Патент на изобретение №2258062
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ НИТРОМЕТАНА
(57) Реферат:
Предложен способ получения нитрометана путем взаимодействия раствора монохлорацетата натрия с раствором нитрита натрия в водной среде при повышенной температуре в две стадии. На первой стадии поддерживают рН 3,5-4,5, а на второй стадии водный раствор нитрометана подвергают прямой перегонке с отделением нитрометана в сепараторе. Технический результат: понижение энергоемкости за счет проведения двух стадий на одном оборудовании, доступность сырья, увеличение выхода нитрометана. 2 з.п. ф-лы, 2 ил.
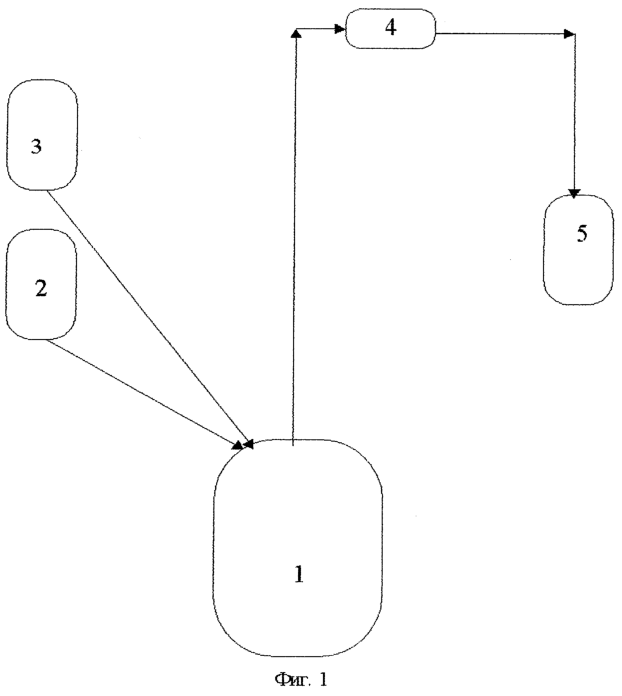
Изобретение относится к технологии органических соединении, а именно к разработке способа получения нитрометана, который широко используется как полупродукт в химической и фармацевтической промышленности, а также как добавка к топливу для форсированных двигателей. Наиболее известным и широко применяемым на практике методом получения нитрометана является взаимодействие хлоруксусной кислоты и нитрита натрия в водной среде и при повышенной температуре. Это метод Кольбе (Е.Ю.Орлова. «Руководство к лабораторным работам по органической химии», М., 1969, стр.241). Процесс осуществляют следующим образом. К водному раствору хлоруксусной кислоты добавляют щелочной агент (углекислый натрий, углекислый калий, аммиак и т.д.) до полной нейтрализации хлоруксусной кислоты. К полученной массе добавляется водный раствор нитрита натрия, и смесь компонентов постепенно нагревается до начала отгонки нитрометана. При 80-85°С, как только начинают выделяться пузырьки углекислого газа, подогрев прекращают. При этой температуре реакция продолжается без внешнего подогрева, и образующийся нитрометан отгоняется с водой в виде тяжелых маслянистых капель. По мере отгонки температура повышается до 100°С. Щелочная среда, возникающая в ходе реакции, приводит к снижению выхода нитрометана, что является недостатком описанного выше способа. Выход нитрометана составляет не более 40% от теории. Известен непрерывный способ получения нитрометана путем взаимодействия диметилсульфата и нитрита натрия в водной среде в каскаде из четырех реакторов, при температуре от 40° С до 115°С, с выделением целевого продукта азеотропной отгонкой при рН-5-7 и последующей сепарацией дистиллята (Патент №2045514, RU, авторы Белоусов Ю.М и др.). Недостатком данного метода получения нитрометана является сложное аппаратурное оформление технологического процесса, большие энергетические затраты, большое количество примесей в нитрометане (нитрометан, полученный этим способом, содержит до 10% метанола, от которого приходится избавляться на сложных ректификационных колоннах и который создает дополнительные экологические проблемы, метанол – яд). Известен способ получения нитрометана из производных хлоруксусной кислоты, щелочного реагента и нитрита натрия в водной среде и при повышенной температуре (Патент №2154627, RU авторы Гареев Г.А и др.), по которому хлоруксусную кислоту применяют в свободном виде, а щелочного реагента берут в количествах, необходимых для обеспечения кислотности среды в пределах рН 6-8, и процесс ведут в условиях постоянной отгонки образующегося нитрометана в виде азеотропа с водой. Отделение нитрометана от воды осуществляется путем азеотропной сушки, а отделение от других примесей – путем ректификации на перегонной установке. Взято за прототип. Недостатками данного способа (прототипа) является следующее: 1. При ведении процесса синтеза нитрометана в реакторе образуются окислы азота, которые через эжекцию выводятся из реактора и поступают на абсорбционную установку для их улова и нейтрализации, т.е. требуются дополнительные энергозатраты, оборудование, затраты на утилизацию образующейся кислоты. 2. Полученный этим способом нитрометан имеет очень кислую среду (рН=1-2) и не может быть использован как добавка к топливам для форсированных и авиамодельных двигателей. Для его нейтрализации нужны дополнительные щелочные реагенты, оборудование, энергозатраты. Целью предлагаемого изобретения является: получение нитрометана простым, экологически безопасным способом, с наименьшими энергозатратами и с выходом нитрометана 50-55%. Для достижения такого технического результата нитрометан получают путем взаимодействия монохлорацетата натрия с нитритом натрия в водной среде при повышенной температуре в две стадии. На первой стадии водный раствор монохлорацетата натрия дозируют в водный раствор нитрита натрия и непрерывно отгоняют образующийся водный раствор нитрометана и затем сразу же стабилизируют его ортофосфорной кислотой до рН 3,5-4,5, а на второй стадии водный раствор нитрометана подвергают прямой перегонке с отделением нитрометана в сепараторе. Растворимость нитрометана в 100 весовых частях воды 9,5 г (при 20°C), 12,2 г (при 60°С). Синтез нитрометана проводят путем дозирования раствора ацетата натрия в раствор нитрита натрия, при повышенной температуре с одновременной отгонкой нитрометана в виде водного раствора при температуре в кубе 