Патент на изобретение №2263106
|
||||||||||||||||||||||||||
(54) (S)-МЕТИЛ 3-(3-НИТРОФЕНИЛ) ПЕНТАНОАТ
(57) Реферат:
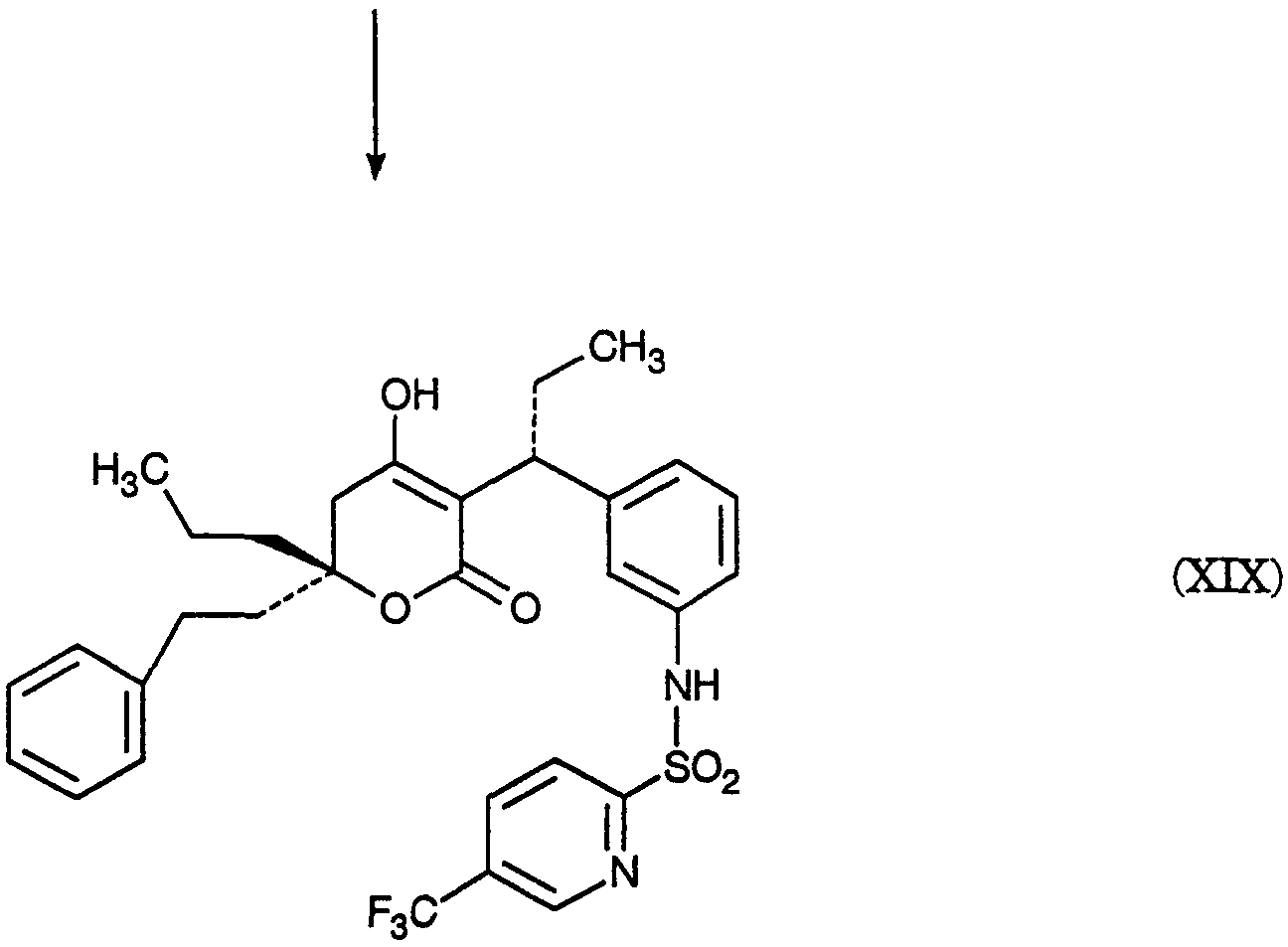
Изобретение относится к новому промежуточному соединению, а именно (5)-Метил 3-(3-нитрофенил) пентаноату, который применяется для получения [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил-6-пропил-2Н-пиран-3-ил]пропил] фенил]-5-(трифторметил)-2-пиридинсульфонамида, представляющего собой ингибитор протеазы, пригодный для лечения людей, инфицированных вирусом ВИЧ. Технический результат – получение нового промежуточного соединения для нового ингибитора протеазы.
