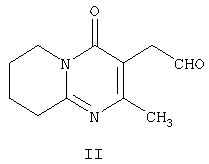
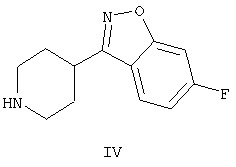
которое обладает нейролептическими свойствами. Способ включает конденсацию (2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2а]пиримидин-3-ил)ацетальдегида формулы II и (6-фтор-3-пиперидинил)-1,2-бензизоксазола формулы IV
 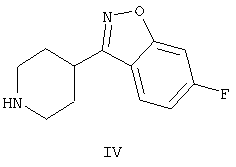
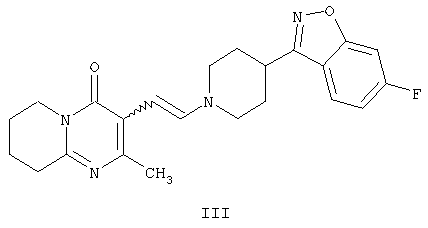
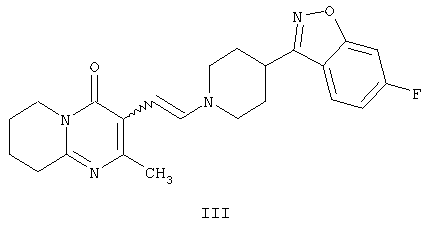
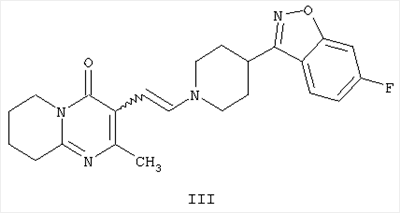
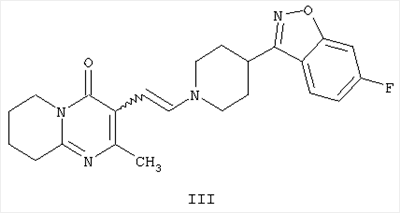
с получением промежуточного енамина, представляющего собой 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)-пиперидин-1-ил]винил}-2-метил-6,7,8,9-тетрагидропиридо-[1,2-а]пиримидин-4-он формулы III
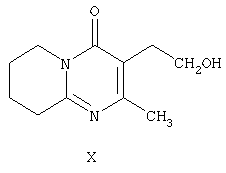
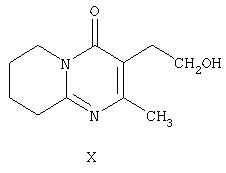
и последующее восстановление такого енамина в присутствии гидрида. Заявлены новые промежуточные соединения формулы II и формулы III, а также описан способ получения соединения формулы II, включающий окисление 3-(2-гидроксиэтил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она формулы Х
Способ по настоящему изобретению характеризуется высокой воспроизводимостью при масштабном производстве и представляет собой уникальное сочетание простоты синтеза и соответствия требованиям снижения стоимости, безопасности и охраны окружающей среды. 4 н. и 5 з.п. ф-лы.
Область техники, к которой относится изобретение
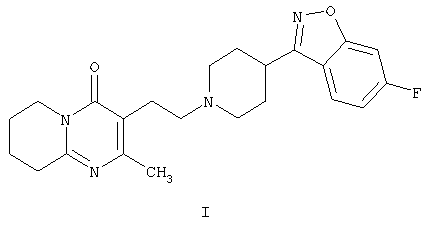
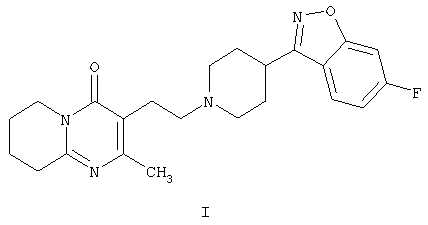
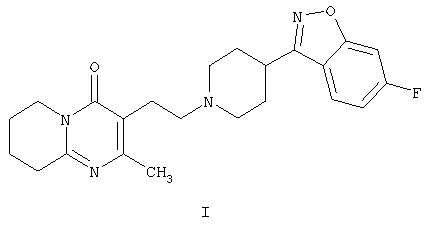
Настоящее изобретение относится к способу получения 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она формулы I
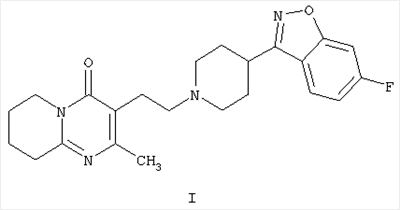
Соединение формулы I, известное под названием рисперидон, является фармацевтически активным благодаря своим нейролептическим свойствам.
