Патент на изобретение №2260011
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА КАРМИНОМИЦИНА ИЛИ ЕГО ГИДРОХЛОРИДА
(57) Реферат:
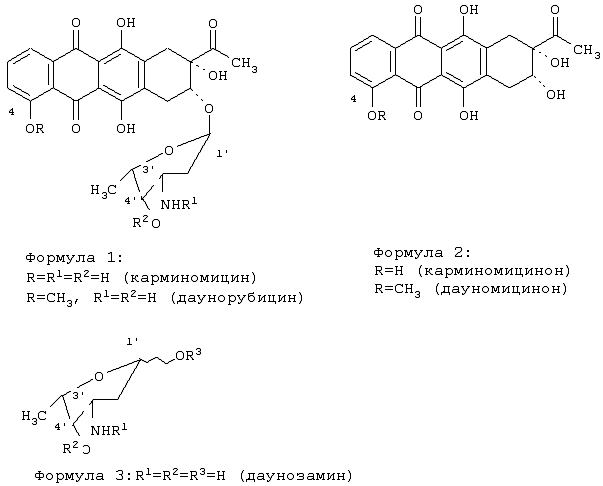
Изобретение относится к способу получения антибиотика карминомицина или его гидрохлорида. Способ включает гликозилирование карминомицинона 1,4-ди-O-замещенным-N-ацилдаунозамином (2,3,6-тридезокси-3-амино-L-ликсо-гексапиранозой) формулы 3 где R1=трифторацетил, N-(9-Н-флуорен-2-илметокси)карбонил, аллилоксикарбонил, R2=ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил, R3=трихлорацетамидил, триалкилсилил, ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил, в присутствии триметилсилилтрифторметансульфоната в смеси безводных апротонных органических растворителей, удаление блокирующих групп и выделение карминомицина в виде основания или гидрохлорида. Гликозил-донор используют в виде смеси
(56) (продолжение): CLASS=”b560m”Chem. Lett. 1984, p.501-504. US 4965352 A 23.10.1990. SU 508076 A, 05.10.1976. CASSINELLI G. et al. “Preparation and biological evaluation of 4-O-demethyldaunorubicin (Carminomycin I) and its 13-dihydro derivative” J. of antibiotics, 1978, v.31, №, p.178-184.
Изобретение относится к фармацевтической промышленности и касается способа получения антибиотика карминомицина или его гидрохлорида. Карминомицин (формула 1, R=R1=R2=Н) относится к группе антрациклиновых антибиотиков, зарекомендовавших себя как высокоэффективные противоопухолевые и противолейкозные препараты, широко используемые в клиниках РФ и за рубежом. Антибиотик карминомицин является оригинальным отечественным антибиотиком, который был впервые получен путем биосинтеза в Институте по изысканию антибиотиков АМН СССР. Карминомицин успешно прошел клинические испытания, и был разрешен к применению в СССР и РФ. Карминомицин рекомендован при следующих заболеваниях: саркома мягких тканей (лейкомиосаркома, рабдомиосаркома, липосаркома и др.); ретикулосаркома и лимфосаркома; рак молочной железы и хорионэпителиома матки; острые миелобластные и лимфобластные лейкозы. Антибиотик имеет широкие показания при лечении злокачественных новообразований у взрослых и детей и применяется как индивидуальный препарат, так и в комбинациях с другими лекарственными средствами. Известен способ биосинтетического получения карминомицина [а.с. “Способ получения карминомицина” №508076 от 11.11.1975 г, а.с. “Способ получения карминомицина” №543257 от 21.09.1976 г.]. Недостатком этого метода является присутствие сопутствующей примеси – 13-дигидрокарминомицина, низкая продуктивность штамма-продуцента Actinomadura carminata по сравнению с продуктивностью штаммов противоопухолевых антибиотиков антрациклиновой группы, сложность процесса выделения и очистки. Известен способ получения карминомицина химическим синтезом [Cassinelli G., Grein A., Masi P., Suarato A., Bemardi L., Arcamone F.Н. Preparation and biological evaluation of 4-0-demethyldaunorubicin (carminomycin I) and its 13-dihydro derivative -Journal of Antibiotics, 31, 178-184, 1978]. Гидролизом гликозидной связи даунорубицина получают агликон дауномицинон (формула 2, R=СН3) и моносахарид – даунозамин (формула 3, R1=R2=R3=H). Дауномицинон подвергают деметилированию реакцией с AlClз в кипящем хлористом метилене. Полученный с выходом 62% агликон карминомицинон (формула 2, R=H) гликозилируют 2,3,6-тридезокси-3-трифторацетамидо-4-O-трифторацетил- Техническая задача изобретения – увеличение выхода карминомицина при получении его путем химического синтеза. Техническая задача решается тем, что предложен способ получения карминомицина или его гидрохлорида, включающий гликозилирование карминомицинона гликозил-донором 