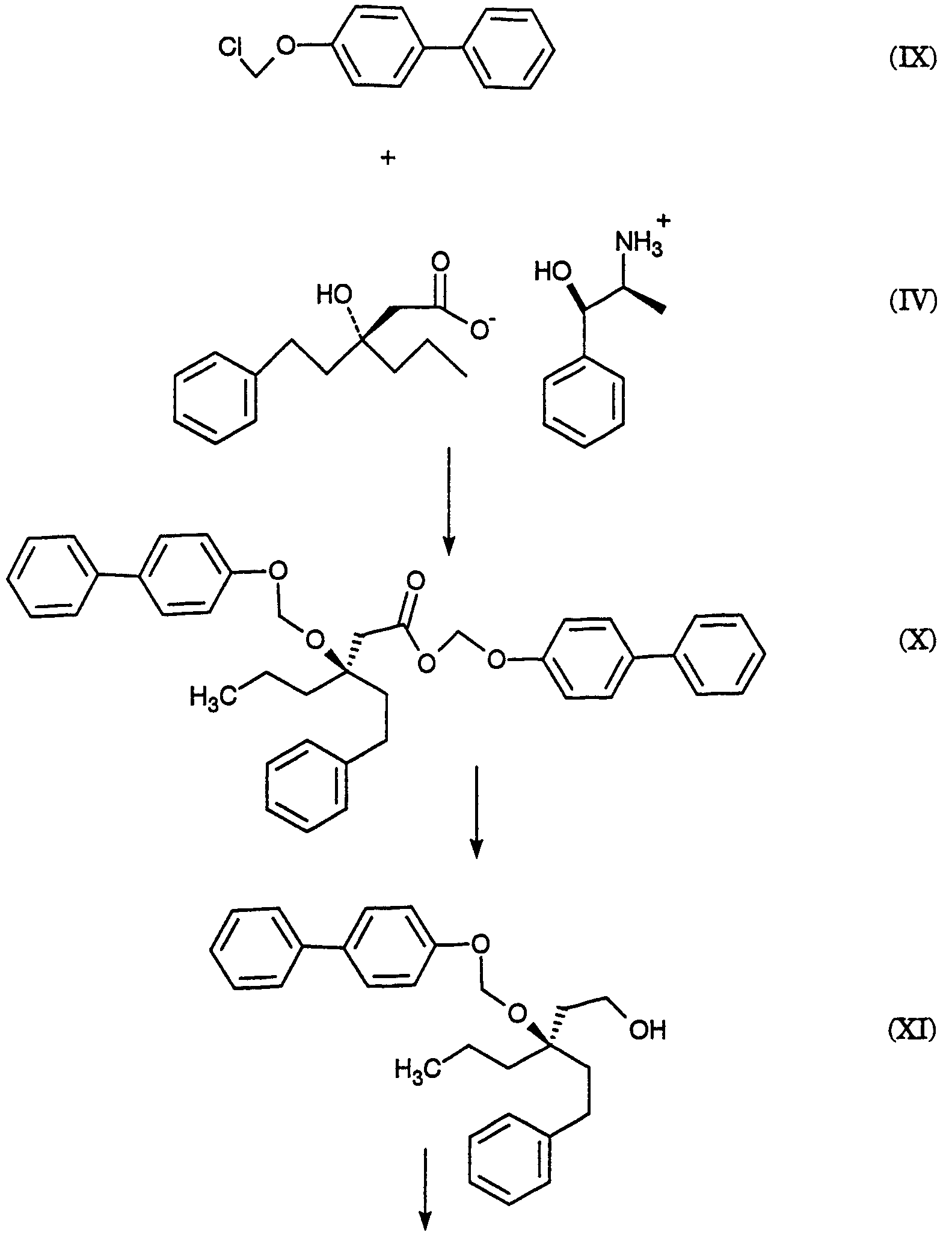
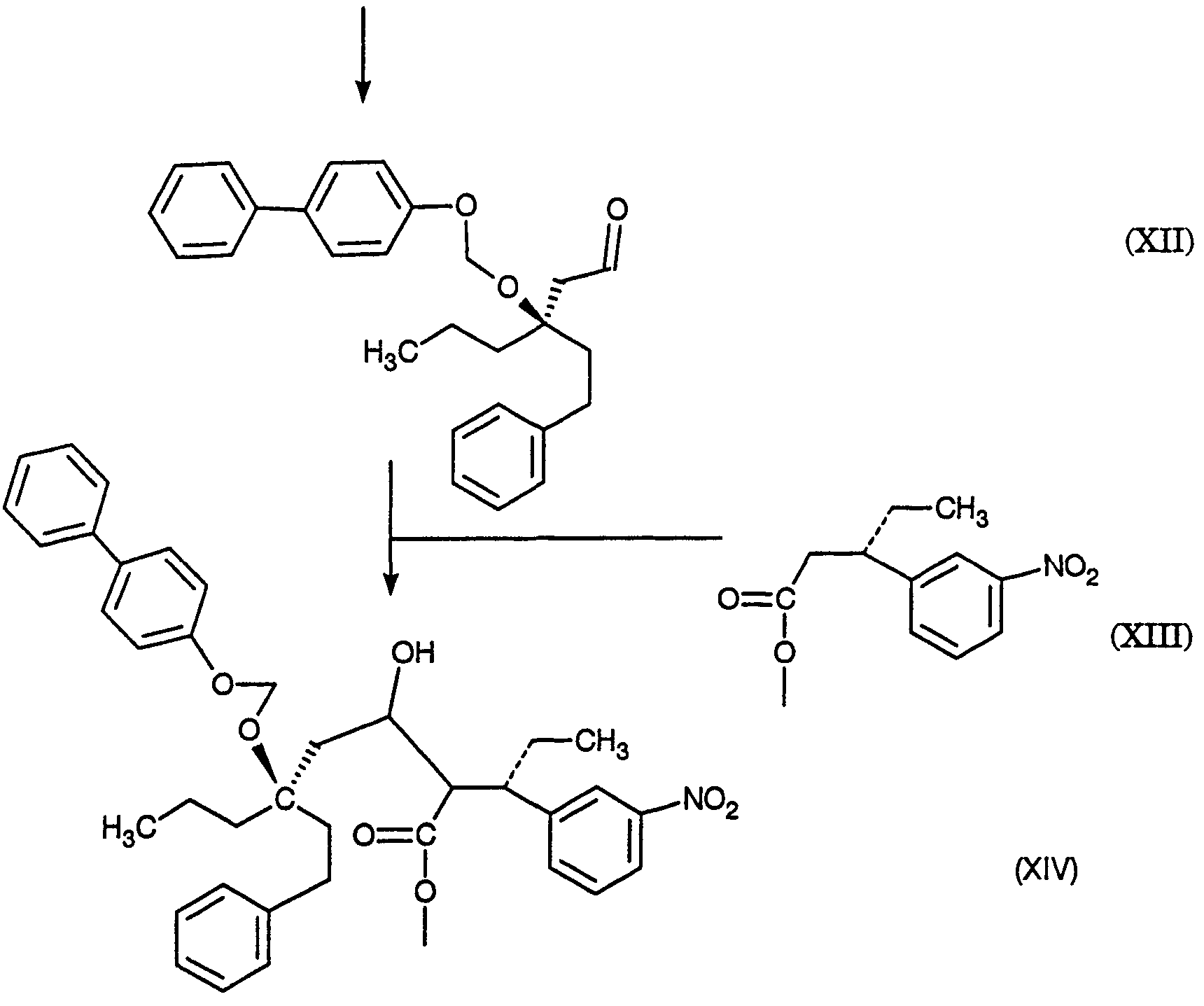
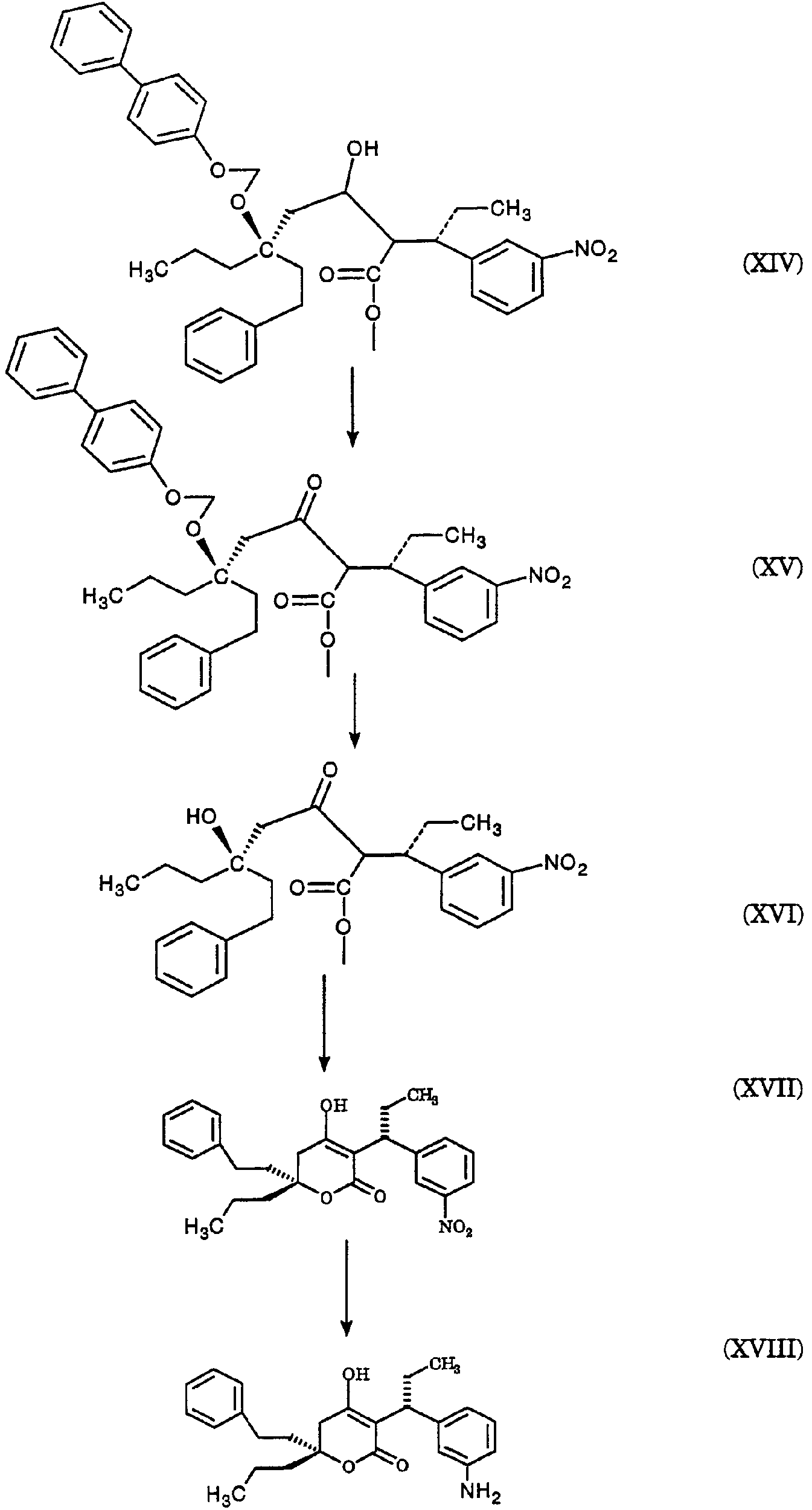
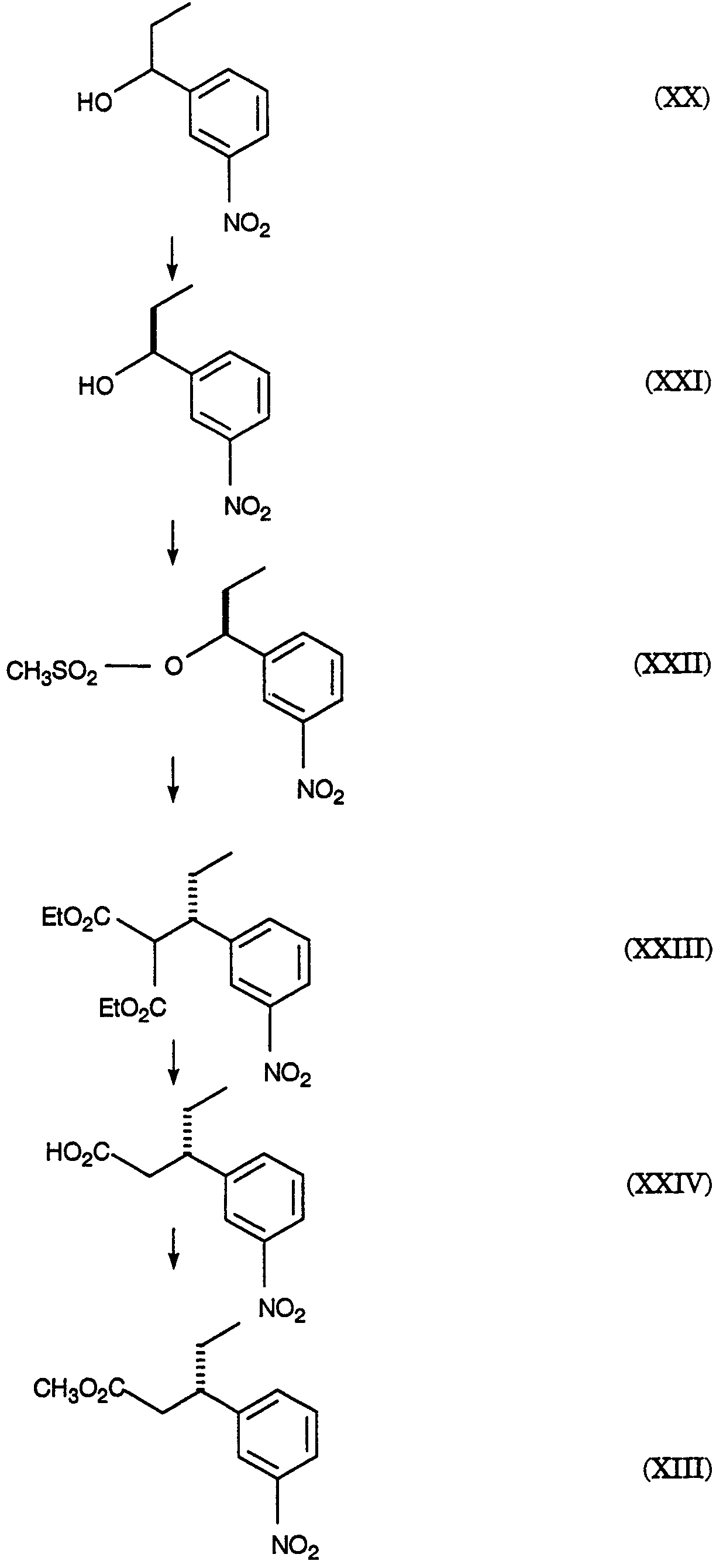
2420-128485/051 (S)-МЕТИЛ 3-(3-НИТРОФЕНИЛ)ПЕНТАНОАТ ОПИСАНИЕ Настоящее изобретение относится к новому промежуточному соединению, используемому для получения [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамида (XIX), который представляет собой ингибитор протеазы, используемый при лечении людей, инфицированных вирусом ВИЧ. [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамид (XIX) может быть получен способом, изложенным в Международных публикациях WO 95/30670 и WO 94/11361. В J. Med. Chem., 39(22), 4349 (1996) описан циклический сложный эфир (VI), но в рацемической форме. В этой статье также описано превращение циклического сложного эфира (VI) в ингибитор протеазы (XIX), но другим синтетическим путем. В J. Am. Chem. Soc., 111, 3627 (1997) описано аминосоединение (XVIII). В Tetrahedron Letters, 34(2), 277-280 (1993) описан способ превращения В J. Med. Chem., 39(23), 4630-4642 (1996) описан несвязанный с настоящим изобретением способ получения соединений, подобных соединениям формул (VI) и (CVI), но в рацемической форме, из исходных веществ, отличающихся от тех, которые используются в настоящем изобретении. В Международной публикации WO 95/14012 заявлено циклическое соединение, подобное циклическим соединениям (VI), (XVII) и (XXV) по настоящему изобретению, но в рацемической форме. Способом по настоящему изобретению данные соединения получают в оптически чистой форме. Настоящее изобретение относится к новому промежуточному продукту – (S)-метил 3-(3-нитрофенил)пентаноату – используемому в получении ингибитора протеазы. Также раскрывается [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамид. [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамид (XIX), который, как известно, представляет собой ингибитор протеазы и используется при лечении людей, инфицированных вирусом ВИЧ, получают, используя новый промежуточный продукт. На схеме С представлены конденсация бифенильного соединения (IX) с солью (IV) с получением сложного эфира простого эфира (X) и превращения сложного эфира простого эфира (X) в соответствующий спирт (XI) и соответствующий альдегид (XII). Также описано взаимодействие оптически чистого сложного нитроэфира (XIII) с альдегидом (XII) с получением соответствующего простого нитроэфира (XIV); см. также примеры 1-4. На схеме D представлены превращения простого нитроэфира (XIV) в соответствующий нитрокетон (XV), соответствующий нитроспирт (XVI), соответствующий нитро- На схеме Е представлено получение оптически чистого сложного нитроэфира (XIII), который используется на схеме С. Схема Е раскрывает оптическое разделение рацемического 1-(3-нитрофенил)пропанола (XX) с получением соответствующего оптически чистого 1-(3-нитрофенил)пропанола (XXI) и его превращение в соответствующий метилсульфонат (XXII), соответствующий сложный диэфир (XXIII), соответствующую нитрокислоту (XXIV) и соответствующий оптически чистый сложный нитроэфир (XIII); см. также получения 1-5. Кислота (III) образует основные аддитивные соли при взаимодействии с основаниями достаточной силы. Фармацевтически приемлемые соли включают соли как с неорганическими, так и с органическими основаниями. Фармацевтически соли являются предпочтительнее, чем кислоты, так как соли лучше растворимы в воде и в большей мере кристаллические. Предпочтительные фармацевтически приемлемые соли включают соли следующих оснований, например, гидроксида, аммиака, трометамина (TEAM), 