Другим воплощением настоящего изобретения являются соединения формулы II, (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегид, и формулы III, 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]винил}-2-метил-6,7,8,9-тетрагидропиридо-[1,2-а]пиримидин-4-он. Эти соединения являются промежуточными производными для синтеза соединения формулы I. Соединения II и III не описаны ранее.
Уровень техники
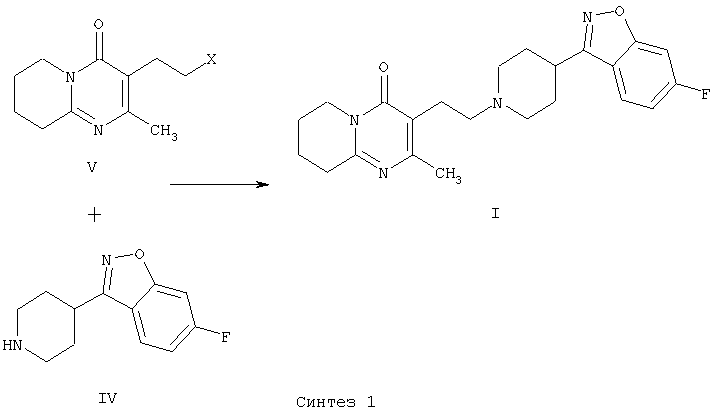
В патенте ЕР 0196132 раскрыты различные методики для получения 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (I), которые включают циклизацию различных промежуточных производных за счет межмолекулярной конденсации различных функциональных групп с образованием системы 2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она. Общий признак большинства этих методик заключается в использовании слишком сложных промежуточных соединений и реакций с неизвестным механизмом, что несомненно влияет на общую стоимость всего способа.
В патенте ЕР 0196132 получают 3-{2-[4-(6-фторбензо[d]-изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-он (I) с использованием другой методики, которая включает образование C-N связи за счет межмолекулярной реакции N-алкилирования 6-фтор-3-(4-пиперидинил)-бензо[d]изоксазола (IV) с использованием 3-(2-хлорэтил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она или аналогичных производных, содержащих другие уходящие группы (V) (Синтез 1).
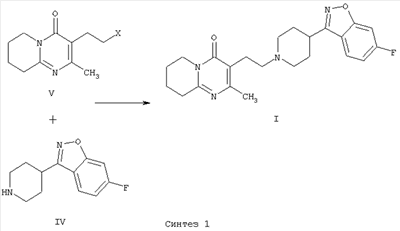
В патентах ES 2006888, ES 2006889 и ES 2050069 описаны различные методики получения 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо-[1,2-а]пиримидин-4-она (I), в которых общий признак заключается в использовании конечной стадии межмолекулярной циклизации различных типов родственных промежуточных соединений с образованием изоксазольного кольца, содержащегося в конечном продукте.