1,4-ди-O-замещенным-3-N-ацилдаунозамином в присутствии катализатора в смеси безводных апротонных органических растворителей, удаление блокирующих групп и выделение карминомицина в виде основания или гидрохлорида, при этом в качестве катализатора используется триметилсилилтрифторметансульфонат, в качестве гликозилдонора используется 1,4-ди-O-замещенный-3-N-ацилдаунозамин (2,3,6-тридезокси-3-амино-L-ликсо-гексапираноза), где R1=трифторацетил или N-(9-Н-флуорен-2-илметокси)карбонил или аллилоксикарбонил, преимущественно N-(9-Н-флуорен-2-илметокси)карбонил или трифторацетил, R2=ацетил или трифторацетил или 4-нитробензоил или аллилоксикарбонил, преимущественно ацетил, R3=трихлорацетамидил или триалкилсилил или ацетил или трифторацетил или 4-нитробензоил или аллилоксикарбонил, преимущественно ацетил, в качестве безводных апротонных органических растворителей используют диоксан-метиленхлорид или диоксан-хлороформ или диоксан-метиленхлорид-диэтиловый эфир или диоксан-ацетон или диоксан-хлороформ-диэтиловый эфир или диоксан-тетрагидрофуран-метиленхлорид, преимущественно диоксан-метиленхлорид, при этом реакция проводится в присутствии молекулярных сит (преимущественно 4Å), удаление блокирующих групп проводят щелочным агентом. В качестве гликозил-донора используют смесь Для защиты 3-амино и 4-O-гидроксильной групп даунозамина используют сочетание блокирующих групп, устойчивых в слабокислых средах. Введение защитных и активирующих групп проводили по стандартным методикам, используемых в химии углеводов [H.M.I.OSBORN, Best Synthetic methods, Carbohydrates, Academic press, 2003; Handbook of Reagents for Organic Synthesis “Activating Agents and Protective Groups”, edited by A.J.Pearson, W.J. Roush, JOHN WILEY & SONS, 1999]. Частный случай в виде использования 1,4-ди-O-замещенного-N-ацилдаунозамина, содержащего в положениях 1-O- и 4-O- одну и ту же группу ацильного типа (формула 3, где R2=R3=ацильная группа) преимущественно ацетильную, позволяет упростить синтез гликозил-донора. Используя эти соединения (формула 3, где R2=R3=ацильная группа), также возможна замена активирующей ацильной группы R3 на другую активирующую группу путем ее селективного гидролиза до гидроксильной группы (с сохранением защитных групп R1, R2), с последующим введением другой активирующей группы (R3) [H.M.I.OSBORN, Best Synthetic methods. Carbohydrates, Academic press, 2003, p.164]. Предлагаемый способ позволяет увеличить выход карминомицина в 3 раза по сравнению с прототипом. Вспомогательные средства: Триметилсилилтрифторметансульфонат, диэтиламин, молекулярные сита 4Å, триэтиламин, 4-(диметиламино)пиридин были коммерческим продуктами фирмы Aldrich (США). N-(9Н-Флуорен-2-илметоксикарбонил)сукцинимид был коммерческим продуктом фирмы Merck (Германия). Тонкослойную хроматографию осуществляли на пластинках с силикагелем G60 (Merck) в смесях растворителей: петролейный эфир-этилацетат, 1:1 (А); хлороформ-метанол-муравьиная кислота, 20:1:0.1 (Б); хлороформ-метанол-25%-ный водный аммиак, 7:3:0.01 (В); хлороформ-метанол-муравьиная кислота, 7:1:0.1 (Г). Для препаративной очистки использовали колоночную хроматографию на силикагеле Merck G60 с размером частиц 0.040-0.063 mm. ВЭЖХ проводили на приборе Shimadzu HPLC LC 10 на колонке Diaspher С 18 №1914, элюент: 0.01н Н3PO4 – MeCN, pH 2.6, в градиенте ацетонитрила от 10% до 90%, скорость потока 1.1 мл/мин. Регистрация велась на длине волны 254 нм, при температуре 20°С. Оптическое вращение [ Пример 1. Карминомицин гидрохлорид. Карминомицинон (0.500 г, 1.30 ммоль) растворяли в смеси сухой хлористый метилен – сухой диоксан (4:1, 375 мл), добавляли 1,4-ди-O-ацетил-3-N-(9-Н-флуорен-2-илметокси)карбонилдаунозамин ( Выход 250 мг (35% в расчете на карминомицинон), Rf(Г) 0.22, Rt 16.20. Тпл 185-187°С (с разложением) 1Н-ЯМР (Py-d5): 7.835 (d, 1Н, 7.33 Hz, H-1); 7.62 (t, 1H, 7.81 Hz, H-2); 7.325 (d, 1H, 8.30 Hz, H-3); 5.82 (s, 1H, W7 Hz, Н-1′); 5.35 (m, 1H, H-7); 4.70 (q, 1H, 6.65 Hz, H-5′); 4..50 (s, 1H, W7 Hz, H-4′); 4.39 (m, 1H, H-3′); 3.60d, 3.42d (2H, 17 Hz, H-10), 2.75 (m, 1H, H-2′); 2.75 (m, 2H, 2H-8); 2.57 (s, 3Н, 3Н-14); 2.80m, 2.45dd, (2H, Jgem 14 Hz, 4.3Hz, 2H-H-2′); 1.43(d, 3H, 6.65 Hz, 3H-6′)
[ Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [ Пример 2. Карминомицин гидрохлорид. Карминомицинон (0.520 г, 1.35 ммоль) растворяли в смеси сухой хлористый метилен – сухой диоксан (4:1, 600 мл), добавляли 1,4-ди-O-ацетил-3-N-трифторацетилдаунозамин ( Выход 230 мг (31% в расчете на карминомицинон), Rf(Г) 0.22, Rt 16.201. Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [ Пример 3. Карминомицин гидрохлорид. Синтез ведется аналогично предыдущему примеру, исходя из 100 мг (0.26 ммоль) карминомицинона, 102 мг (0.31 ммоль, 1.2 экв) 1,4-ди-O-ацетил-N-трифторацетилдаунозамина (чистый Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [ Пример 4. 1,4-Ди-O-ацетил-3-N-(9-Н-флуорен-2-илметокси)карбонилдаунозамин ( Даунозамин гидрохлорид (1.00 г, 5.45 ммоль) растворяли в смеси диоксан – вода (14 мл, 1:1), добавляли Et3N до рН 8, порциями добавляли сухой Н-(9Н-флуорен-2-илметоксикарбонил)сукцинимид (2.20 г, 6.54 ммоль), поддерживая рН 8 добавлением Et3N. Реакционную смесь перемешивали 20 часов, добавляли 30 мл воды, частично упаривали диоксан. Выпавший осадок отфильтровывали и высушивали. Осадок растворяли в этилацетате и добавляли петролейный эфир. Выпавший осадок отфильтровывали, упаривали с пиридином и использовали в следующей стадии без дополнительной очистки. 3-N-(9-Н-Флуорен-2-илметокси)карбонилдаунозамин [2 г, Rf=0.75 (В)] растворяли в сухом пиридине (20 мл) и добавляли уксусный ангидрид (1.03 мл, 10.9 ммоль) и каталитическое количество 4-(диметиламино)пиридина (30 мг). Реакционную смесь перемешивали в течение 20 часов, добавляли 1 мл метанола, перемешивали в течении 1 часа и упаривали досуха. Остаток растворяли в этилацетате и добавляли петролейный эфир. Выпавший осадок отфильтровывали, высушивали. Выход 1.97 г (80% в расчете на даунозамин). Соотношение аномеров Пример 5. 1,4-Ди-O-ацетил-3-N-трифторацетилдаунозамин ( Даунозамин гидрохлорид (4) (1.26 г, 6.87 ммоль) растворяли в сухом МеОН (20 мл), добавляли Et3N (2.87 мл, 2.6 ммоль) и этиловый эфир трифторуксусной кислоты (1.63 мл, 13.73 ммоль). Реакционную смесь перемешивали в течение 3 ч, затем упаривали. Остаток растворяли в горячем ацетоне (40 мл), выпавший осадок отфильтровывали. Раствор упаривали, остаток повторно упаривали из сухого пиридина и использовали без дополнительной очистки в следующей стадии. 3-N-Трифторацетилдаунозамин (1.42 г, Rf 0.70, В) растворяли в абсолютном пиридине (30 мл), добавляли Ас2О (1.8 мл) и каталитическое количество 4-(диметиламино)пиридина (20 мг). Реакционную смесь перемешивали в течение 48 ч, после чего охлаждали до 0°С и добавляли МеОН (1 мл). Реакционную смесь перемешивали в течение 1 ч, затем упаривали. Остаток делили флэш-хроматографией на силикагеле, петролейный эфир – этилацетат, используя градиент этилацетата от 20% до 50%. Выделяли три фракции: Пример 6. Карминомицинон. К раствору дауномицинона (2.50 г, 6.28 ммоль) в сухом дихлорэтане (300 мл) добавляли AlCl3 (2.51 г, 18.84 ммоль). Реакционную смесь кипятили в течение 1.5 ч, затем добавляли свежую порцию AlCl3 (2.51 г, 18.84 ммоль) и кипятили в течение 1.5 ч, процедуру повторяли еще раз (всего добавляли 8 г AlCl3), после чего выливали реакционную