2-амино-2-(гидроксиметил)-1,3-пропандиола, (1R,2S)-норэфедрина, (1S,2R)-норэфедрина, (R)-2-амино-2-фенилэтанола, (S)-2-амино-2-фенилэтанола, (R)-1-фенилэтиламина и (S)-1-фенилэтиламина. Наиболее предпочтительной солью является соль (1R,2S)-норэфедрина. [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамид (XIX), соединение примера 9 (СОЕДИНЕНИЕ), как известно, является полезным при лечении людей, инфицированных ВИЧ, см. Международные публикации WO95/30670 и WO 94/11361. Это СОЕДИНЕНИЕ ингибирует ретровирусные протеиназы и таким образом ингибирует репликацию вируса. СОЕДИНЕНИЕ по настоящему изобретению полезно для ингибирования ретровирусной протеазы человека. СОЕДИНЕНИЕ полезно для лечения людей, инфицированных ретровирусом человека, таким как вирус иммунодефицита человека (штаммы ВИЧ-1 или ВИЧ-2) или вирусами Т-клеточного лейкоза человека (ВТЛЧ-I или ВТЛЧ-II), которые приводят к синдрому приобретенного иммунодефицита (СПИД) и/или родственным заболеваниям. Пациенты, которым показано такое лечение, представляют собой людей (1), инфицированных одним или несколькими штаммами ретровируса человека, что определено по наличию или измеряемого вирусного антитела, или антигена в плазме крови, или (2) в случае ВИЧ, имеющих или асимптоматическое ВИЧ инфицирование, или симптоматическое СПИД характеристичное инфицирование, как, например, (а) диссеминированный гистоплазмоз, (b) изопсориаз, (с) бронхиальный и пульмональный кандидоз, включая пневмоциститную пневмонию, (d) лимфома не Ходгкиновского типа или (е) саркома Капоши, или которым меньше шестидесяти лет; или имеющих абсолютное число СВ4+лимфоциты меньше 500/мм3 в периферической крови. Лечение состоит в поддержании СОЕДИНЕНИЕМ по данному изобретению уровня ингибирования у пациента на протяжении всего времени. СОЕДИНЕНИЕ по настоящему изобретению полезно для лечения пациентов, инфицированных вирусом иммунодефицита человека (ВИЧ), что приводит к появлению синдрома приобретенного иммунодефицита (СПИД) и родственным заболеваниям. С этой целью такие соединения могут вводиться пероральным, внутриназальным, чрескожным, подкожным и парентеральным (включая внутримышечное и внутривенное введения) путями в дозах от 0,1 мг до 100 мг/кг веса тела в сутки. Предпочтительно пероральное введение соединения. Специалистам в данной области известно, каким образом СОЕДИНЕНИЕ вводят в состав рецептуры подходящих фармацевтических лекарственных форм. Примеры лекарственных форм включают пероральные препаративные формы, такие как таблетки или капсулы, или парентеральные препаративные формы, такие как стерильные растворы. Когда соединение согласно данному изобретению вводится перорально, эффективные количества составляют от примерно 0,1 мг до примерно 100 мг на кг веса тела в сутки. Предпочтительно, чтобы эффективное количество составляло от примерно 10 до примерно 100 мг на кг веса тела в сутки. Более предпочтительно, чтобы количество составляло от примерно 30 мг до примерно 90 мг на кг веса тела. Предпочтительно, чтобы соединение вводили от 2 до 5 раз в сутки, более предпочтительно 3 раза в сутки. Предпочтительно, чтобы вводимая доза составляла от примерно 2700 мг/сутки до примерно 4500 мг/сутки. Для перорального введения могут быть получены или твердые, или жидкие лекарственные формы. Предпочтительно, чтобы соединение вводилось в твердой лекарственной форме, более предпочтительно в виде капсулы. При парентеральном введении соединений по данному изобретению оно может быть осуществлено путем инъекции или путем внутривенной инфузии. Эффективное количество составляет от примерно 0,1 мг до 100 мг/кг веса тела в сутки. Растворы для парентерального введения получают путем растворения соединений по