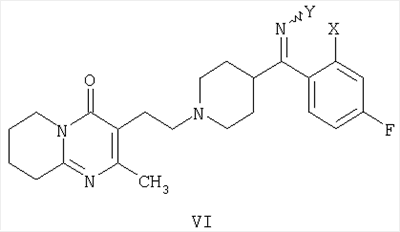
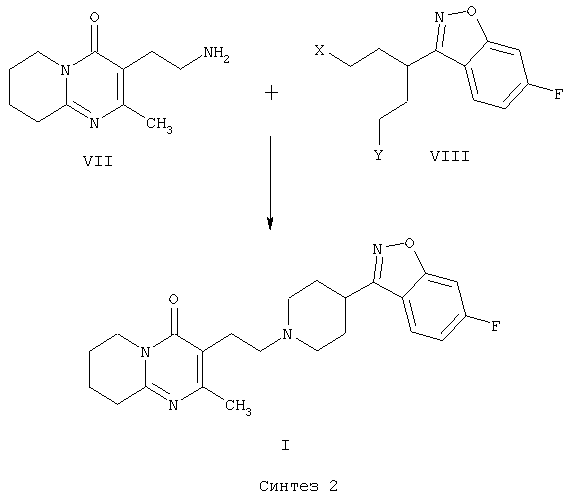
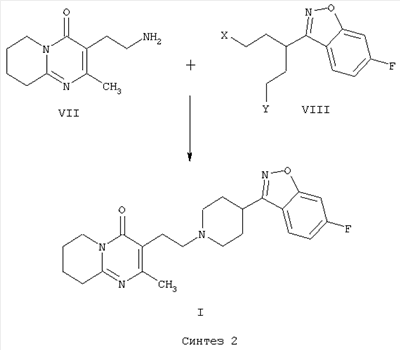
В патенте ES 2074966 описана методика получения 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (I), основанная на конечной стадии циклизации пиперидинового кольца за счет двойной межмолекулярной реакции N-алкилирования 3-(2-аминометил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (VII) с использованием пентанподобного соединения, содержащего бензо[d]изоксазольную систему и две уходящие группы в положении 1 и 5 соединения общей формулы VIII (Синтез 2).
Одним из исходных соединений, используемых для получения промежуточного производного VIII, является 4-тетрагидропиранкарбонилхлорид (IX)
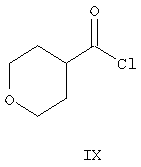
который не выпускается фирмами. Кроме того, способ получения соединения VIII из соединения IX включает пять стадий (ES 2074966).
Раскрытие изобретения
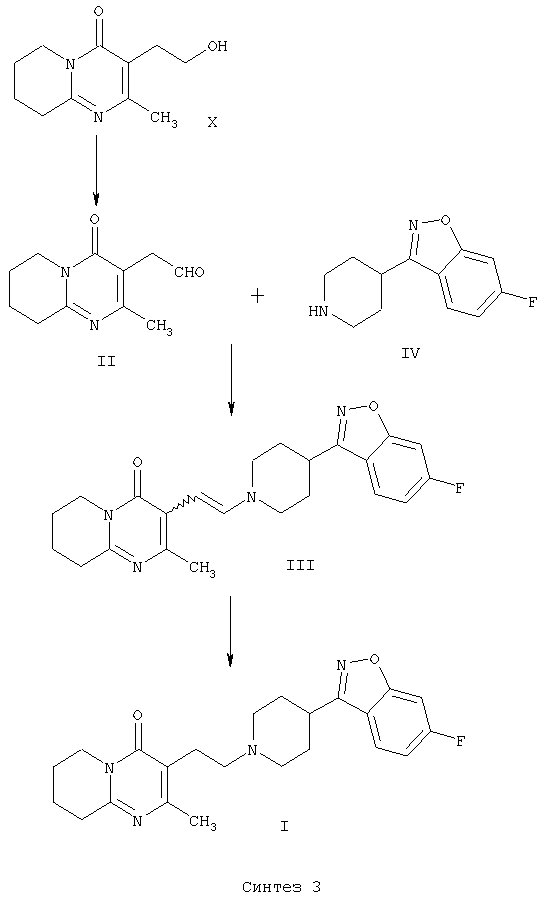
В настоящем изобретении описан альтернативный способ получения 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (I), который показан на схеме “Синтез 3”.