100-110°С, затем проводят его прямую перегонку при температуре в кубе 80-100°С, при этом нитрометан отделяется от воды в сепараторе. Полученный после сепаратора нитрометан стабилизируют ортофосфорной кислотой до рН-4,7-5,2 и осушают силикагелем или ортофосфорной кислотой до содержания в нем влаги 0,01-1,0% мас. Проведение синтеза нитрометана в две стадии позволяет непрерывно отгонять образовывающийся водный раствор нитрометана, не давая необратимо разложиться нитрометану в щелочной среде, стабилизировать его ортофосфорной кислотой (перевести в подкисленное состояние) до рН 3,5-4,5. При рН менее 3,5 увеличивается расход ортофосфорной кислоты, а стабильность остается прежней. При рН более 4,5 стабильность нитрометана недостаточная. Для реализации заявляемого способа используют существующее серийное оборудование. Предложенный способ получения ниторометана проверен практически, схематически показан на чертежах (фиг.1, фиг.2) и включает следующие стадии. 1. Приготовление водного раствора монохлорацетата натрия. В аппарате приготовления раствора ацетата натрия из 300 л артезианской воды и 150 кг монохлорацетата натрия приготовили раствор. 2. Приготовление водного раствора нитрита натрия. В аппарате приготовления раствора нитрита натрия из 260 л артезианской воды и 100 кг нитрита натрия приготовили раствор нитрита натрия. Перед синтезом раствор сливают в реактор синтеза. Синтез нитрометана В раствор нитрита натрия, предварительно подогретый до 101-103°С, сдозировали раствор монохлорацетата натрия. Через 15-20 минут с момента начала дозировки монохлорацетата натрия началась отгонка из реактора паров водного раствора нитрометана, в ходе дозировки рабочая температура в реакторе установилась в пределах 101-110°С. Регулируя температуру в реакторе, достигли полного вывода нитрометана из реакционной массы. После конденсации в холодильнике водный раствор нитрометана собрали в сборнике водного раствора, где подвергли немедленной стабилизации (подкислению ортофосфорной кислотой до рН 3,5-4,5). Стабилизацию (подкисление ортофосфорной кислотой) нитрометана и водного раствора нитрометана производят сразу же при проведении синтеза и прямой перегонки, т.к. в щелочной среде происходит необратимое разложение нитрометана. После того как водный раствор нитрометана стабилизировали, его можно накапливать, хранить или подвергнуть перегонке с получением нитрометана. 4. Отгонка водного слоя. Перегонку водного раствора нитрометана проводили на том же оборудовании, что и синтез, с использованием сепаратора для отделения нитрометана. Водный слой подвергли перегонке в аппарате для перегонки (он же реактор на 1 стадии синтеза) с получением нитрометана-сырца, водного слоя и кубового остатка. Прямую перегонку проводили при температуре в кубе 80-100°С. Нитрометан, полученный в результате отгонки водного раствора, стабилизировали ортофосфорной кислотой до рН 4,7-5,2 и осушили силикагелем или ортофосфорной кислотой до содержания в нем влаги 0,01-1,0% мас. Преимущества данного способа получения нитрометана: 1. Простое аппаратурное оформление (обе стадии технологического процесса проводятся на одном оборудовании), низкая энергоемкость, доступность сырья, т.к. все сырье производится в России. 2. Полученый нитрометан не имеет примесей, кроме воды, что не требует его дальнейшей очистки путем сложной ректификации. Воду удаляют до содержания 0,5-1% мас., пропуская его через силикагель, при необходимости получения нитрометана с влагой 0,01-0,4% мас. его осушают ортофосфорной кислотой, которая затем отделяется и используется в дальнейшем для стабилизации водного раствора нитрометана при синтезе. 3. За счет применения 2-стадийного процесса нитрометан, образующийся во время синтеза, немедленно выводится из аппарата, не успев разложиться в щелочной среде, за счет чего увеличивается его выход до 50-55% от теории. Полученный таким способом нитрометан соответствует требованиям ТУ 2471-006-20992222-2002.
Формула изобретения
1. Способ получения нитрометана путем взаимодействия раствора монохлорацетата натрия с раствором нитрита натрия в водной среде при повышенной температуре, отличающийся тем, что на первой стадии водный раствор монохлорацетата натрия дозируют в водный раствор нитрита натрия и непрерывно отгоняют образовавшийся водный раствор нитрометана, стабилизируют его ортофосфорной кислотой до рН 3,5-4,5, а на второй стадии водный раствор нитрометана подвергают прямой перегонке с отделением нитрометана в сепараторе. 2. Способ по п.1, отличающийся тем, что отгонку водного раствора нитрометана ведут при температуре в кубе 100-110°С, прямую перегонку при температуре в кубе 80-100°С, а полученный при этом нитрометан стабилизируют ортофосфорной кислотой до рН 4,7-5,2. 3. Способ по п.1, отличающийся тем, что полученный после сепаратора нитрометан осушают силикагелем или ортофосфорной кислотой до содержания в нем влаги 0,01-1,0 мас.%.
РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 02.03.2008
Извещение опубликовано: 10.03.2010 БИ: 07/2010
|
||||||||||||||||||||||||||