данному изобретению в водном носителе и стерилизации раствора фильтрованием перед его помещением в подходящие герметично укупориваемые пузырьки или ампулы. Парентеральные суспензии получают в основном таким же способом за исключением того, что используют стерильный суспензионный носитель и соединения по данному изобретению стерилизуют с использованием этиленоксида или подходящего газа перед их суспендированием в носителе. Конкретный путь введения, дозировка или частота введения легко могут быть определены специалистом в данной области и будут зависеть от возраста, веса, общего физического состояния и/или от других клинических симптомов, характерных для данного пациента, подвергающегося лечению. Определения и условные обозначения Определения и пояснения, приведенные ниже, даны для терминов, используемых в описании и в формуле изобретения. Определения Все температуры приведены в градусах Цельсия. ТСХ обозначает тонкослойную хроматографию. ВЭЖХ относится к жидкостной хроматографии под высоким давлением; С-8 колонка 4,6×250 мм Zorbax, подвижная фаза А = метанол, подвижная фаза В = 6,5 г гидроксида трет-бутиламмония в воде, рН доведено до 4,0 уксусной кислотой, градиентно от 65/35 А/В до 70/30 А/В в течение 20 минут, затем изократически (не меняя соотношения) 70/30 А/В в течение 5 минут, затем градиентно до 90/10 А/В в течение 20 минут; скорость потока 1,0 мл/мин; УФ-детектирование при 254 нм. ТГФ обозначает тетрагидрофуран. ДМФ обозначает диметилформамид. МТВЕ обозначает метил-трет-бутиловый простой эфир. ДМСО обозначает диметилсульфоксид. Солевой раствор представляет собой насыщенный водный раствор хлорида натрия. Хроматография (колоночная и флэш-хроматография) относится к очистке/отделению соединений, выраженной в виде (носитель, элюент). Понятно, что подходящие фракции объединяют и концентрируют с получением целевого(ых) соединения(й). СМР относится к спектроскопии С-13 ядерного магнитного резонанса, химические сдвиги приведены в м.д. ( ЯМР относится к спектроскопии ядерного (протонного магнитного резонанса), химические сдвиги приведены в м.д. ( МС относится к масс-спектроскопии, выраженной в виде единиц m/e, m/z или масса/заряд. [М+Н]+ относится к положительному иону исходного вещества плюс атом водорода. ЭУ относится к электронному удару. ХИ относится к химической ионизации. FAB относится к бомбардировке быстрыми атомами. Эфир относится к диэтиловому эфиру. Фармацевтически приемлемый относится к таким свойствам и/или веществам, которые приемлемы для пациента с фармакологической/токсикологической точки зрения и для производящего химика-фармацевта с физической/химической точки зрения в отношении композиции, рецептуры, стабильности, переносимости пациентом и биодоступности. Когда используют пары растворителей, соотношение используемых растворителей представлено объем/объем (об/об). Когда используется растворимость твердого вещества в растворителе, то соотношение твердого продукта к растворителю указано вес/объем (в/об). СОЕДИНЕНИЕ относится к [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамиду (XIX). Алкил относится к С1-С4 алкилу, включая как линейные, так и изомеры с разветвленной цепью. W1 относится к этилу и трет-бутилу. Примеры Без дополнительного уточнения предполагается, что специалист в данной области может с использованием предшествующего описания использовать на практике настоящее изобретение во всей его полноте. В следующих подробных примерах описывается, каким образом получают различные соединения и/или осуществляют различные способы по изобретению, и они предназначены только для иллюстрации, а не для ограничения изобретения. Специалисты в данной