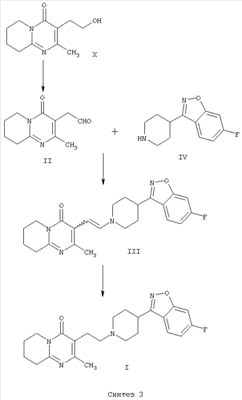
Соединение формулы II по настоящему изобретению, (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегид, получают из 3-(2-гидроксиэтил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (X) (получение этого соединения описано в статье H.Fujita et al., Ann.Rep. Sankyo Res.Lab. 1977, т.29, стр.75-78).
Енамин формулы III, 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]винил}-2-метил-6,7,8,9-тетрагидропиридо-[1,2-а]пиримидин-4-он, получают из альдегида формулы II конденсацией с 6-фтор-3-(4-пиперидинил)бензо[d]изоксазолом формулы IV (получение соединения IV описано в патенте ES 8405791).
Авторами доказано, что енамин формулы III легко восстанавливается с образованием 3-{2-[4-(6-фторбензо[d]-изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо-[1,2-а]пиримидин-4-она (I) в присутствии восстанавливающего агента.
Другой способ заключается в непосредственном и преимущественном образовании конечного продукта, 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (I), из (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегида (II) и 6-фтор-3-(4-пиперидинил)бензо[d]изоксазола (IV) в условиях восстановительного аминирования, то есть с использованием одностадийного способа синтеза.
Способ по настоящему изобретению представляет собой уникальное сочетание простоты синтеза и соответствия требованиям снижения стоимости, безопасности и охраны окружающей среды при получении активной субстанции рисперидона (I). Химический способ характеризуется высокой воспроизводимостью при масштабировании процесса благодаря простоте синтеза и высокому выходу продуктов на всех стадиях, при этом получают конечный продукт высокого качества.
Соединения формул II и III ранее не описаны и являются частью настоящего изобретения.
Осуществление изобретения
Для окисления 3-(2-гидроксиэтил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (X) с образованием (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2а]пиримидин-3-ил)ацетальдегида (II) используют предпочтительно методики, основанные на применении диметилсульфоксида в присуствии электрофильного агента, такого как дициклогексилкарбодиимид, уксусный ангидрид, трифторуксусный ангидрид, триоксид серы или предпочтительно оксалилхлорид. Реакцию проводят в инертном растворителе, предпочтительно в хлористом метилене, в присутствии основания, предпочтительно триэтиламина, при температуре в диапазоне от -78°С до комнатной температуры.
Для получения промежуточного производного енамина III проводят реакцию конденсации (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]-пиримидин-3-ил)ацетальдегида (II) и 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (IV) или любой его соли, причем реакцию проводят в инертном растворителе, предпочтительно толуоле, при температуре в диапазоне от 20°С до 150°С, предпочтительно при температуре кипения растворителя (при нагревании с обратным холодильником). Реакцию можно проводить в присутствии катализатора, то есть при добавлении кислот или оснований. Выход реакции можно повысить с помощью сдвига химического равновесия путем удаления из системы образующейся в ходе реакции воды, например, при использовании молекулярных сит или других осушителей. Воду предпочтительно удаляют из реакционной смеси с помощью азеотропной перегонки.
Для получения конечного соединения I восстановление промежуточного енамина III можно проводить в присутствии различных гидридов, таких как гидрид натрия, гидрид калия, гидрид магния, гидрид кальция, боргидрид натрия, цианоборгидрид натрия, триацетоксиборгидрид натрия, гидрид лития, гидрид лития-алюминия, гидрид натрия-алюминия, гидрид алюминия, гидрид натрия-бис(2-метоксиэтокси)алюминия, гидрид алюминия-моно(С1-4алкокси)-алюминия, гидрид лития-ди(С1-4алкокси)алюминия, гидрид натрия-диэтилалюминия или их смеси, по выбору в присутствии соли, основания или неорганической кислоты. Восстановление можно также проводить с использованием борана или диборана по выбору в присутствии амина. Предпочтительно используют цианоборгидрид натрия или боргидрид натрия в присутствии уксусной кислоты. Реакцию можно проводить в различных слабо полярных растворителях, таких как тетрагидрофуран (ТГФ), этиловый эфир, трет-бутилметиловый эфир, смеси толуола и ТГФ и т.п., или в полярных растворителях, таких как этанол, метанол, изопропанол, бутанол, или в других высококипящих спиртах, таких как вода, смеси бутанола и воды и т.п., при этом наиболее подходящим является этанол. Температуру реакции можно поддерживать в диапазоне от -20 до 80°С, предпочтительно приблизительно 25°С.