смесь в воду (400 мл), подкисляли водную фракцию 4н HCl до рН 3. Отделяли водный слой и нагревали его до кипения. Выпавший осадок отфильтровывали, промывали водой, высушивали. Органическую фракцию высушивали, частично упаривали и оставляли кристаллизоваться сначала при комнатной температуре в течение 4 часов, а затем при 4°С в течение 20 часов. Выпавший осадок отфильтровывали, объединяли с осадком из водной фракции и высушивали до постоянного веса. Выход 2.05 г (85%), Rf(A) 0.37, Rf(B) 0.47. Пример 7. Карминомицин гидрохлорид. Карминомицинон (2.60 г, 6.77 ммоль) растворяли в смеси сухой хлористый метилен – сухой диоксан (4:1, 1.8 л), добавляли 1,4-ди-O-ацетил-3-N-трифторацетилдаунозамин ( Выход 1.23 г (33% в расчете на карминомицинон), Rf(Г) 0.22, Rt 16.203. Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [ Способ получения антибиотика карминомицина или его гидрохлорида R1=трифторацетил, N-(9-Н-флуорен-2-илметокси)карбонил, аллилоксикарбонил; R2=ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил; R3=трихлорацетамидил, триалкилсилил, ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил.
Формула изобретения
1. Способ получения карминомицина или его гидрохлорида, включающий гликозилирование карминомицинона гликозил-донором 1-,4-O-,3-N-тризамещенным даунозамином в присутствии катализатора в смеси безводных апротонных органических растворителей, удаление блокирующих групп и выделение карминомицина в виде основания или гидрохлорида, отличающийся тем, что в качестве катализатора используют триметилсилилтрифторметансульфонат, в качестве гликозил-донора используют 1,4-ди-O-замещенный-3-N-ацилдаунозамин (2,3,6-тридезокси-3-амино-L-ликсо-гексапираноза), где R1 – трифторацетил, N-(9-Н-флуорен-2-илметокси)карбонил, аллилоксикарбонил, R2 – ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил, R3 – трихлорацетамидил, триалкилсилил, ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил, в качестве безводных апротонных органических растворителей используют диоксан-метиленхлорид, диоксан-хлороформ, диоксан-метиленхлорид-диэтиловый эфир, диоксан-ацетон, диоксан-хлороформ-диэтиловый эфир, диоксан-тетрагидрофуран-метиленхлорид, при этом реакцию проводят в присутствии молекулярных сит, удаление блокирующих групп проводят щелочным агентом. 2. Способ по п.1, отличающийся тем, что гликозил-донор используют в виде смеси 3. Способ по п.1, отличающийся тем, что реакцию проводят при температуре не выше 10°С. 4. Способ по п.1, отличающийся тем, что реакцию проводят в атмосфере осушенного воздуха или осушенного инертного газа. 5. Способ по п.1, отличающийся тем, что удаление защитных групп проводят в присутствии гидроксидов щелочных металлов в водно-органической среде: вода – метанол, вода – диоксан, вода – тетрагидрофуран, вода – ацетон.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 28.02.2006
Извещение опубликовано: 10.02.2007 БИ: 04/2007
NF4A Восстановление действия патента СССР или патента Российской Федерации на изобретение
Дата, с которой действие патента восстановлено: 20.05.2007
Извещение опубликовано: 20.05.2007 БИ: 14/2007
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 28.02.2008
Извещение опубликовано: 27.02.2010 БИ: 06/2010
|
||||||||||||||||||||||||||
 и
и  изомеров или в виде индивидуальных
изомеров или в виде индивидуальных 
 max 236, 256, 293, 464, 478, 492, 512, 525 нм (в этаноле)
max 236, 256, 293, 464, 478, 492, 512, 525 нм (в этаноле) l триметилсилилтрифторметансульфоната. Выход гидрохлорида карминомицина – 38 мг (28% в расчете на карминомицинон).
l триметилсилилтрифторметансульфоната. Выход гидрохлорида карминомицина – 38 мг (28% в расчете на карминомицинон). Rf
Rf