области сразу узнают соответствующие изменения в способах, как в отношении реагентов, так и реакционных условий и методик. Получение 1. Разделение (±)-1-(3-нитрофенил)пропанола (XX) путем превращения в (S)-1-(3-нитрофенил)пропанол (XXI) и (R)-1-(3-нитрофенил)пропанол ацетат К (±)-1-(3-нитрофенил)пропанолу (XX, 24,00 г, 0,13 моль) в МТВЕ (240 мл) добавляют PS-30 липазу на целите в качестве носителя (Amano, 24 г) и изопропенилацетат (22,00 мл, 0,20 моль). Смесь перемешивают в течение 2 дней при 20-25°С. По окончании этого времени катализатор удаляют фильтрованием, слой катализатора промывают эфиром и смесь концентрируют при пониженном давлении с получением смеси ацетат-спирт. Разделение этой смеси хроматографически на силикагеле дает (R)-1-(3-нитрофенил)пропанол ацетат (13,03 г) [ Получение 2. Мезилат (S)-1-(3-нитрофенил)пропанола (XXII) К смеси (S)-1-(3-нитрофенил)пропанола (XXI, получение 1, 1 г, 5,5 ммоль) в хлористом метилене (20 мл) добавляют диизопропилэтиламин (1,07 г, 8,3 ммоль). Смесь охлаждают до -20° и добавляют метансульфонилхлорид (0,69 г, 6,02 ммоль). Реакционную смесь выдерживают при -20° в течение 10 минут, затем выдерживают при 0° в течение 40 минут. Реакционную смесь разбавляют хлористым метиленом, добавляют бикарбонат натрия (5%) и разделяют фазы. Хлористый метилен упаривают, получая указанное в заголовке соединение, [ Получение 3. (S)-диметил 1-[1-(3-нитрофенил)пропил]малонат (XXIII) Раствор этоксида натрия (1,0 М) получают, растворяя металлический натрий (1,27 г, 0,055 моль) в абсолютном этаноле (55 мл). К вышеуказанному раствору при 0° добавляют диэтилмалонат (8,84 г, 0,055 моль). К полученному раствору малоната натрия (6,4 мл, 6,4 ммоль) при -20° добавляют по каплям мезилат (S)-1-(3-нитрофенил)пропанола (XXII, получение 2, 1,43 г, 5,5 ммоль). Через 2 часа при 20-25° к реакционной смеси добавляют дополнительную аликвоту малоната натрия (5 мл, 5,0 ммоль) и затем перемешивают реакционную смесь при 20-25° в течение ночи. Реакционную смесь концентрируют и распределяют между зтилацетатом и соляной кислотой (1 н.). Органическую фазу отделяют и удаляют растворитель, получая сырой продукт, который хроматографируют (силикагель; этилацетат/гексан, 10/90), получая указанное в заголовке соединение, [ Получение 4.(S)-3-(3-нитрофенил)пентановая кислота (XXIV) (S)-диметил 1-[1-(3-нитрофенил)пропил]малонат (XXIII, получение 3, 0,73 г, 2,26 ммоль) нагревают при кипении с обратным холодильником в хлорводородной кислоте (6 н., 10 мл) в течение 18 часов. Реакционную смесь охлаждают и экстрагируют этилацетатом. Этилацетатную фазу промывают водой, отделяют и конденсируют, получая указанное в заголовке соединение, [ Получение 5. Метиловый эфир (±)-3-(3-нитрофенил)пентановой кислоты (XIII) К раствору (±)-3-(3-нитрофенил)пентановой кислоты (XXIV, 30,21 г, 135 ммоль) в метаноле (250 мл) добавляют концентрированную серную кислоту (0,6 мл). Полученную смесь нагревают при кипении с обратным холодильником в течение 3 часов. После охлаждения смесь распределяют между этилацетатом и бикарбонатом натрия (5%-ным водным). Водный слой отделяют и повторно экстрагируют двумя дополнительными порциями этилацетата. Объединенные органические фазы промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют, получая указанное в заголовке соединение, ТСХ (силикагель GF) для уксусная кислота/этилацетат/гексан (2/20/80) Rf=0,54, для этилацетат/гексан (20/80) Rf=0,54; ЯМР (CDCl3, TMC) 0,80, 1,56-1,83, 2,55-2,75, 3,05-3,2, 3,57, 7,4-7,55 и 8,03-8,12 кислота/этилацетат/гексан (2/20/80) Rf=0,54, для этилацетат/гексан (20/80) Rf=0,54; ЯМР (CDCl3, TMC) 0,80, 1,56-1,83, 2,55-2,75, 3,05-3,2, 