Следующие примеры предназначены для иллюстрации настоящего изобретения и не ограничивают его объем.
Пример 1: (2-Метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегид (II)
К перемешиваемой смеси 2,2 мл (25,22 ммоль) оксалилхлорида в 70 мл безводного хлористого метилена при низкой температуре (от -70 до -50°С) в инертной атмосфере добавляют 4,1 мл (57,58 ммоль) диметилсульфоксида. Затем добавляют раствор 5 г (24,02 ммоль) 3-(2-гидроксиэтил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (X) в 30 мл хлористого метилена. Через 30 мин добавляют 14,6 мл (104,75 ммоль) триэтиламина и смесь нагревают в течение 45 мин при комнатной температуре. К смеси добавляют 75 мл воды и органическую фазу декантируют. Водную фазу удаляют хлористым метиленом. Все органические фазы сушат над безводным сульфатом натрия, фильтруют и растворитель упаривают, при этом получают 4,90 г (99%) (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегида (II) в виде масляного остатка. Полученное соединение является достаточно чистым для использования на следующей стадии без дополнительной очистки.
1H ЯМР (CDCl3),  (часть/млн): 9,71 (t, J=1,2 Гц, 1Н, альдегид), 3,93 (t, J=6,0 Гц, 2Н, (часть/млн): 9,71 (t, J=1,2 Гц, 1Н, альдегид), 3,93 (t, J=6,0 Гц, 2Н, 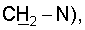 3,66 (d, J=1,2 Гц, 2H, 3,66 (d, J=1,2 Гц, 2H, 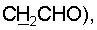 2,90 (t, J=6,8 Гц, 2Н, 2,90 (t, J=6,8 Гц, 2Н, 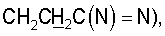 2,22 2,22 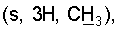 2,00-1,85 (m, 4H, 2,00-1,85 (m, 4H, 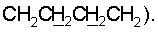
13С ЯМР (CDCl3), (часть/млн): 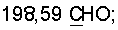 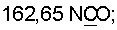 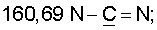 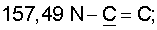 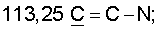 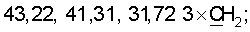 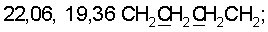 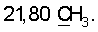
Rf (SiO2, этилацетат/метанол, 9:1)=0,2
Пример 2: 3-{2-[4-(6-Фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]винил}-2-метил-6,7,8,9-тетрагидропиридо[1,2-а]пиримидин-4-он (III)
Смесь 2,96 г (14,33 ммоль) (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегида (II), 3,16 г (14,33 ммоль)6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (IV) и 50 мл толуола переносят в прибор Дина Старка. Смесь доводят до кипения, кипятят с обратным холодильником в течение 3 ч и растворитель удаляют при пониженном давлении, при этом получают 5,70 г (97,3%) 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]винил}-2-метил-6,7,8,9-тетрагидропиридо[1,2-а]пиримидин-4-она (III) в виде твердого вещества ярко-оранжевого цвета. Полученное соединение является достаточно чистым для использования на следующей стадии без предварительной очистки или его по выбору кристаллизуют из органического растворителя, предпочтительно этанола.
