Патент на изобретение №2256666
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ПИРИМИДИНОВОГО НУКЛЕОЗИДА (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ОПУХОЛЕВЫХ ЗАБОЛЕВАНИЙ И ПРИМЕНЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ
(57) Реферат:
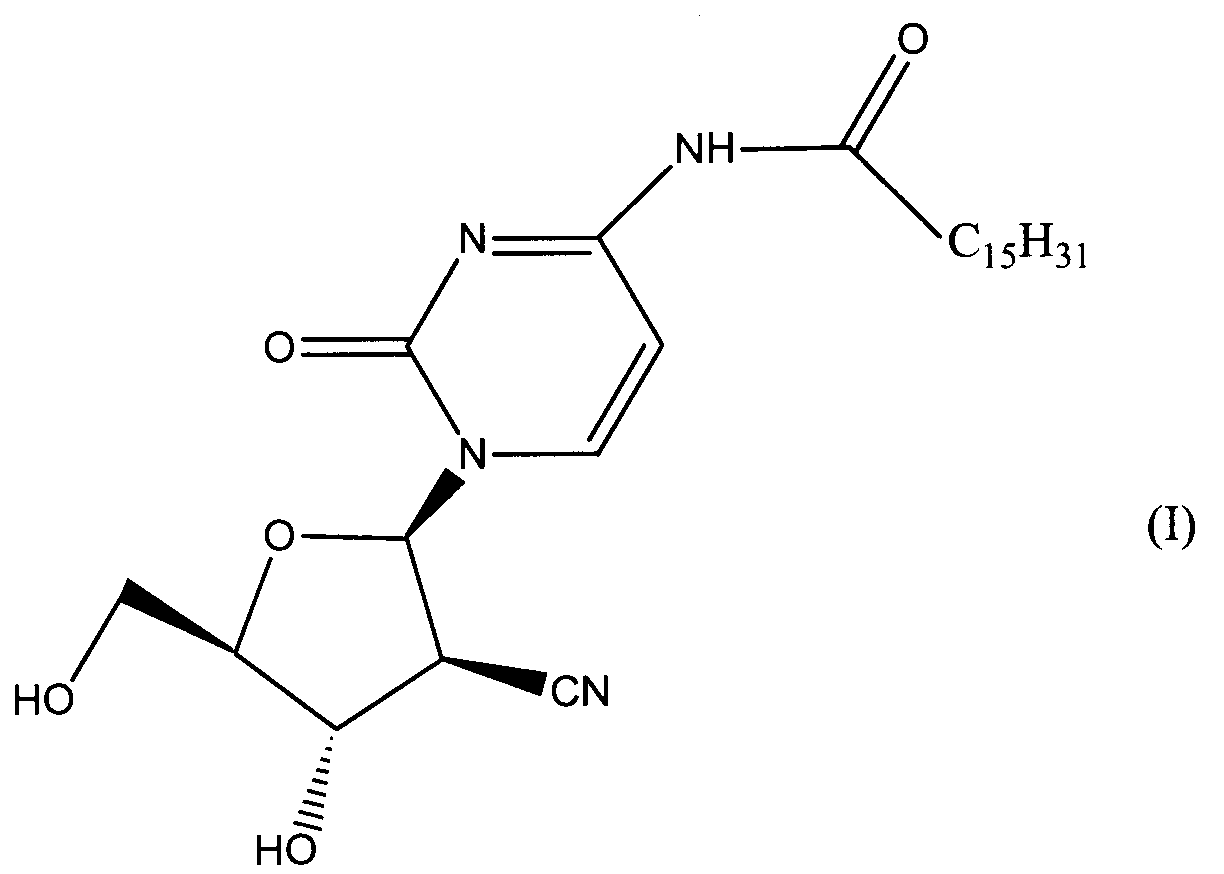
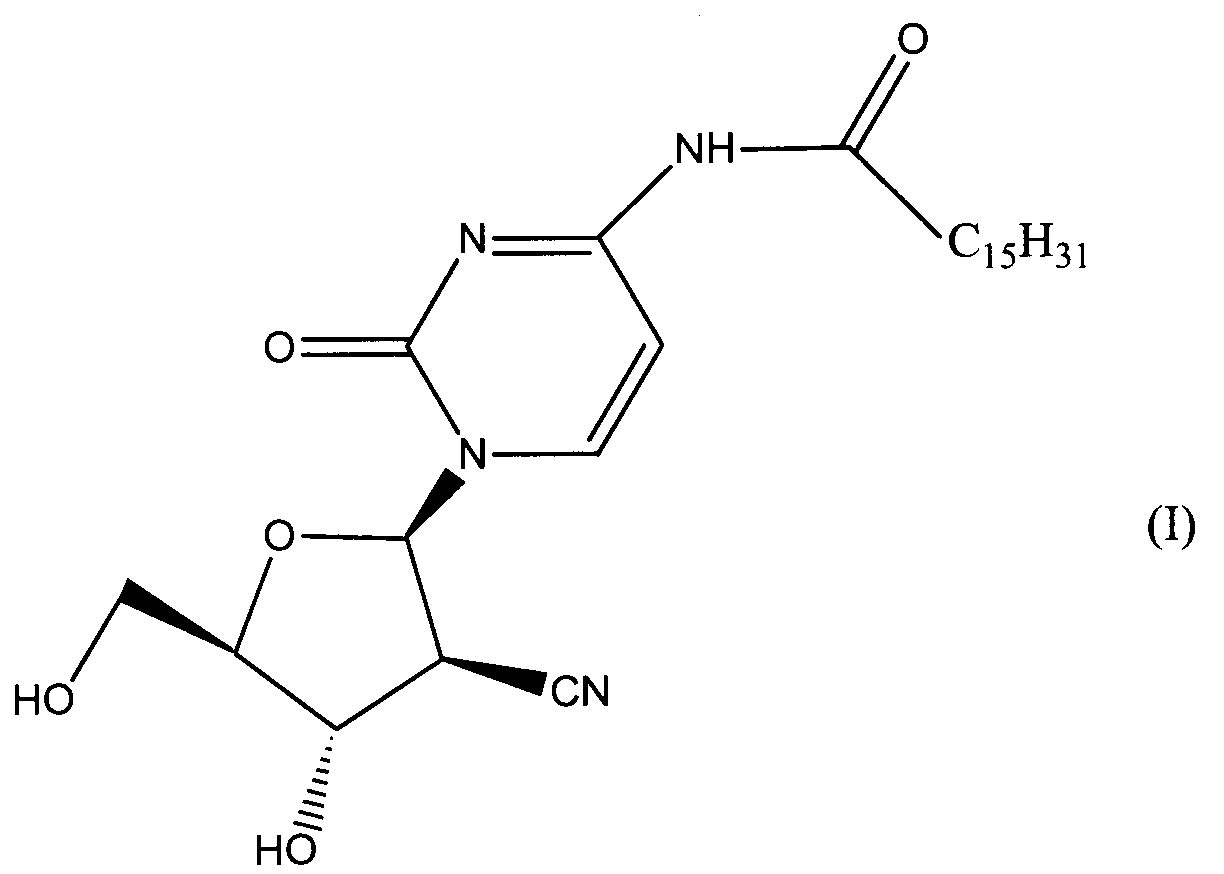
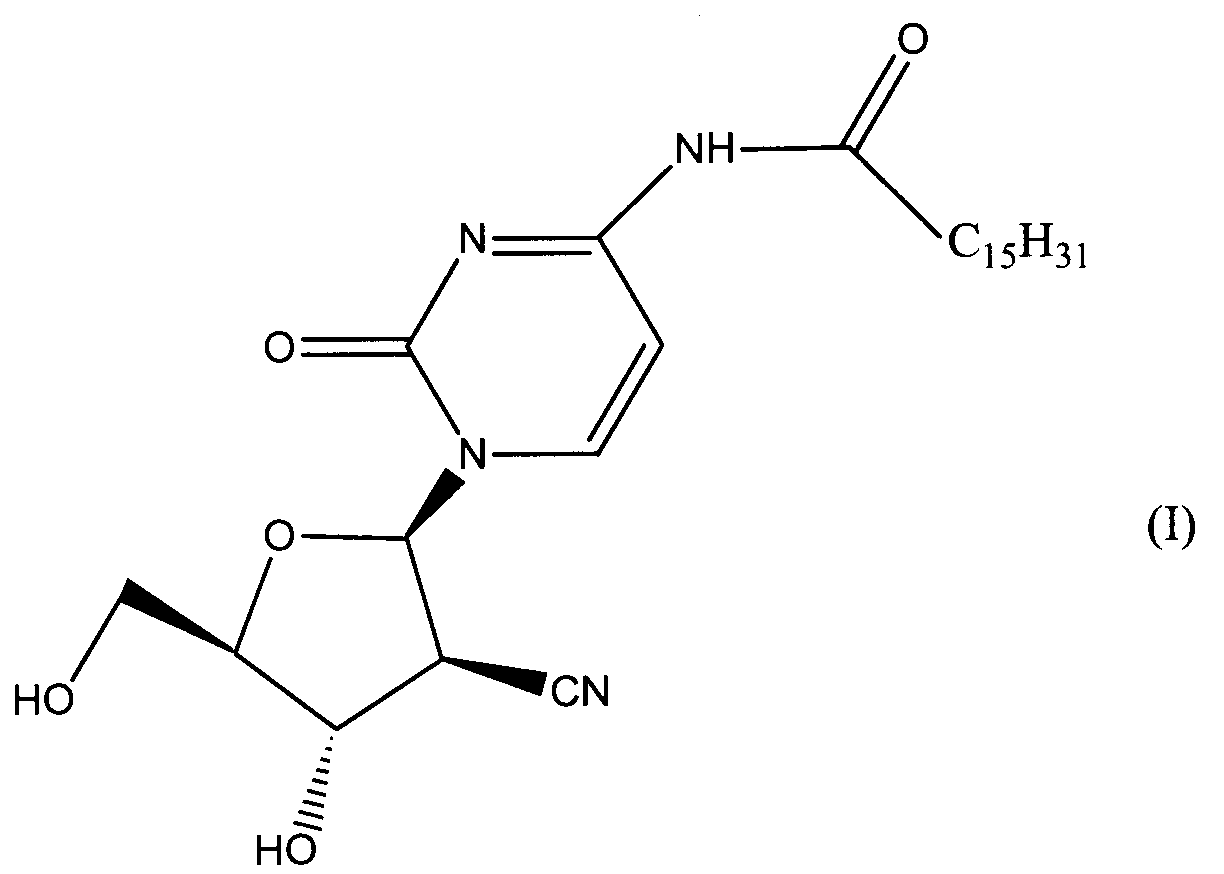