3,57, 7,4-7,55 и 8,03-8,12 5. Получение 6. Метиловый эфир (S)-3-(3-нитрофенил)пентановой кислоты (XIII) Следуя общей методике получения 5 и внося несущественные изменения, но исходя из (S)-3-(3-нитрофенил)пентановой кислоты (XXIV, получение 4) получают указанное в заголовке соединение. TLC Rf=0.54 (20% EtOAc/гексан); 1H ЯМР Пример 1. (R)-(4-фенилфенокси)метил-3-(2-фенилэтил)-3-[(4-фенилфенокси)метокси]гексаноат (X) К суспензии (-)-норэфедриновой соли (R)-3-гидрокси-3-(2-фенилэтил)гексановой кислоты (IV, 25,04 г, 64,62 ммоль) в воде (185 мл) и МТВЕ (185 мл) при 20-25° добавляют водную хлороводородную кислоту (37,5 вес.%, 7,51 г, 77,24 ммоль, 1,20 эквив.), доводя рН от 8,04 до 1.30. Фазы разделяют и водную фазу промывают МТВЕ (185 мл). Органические фазы сушат над сульфатом магния и концентрируют. К данному концентрату затем добавляют толуол (77 мл), N,N-диизопропилэтиламин (96 мл, 551 ммоль, 8,53 эквив.) и 1-хлорметокси-4-фенилбензол (IX, 71,88 г, 328,68 ммоль, 5,09 эквив.). Затем смесь нагревают до 110° и перемешивают при 110-117° в течение 5 часов. Смесь охлаждают до 65° и добавляют метанол (800 мл). Полученную суспензию охлаждают до -30° и продукт собирают вакуумным фильтрованием, промывают метанолом (200 мл) и сушат, получая сырой продукт. Аналитически чистый образец получают путем хроматографии (этилацетат/гексан) с последующей кристаллизацией, получая указанное в заголовке соединение, т.пл.=104,0-105,5°; 142,04, 140,71, 140,41, 135,85, 134,56, 128,75, 128,68, 128,35, 128,29, 128,09, 126,97, 126,81, 126,73, 125,78, 116,13, 87,34, 85,20, 80,40, 41,19, 38,80, 38,61, 29,73, 16,74, 14,35 Пример 2. (R)-3-(2-Фенилэтил)-3-[(4-фенилфенокси)метокси]гексанол (XI) К суспензии сырого (R)-(4-фенилфенокси)метил-3-(2-фенилэтил)-3-[(4-фенилфенокси)метокси]гексаноата (X, пример 1, 56,5 вес.%, 49.32 г, 46,38 ммоль) в толуоле (500 мл) добавляют раствор диизобутилалюминийгидрида в толуоле (1,52 М, 85 мл, 129,2 ммоль, 2,79 эквив.) при поддержании температуры -20°. Смесь медленно нагревают до 1° в течение 2,5 часов, затем перемешивают в течение 1/2 часа. Добавляют ацетон (8,0 мл, 108,5 ммоль, 2,34 эквив.) и смесь подают через канюлю в раствор при температуре 18° моногидрата лимонной кислоты (136 г, 647,2 ммоль, 14,0 зквив.) в воде (433 мл) при контролируемом экзотермическом подъеме температуры до 28°, промывая толуолом (100 мл). Смесь перемешивают при 20-25° в течение 1,5 часов и нерастворимые примеси удаляют вакуумным фильтрованием, промывая толуолом. Фазы фильтрата разделяют и водную фазу промывают толуолом (2×300 мл). Органические фазы сушат над сульфатом магния, затем промывают водным гидроксидом натрия (0,5 М, 2×500 мл). Органические фазы концентрируют до 137 г чистого веса и добавляют метанол (250 мл). Полученную суспензию концентрируют и добавляют метанол (250 мл). Смесь снова концентрируют и добавляют метанол (250 мл). Суспензию охлаждают до -60° и нерастворимые продукты удаляют фильтрованием. Фильтрат концентрируют до 60 г чистого веса, добавляют гексан (500 мл) и смесь концентрируют до 22 г чистого веса. Добавляют гексан (500 мл) и смесь снова концентрируют до 40 г чистого веса. Добавляют хлористый метилен (25 мл) с последующим медленным добавлением гексана (500 мл) и пентана (250 мл) при охлаждении до -55°. Продукт собирают вакуумным фильтрованием, промывают пентаном (200 мл) и сушат в токе азота, получая целевой продукт. Аналитически чистый образец получают хроматографией (этилацетат/гексан) с последующей кристаллизацией (хлористый метилен/гексан), получая указанное в заголовке соединение, т.пл.