1H ЯМР (CDCl3), (часть/млн): 7,85 (d, J=14 Гц, 1Н, 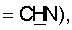 7,67 (dd, J1=8,8 Гц, J2=4,8 Гц, 1Н, 7,67 (dd, J1=8,8 Гц, J2=4,8 Гц, 1Н, 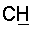 ароматический), 7,25 (dd, J1=8,4 Гц, J2=2,0 Гц, 1Н, ароматический), 7,25 (dd, J1=8,4 Гц, J2=2,0 Гц, 1Н, 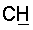 ароматический), 7,07 (m, 1Н, ароматический), 7,07 (m, 1Н, 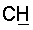 ароматический), 5,16 (d, J=14 Гц, 1Н, ароматический), 5,16 (d, J=14 Гц, 1Н, 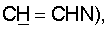 3,97 (t, J=6,4 Гц, 2Н, 3,97 (t, J=6,4 Гц, 2Н, 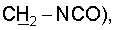 3,65 (d, J=12,8 Гц, 2Н), 3,23 (m, 1H), 2,95 (m, 2H), 2,88 (t, J=6,6 Гц, 2H, 3,65 (d, J=12,8 Гц, 2Н), 3,23 (m, 1H), 2,95 (m, 2H), 2,88 (t, J=6,6 Гц, 2H, 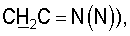 2,34 (s, 3H, 2,34 (s, 3H, 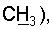 2,11 (m, 4H), 1,97 (m, 2H), 1,88 (m, 2H). 2,11 (m, 4H), 1,97 (m, 2H), 1,88 (m, 2H).
13C ЯМР (CDCl3), (часть/млн): 165,32-162,83, d, JC-F=249 Гц, С ароматический; 163,92-163,79, d, JC-F=13 Гц, С ароматический; 162,83, С ароматический; 160,98, С ароматический; 160,64, С ароматический; 152,60, С ароматический; 143,59, СН енамин; 122,40-122,29, d, JC-F=11 Гц, СН ароматический; 118,42, С ароматический; 117,13-117,1, d, JC-F=2 Гц, С ароматический; 112,53-112,28, d, JC-F=25 Гц, СН ароматический; 97,58-97,31, d, JC-F=27 Гц, СН ароматический; 91,87, СН енамин; 48,16 CH2; 42,55 CH2; 34,43 СН; 31,29 СН2; 29,69 CH2; 22,19 CH2; 22,07 СН3; 19,38 CH2.
Rf (SiO2, этилацетат/метанол, 9:1) =0,21
Rf (SiO2, хлороформ/метанол, 9:1) =0,50
Пример 3: 3-{2-[4-(6-Фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-он (I)
К суспензии 1,39 г (3,42 ммоль) 3-{2-[4-(6-фторбензо[d]-изоксазол-3-ил)пиперидин-1-ил]винил}-2-метил-6,7,8,9-тетрагидро-пиридо[1,2-а]пиримидин-4-она (III) в 22 мл абсолютного этанола и 1 мл ледяной уксусной кислоты при перемешивании при комнатной температуре в защитной атмосфере небольшими порциями в течение 1 ч добавляют 0,254 г (4,04 ммоль) цианоборгидрида натрия. Через 1,5 ч растворитель упаривают и полученный неочищенный продукт растворяют в 250 мл этилацетата. Органическую фазу последовательно промывают 50 мл 1 М водного раствора бикарбоната натрия, 50 мл воды и 50 мл насыщенного раствора хлорида натрия и сушат над безводным сульфатом магния. Полученный раствор фильтруют и растворитель упаривают, при этом получают 1,17 г твердого вещества (83%), которое разбавляют хлористым метиленом и очищают колоночной хроматографией (силикагель). В качестве элюента используют хлористый метилен, метанол и триэтиламин (95:5:1). Фракции, содержащие очищенный продукт, собирают и растворитель упаривают при пониженном давлении, при этом получают 0,83 г (70,9%) 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она (I). По выбору неочищенный продукт очищают кристаллизацией из подходящего органического растворителя, предпочтительно этанола или 4-метил-2-пентанона.