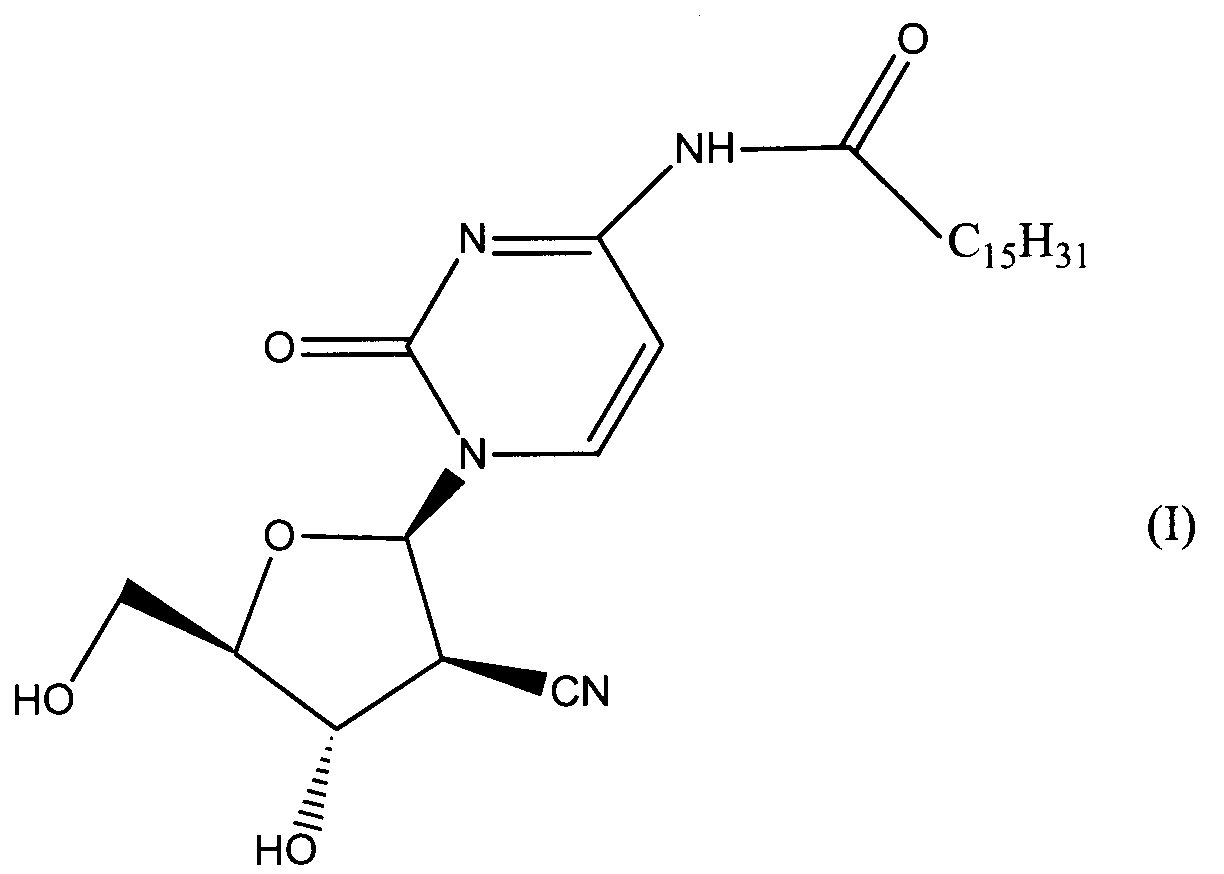
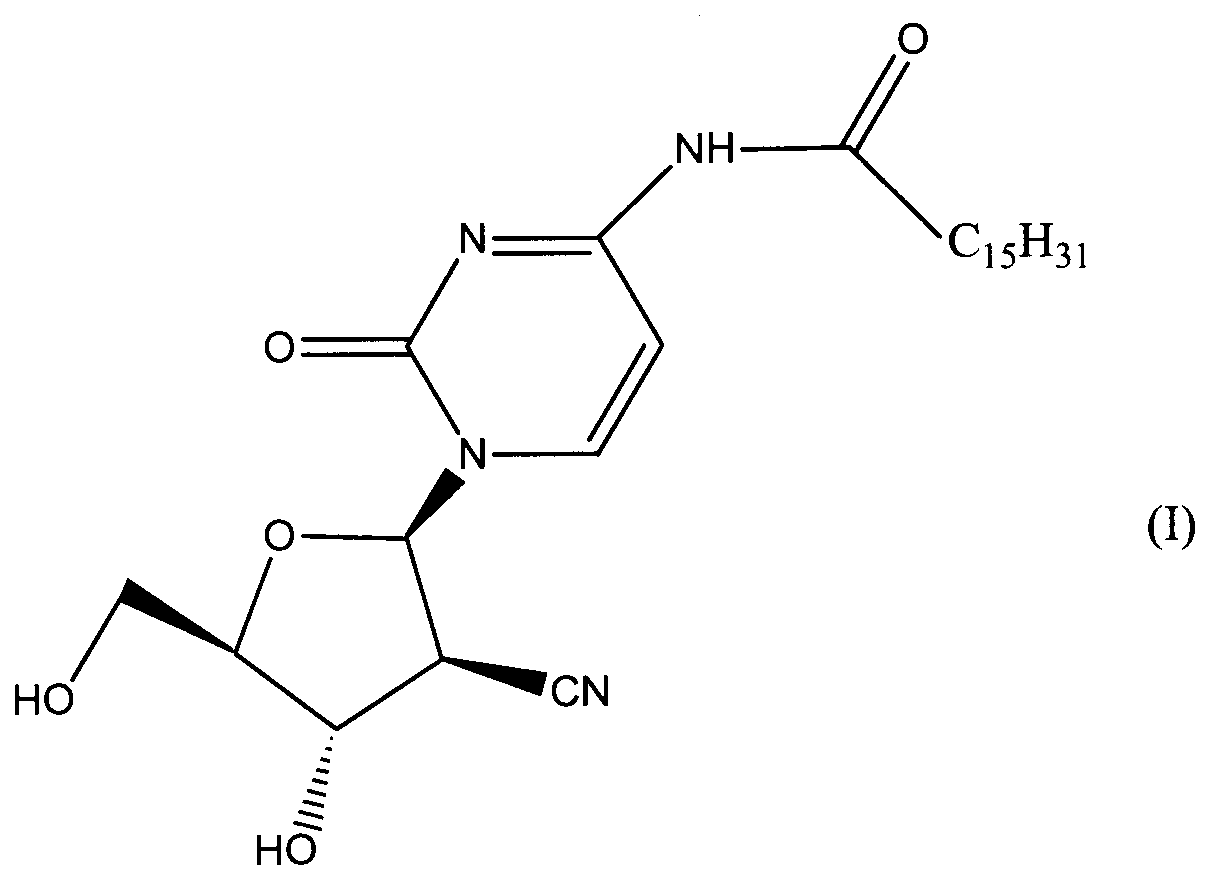
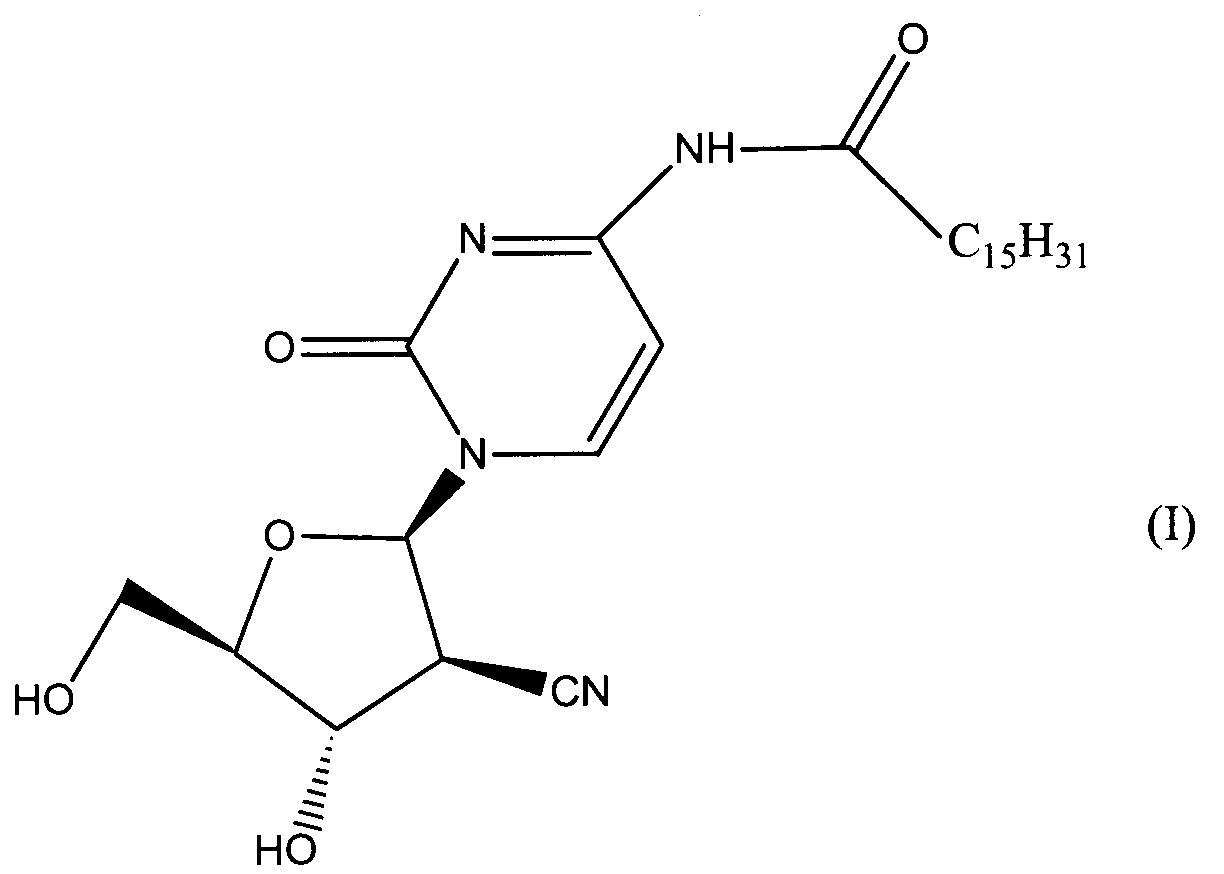
Данное изобретение относится к новым устойчивым кристаллическим формам производного пиримидинового нуклеозида формулы (I), обладающим превосходной противоопухолевой активностью. Изобретение также относится к фармацевтической композиции, обладающей противоопухолевым действием, применению кристаллической формы для получения лекарственного средства и способу профилактики или лечения опухолевых заболеваний. 7 н. 3 з.п. ф-лы, 2 табл., 4 ил.