=49-53°; ТСХ Rf=0,14 (15% этилацетат/гексан); ВЭЖХ время удерживания = 9,18 мин; ЯМР (CDCl3) 7,56-7,07, 5,36, 3,76-3,74, 2,63-2,58, 1,94-1,88, 1,70-1,65, 1,38-1,30, 0,93 Пример 3. (R)-3-(2-фенилэтил)-3-[(4-фенилфенокси)метокси]гексаналь (XII) К смеси сырого (R)-3-(2-фенилэтил)-3-[(4-фенилфенокси)метокси]гексанола (XI, пример 2, 91,1 вес.%, 15.40 г, 34,68 ммоль) в хлористом метилене (47 мл) при 0° добавляют раствор бромида калия (0,4057 г, 3,409 ммоль, 0,098 эквив.) и бикарбоната натрия (1,557 г, 18.53 ммоль, 0,53 эквив.) в воде (20,5 мл), после чего 4-гидрокси-2,2,6,6-тетраметилпиперидинилокси, в виде свободного радикала (0,3060 г, 1,776 ммоль, 0,051 эквив.). Затем добавляют шприцом в течение 1 часа, поддерживая температуру 1-5°, водный гипохлорит натрия (13,4 вес.%/об, 26,6 мл, 47,88 ммоль, 1,38 эквив.). Затем добавляют раствор пентагидрата тиосульфата натрия (0,5182 г, 2,088 ммоль, 0,0602 эквив.) в воде (14 мл). Фазы отделяют при 0° и водную фазу промывают 2×50 мл хлористым метиленом. Органическую фазу сразу же фильтруют через магнезоль (50,25 г) и промывают последний хлористым метиленом (400 мл). Экстракты концентрируют до масла (30 г) и добавляют гексан (500 мл). Смесь концентрируют до 250 г чистого веса и добавляют гексан (100 мл). Смесь концентрируют до 186 г чистого веса и добавляют пентан (300 мл). Полученную суспензию охлаждают до -50° и целевой продукт отфильтровывают в вакууме, промывают охлажденным до -50° пентаном (100 мл) и сушат, получая аналитически чистый твердый продукт указанного в заголовке соединения, т.пл.=47,0-48,5°; ТСХ Rf=0,41 (этилацетат/гексан, 10/90); ВЭЖХ время удерживания = 10,95 мин; ЯМР (CDCl3) 9,79, 7,53, 7,40, 7,26, 7,20-7,08, 5,40, 2,67, 2,65-2,56, 1,99, 1,76, 1,38, 0,93 Пример 4. (3S),(7R)-4-карбометокси-З-(3-нитрофенил)-7-(2-фенилэтил)-7-[(4-фенилфенокси)метокси]декан-5-ол (смесь диастреомеров при С-4 и С-5) (XIV) К смеси (S)-метил 3-(3-нитрофенил)пентаноата, также известного как метиловый эфир (S)-3-(3-нитрофенил)пентановой кислоты (XIII, получение 6, 3,78 г, 15,932 ммоль) в ТГФ (55 мл) при -80° добавляют раствор гексаметилдисилазида натрия в ТГФ (0,935 М, 17,5 мл, 16,36 ммоль, 1,027 эквив.) в течение 7 минут при поддержании температуры от -80° до -85°. Полученную смесь затем нагревают до -74° и перемешивают при от -74 до -76° в течение 18 минут. Смесь охлаждают до -90° и добавляют раствор (R)-3-(2-фенилэтил)-3-[(4-фенилфенокси)метокси]гексаналя (XII, пример 3, 6,50 г, 16,147 ммоль, 1,013 эквив.) в ТГФ добавляют в течение 10 минут при поддержании температуры от -85 до -90° и споласкивают ТГФ (20 мл). Затем смесь нагревают до -71° и добавляют насыщенный водный раствор хлорида аммония (90 мл), после чего воду (90 мл) и МТВЕ (90 мл) и смесь нагревают до 20-25°. Фазы разделяют и водную фазу промывают МТВЕ (90 мл). Экстракты сушат над сульфатом магния и концентрируют до масла. Аналитически чистый образец получают хроматографией (этилацетат/гексан), получая указанное в заголовке соединение, ТСХ Rf=0,16, 0,24 (этилацетат/гексан, 10/90); ВЭЖХ время удерживания = 12,52, 12,68, 12,97 мин; МС (электрораспыление, ацетат натрия) m/z (относительная интенсивность) 662,5 (100). Пример 5. (3S),(7R)-4-карбометокси-3-(3-нитрофенил)-7-(2-фенилэтил)-7-[(4-фенилфенокси)метокси]декан-5-он (смесь диастреомеров при С-4) Раствор (3S),(7R)-4-карбометокси-3-(3-нитрофенил)-7-(2-фенилэтил)-7-[(4-фенилфенокси)метокси]декан-5-ола (XIV, пример 4, 11,12 г, 79,0 вес.