1H ЯМР (CDCl3), (часть/млн): 7,71 (dd, J1=8,8 Гц, J2=5,2 Гц, 1Н, СН ароматический), 7,23 (dd, J1=8,6 Гц, J2=2,2 Гц, 1Н, СН ароматический), 7,05 (ddd, J1=8,8 Гц, J2=8,6 Гц, J3=2,2 Гц, 1Н, СН ароматический), 3,93 (t, J=6,2 Гц, 2Н, СН2), 3,18 (d, J=11,6 Гц, 2Н, СН2), 3,08 (m, 1Н, СН), 2,87 (t, J=6,6 Гц, 2Н, СН2), 2,77 (m, 2Н, СН2), 2,55 (m, 2Н, CH2), 2,31 (s, 3Н, СН2), 2,27 (m, 2H, СН2), 2,09 (m, 4H, 2×СН2), 1,96 (m, 2H, СН2), 1,89 (m, 2H, СН2).
13С ЯМР (CDCl3), (часть/млн): 165,26-166,77, d, JC-F=249 Гц, С ароматический; 163,88-163,75, d, JC-F=14 Гц, С ароматический; 162,56; 161,09, С ароматический; 158,39, С ароматический; 155,87, С ароматический; 122,67-122,55, d, JC-F=11 Гц, С ароматический; 119,27, С ароматический; 117,28, С ароматический; 112,35-112,10, d, JC-F=25 Гц, СН ароматический; 97,48-97,21, d, JC-F=27 Гц, СН ароматический; 56,68 CH2; 53,35 CH2; 42,66 CH2; 34,59 СН; 31,41 СН2; 30,53 СН2; 23,74 СН2; 21,95 CH2; 21,24 СН2; 19,20 CH2.
Формула изобретения
1. Способ получения 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]этил}-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она формулы I
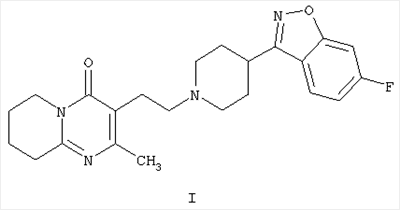
отличающийся тем, что проводят конденсацию (2-метил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-3-ил)ацетальдегида формулы II
и 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола формулы IV
или любой его соли с образованием промежуточного производного енамина, представляющего собой 3-{2-[4-(6-фторбензо[d]изоксазол-3-ил)пиперидин-1-ил]-винил}-2-метил-6,7,8,9-тетрагидропиридо[1,2-а]пиримидин-4-он формулы III
с последующим его восстановлением в присутствии гидрида, по выбору в присутствии соли, кислоты или неорганического основания, и в присутствии подходящего инертного растворителя, причем реакции проводят с выделением промежуточного производного формулы III или без его выделения в одну стадию.
2. Способ по п. 1, отличающийся тем, что восстановление реакционноспособного производного енамина формулы III проводят в присутствии гидрида или борана в подходящем растворителе.
3. Способ по п. 2, отличающийся тем, что гидридом является цианоборгидрид натрия или боргидрид натрия, по выбору в присутствии уксусной кислоты.
4. Способ по п. 2, отличающийся тем, что подходящим растворителем предпочтительно является этанол.
5. Енамин формулы III
предназначенный для использования в качестве реакционноспособного промежуточного производного в синтезе соединения формулы I
6. Альдегид формулы II
предназначенный для использования в качестве ключевого промежуточного производного в синтезе соединения формулы I
7. Способ получения соединения формулы II
отличающийся тем, что осуществляют окисление 3-(2-гидроксиэтил)-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-она формулы Х
8. Способ по п. 7, отличающийся тем, что окисление проводят с использованием диметилсульфоксида в присутствии электрофильного агента, выбранного из группы, включающей оксалилхлорид, дициклогексилкарбодиимид, уксусный ангидрид, трифторуксусный ангидрид и триоксид серы.
9. Способ по п. 8, отличающийся тем, что реакцию проводят предпочтительно с использованием оксалилхлорида в качестве электрофильного агента в присутствии триэтиламина, и с использованием хлористого метилена в качестве реакционноспособного растворителя.
TH4A – Переиздание описаний изобретений к патентам Российской Федерации
Причина переиздания: Коррекция библиографической части, текстов описания и формулы изобретения
Извещение опубликовано: 20.06.2006 БИ: 17/2006
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 04.03.2009
Извещение опубликовано: 27.12.2010 БИ: 36/2010
|
|