%, 13,73 ммоль) в хлористом метилене (530 мл) добавляют к основной смеси хлорхромата пиридиния (16,099 г, 74,685 ммоль, 5.44 эквив.), ацетата натрия (6,984 г, 85,14 ммоль, 6,20 эквив.) и флорисиля (5,181 г) при поддержании температуры менее 11°. Смесь нагревают до 21° и перемешивают при 20-25° в течение 20 часов. Полученную суспензию фильтруют через магнезоль (47,7 г) и промывают хлористым метиленом (375 мл). Фильтрат концентрируют до масла. Аналитически чистый образец указанного в заголовке соединения получают хроматографией (этилацетат/гексан): ТСХ Rf=0,34 (этилацетат/гексан, 10/90); ВЭЖХ время удерживания = 13,02, 13,23 мин; ЯМР (CDCl3) 8,05-8,01, 7,60-7,00, 5,37, 5,21, 4,03, 3,94, 3,75, 3,58-3,43, 3,39, 2,96, 2,78-1,37, 1,20, 0,91, 0,71-0,61 Пример 6. (3S),(7R)-4-карбометокси-7-гидрокси-3-(3-нитрофенил)-7-(2-фенилэтил)-декан-5-он (смесь диастреомеров при С-4) (XVI) К смеси (3S),(7R)-4-карбометокси-3-(3-нитрофенил)-7-(2-фенилэтил)-7-[(4-фенилфенокси)метокси]декан-5-она (XV, пример 5, 9,14 г, 83,7 вес.%, 11,995 ммоль) в ТГФ (20 мл) при 23° добавляют раствор серной кислоты в метаноле (0,524 М, 20 мл, 10,48 ммоль, 0,87 эквив.). Смесь оставляют на 22 часа при 23°, затем добавляют раствор бикарбоната натрия (3,52 г, 41,90 ммоль, 3,49 эквив.) в воде (50 мл), затем МТВЕ (50 мл). Фазы разделяют и водную фазу промывают МТВЕ (30 мл). Объединенные органические фазы промывают водным гидроксидом натрия (0,5М, 2×50 мл) при 5°, затем водой (2×10 мл), затем дважды смесью насыщенного водного хлорида аммония (15 мл) и воды (35 мл). Органические фазы сушат над сульфатом магния и концентрируют до масла. Аналитически чистый образец указанного в заголовке соединения получают хроматографией (этилацетат/гексан): ТСХ Rf=0,39 (этилацетат/гексан, 25/75); ВЭЖХ время удерживания = 8,15, 8,50 мин; ЯМР (CDCl3) 8,15-7,85, 7,48-7,01, 3,99, 3,92, 3,78, 3,50-3,39, 3,38, 3,32-1,21, 0,82 и 0,74-0,67 Пример 7. [3 Раствор водного гидроксида натрия с температурой 4° (1 М, 11,4 мл, 11,4 ммоль, 1,89 эквив.) в метаноле (35 мл) добавляют к сырому (3S),(7R)-4-карбометокси-7-гидрокси-3-(3-нитрофенил)-7-(2-фенилэтил)декан-5-ону (смесь диастереомеров при С-4) (XVI, пример 6, 73,3 вес.%, 3,740 г, 6,018 ммоль) и промывают метанолом (45 мл) при поддержании температуры <5°. Смесь интенсивно перемешивают до растворения большей части сырого масла, затем перемешивают со средней скоростью при 0-5° в течение 67 часов. Смесь охлаждают до -5° и добавляют гексан (90 мл). Фазы разделяют при <5° и органическую фазу промывают при <5° смесью метанола (50 мл) и воды (7 мл). рН объединенной водной фазы доводят от 12,55 до 6,24 при <5° уксусной кислотой (1,52 г, 25,31 ммоль, 4,21 эквив.). Водную фазу концентрируют, экстрагируют хлористым метиленом (2×40 мл), сушат над сульфатом магния и концентрируют, получая сырой продукт. К образцу сырого продукта (0,401 г) добавляют эфир (1,0 мл). Полученную суспензию охлаждают до -30° и осадок собирают фильтрованием в вакууме, промывают холодным эфиром и сушат в токе азота, получая указанное в заголовке соединение, ТСХ Rf=0,49 (этилацетат/гексан, 1/1); ВЭЖХ время удерживания = 6,93 мин; ЯМР (CDCl3/CD3OD, 1/1) 8,08, 7,80, 7,56, 7,22, 7,07-6,88, 3,98, 3,33-3,30, 2,50-2,37, 1,92-1,70, 1,58-1,50, 1,22-1,14, 0,76 и 0,72 Пример 8. [3 К раствору [3 Пример 9. [R-(R*,R*)]-N-[3-[1-[5,6-дигидро-4-гидрокси-2-оксо-6-(2-фенилэтил)-6-пропил-2Н-пиран-3-ил]пропил]фенил]-5-(трифторметил)-2-пиридинсульфонамида (XIX) К смеси [3 СХЕМА С СХЕМА С – продолжение СХЕМА D СХЕМА D – продолжение СХЕМА Е
Формула изобретения
(S)-Метил 3-(3-нитрофенил) пентаноат.
|
||||||||||||||||||||||||||
 -гидроксикарбонильного соединения в кольцо, аналогичное кольцу в формулах (VI) и (CVI).
-гидроксикарбонильного соединения в кольцо, аналогичное кольцу в формулах (VI) и (CVI).  ,
, ) в слабом поле относительно ТМС (тетраметилсилана).
) в слабом поле относительно ТМС (тетраметилсилана).