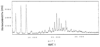
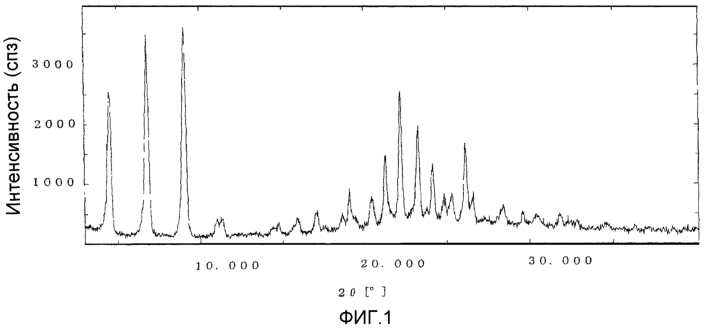
Область техники Данное изобретение относится к кристаллическим формам производного пиримидинового нуклеозида, которое обладает превосходной противоопухолевой активностью; к фармацевтическим композициям (предпочтительно противоопухолевым агентам), содержащим указанную кристаллическую форму в качестве активного ингредиента; к применению указанной кристаллической формы при получении указанных фармацевтических композиций; и к способу профилактики или лечения заболеваний (предпочтительно, опухоли), который включает введение теплокровному животному (предпочтительно, человеку), при необходимости такой профилактики или лечения, фармакологически эффективного количества указанной кристаллической формы. Предпосылки изобретения Производное пиримидинового нуклеозида формулы (I) (далее обозначенное как соединение (I)) описано в патенте Японии №2569251. Соединение (I) обладает превосходной противоопухолевой активностью и рассматривается как агент для лечения или профилактики опухолей. Для получения соединения (I), более приспособленного для применения в качестве лекарственного средства, необходима лучшая устойчивость при хранении, большая простота обращения и так далее. Описание изобретения Авторы данного изобретения изучили устойчивость и другие подобные свойства соединения (I) и преуспели в получении кристаллов соединения (I). Эти кристаллы имеют значительно лучшую устойчивость при хранении и простоту обращения по сравнению с порошком соединения (I), полученным в примере 1, представленном в описании к патенту Японии №2569251. Эти кристаллы демонстрируют превосходный фармакокинетический профиль, такой как оральная абсорбция и подобные свойства, и, поэтому, являются применимыми на практике лекарственными средствами. В соответствии с данным изобретением предлагается: (1) кристаллическая форма соединения формулы (I) (2) кристаллическая форма по (1), где соединение формулы (I) является гидратом, (3) кристаллическая форма по (1) или (2), где указанная кристаллическая форма имеет основные пики на расстояниях кристаллической решетки 19,53, 13,03, 9,75, 4,17, 4,00, 3,82, 3,68 и 3,41 Е, определенных рентгенографией порошковым методом с применением медных лучей K (4) кристаллическая форма по (1) или (2), где указанная кристаллическая форма имеет основные пики на расстояниях кристаллической решетки 19,36, 12,87, 9,63, 4,70, 4,64, 4,28, 4,10, 3,92, 3,77 и 3,48 Е, определенных рентгенографией порошковым методом с применением медных лучей K (5) кристаллическая форма по (1) или (2), где указанная кристаллическая форма имеет основные пики на расстояниях кристаллической решетки 19,62, 13,06, 9,82, 4,72, 4,63, 4,56, 4,15, 3,98, 3,93, 3,82, 3,45 и 3,40 Е, определенных рентгенографией порошковым методом с применением медных лучей K (6) кристаллическая форма по (1) или (2), где указанная кристаллическая форма имеет основные пики на расстояниях кристаллической решетки 22,52, 5,17, 4,60, 4,28 и 3,87 Е, определенных рентгенографией порошковым методом с применением медных лучей K (7) фармацевтическая композиция, содержащая кристаллическую форму по любому из (1)-(6) в качестве активного ингредиента, (8) фармацевтическая композиция по (7) для профилактики или лечения опухолей, (9) применение кристаллической формы по любому из (1)–(6) для получения фармацевтической композиции, (10) применение по (9), где фармацевтическая композиция предназначена для профилактики или лечения опухолей, (11) способ профилактики или лечения заболеваний, включающий введение фармакологически эффективного количества кристаллической формы по любому из (1)–(6) теплокровному животному при необходимости такой профилактики или лечения, (12) способ по (11), в котором заболеванием является опухоль, (13) способ по (11) или (12), в котором теплокровным животным является человек. Кристаллическая форма соединения (I) в соответствии с данным изобретением является твердым веществом, которое имеет правильное повторяющееся расположение атомов (или групп атомов) в трехмерной структуре. Кристалл отличается от аморфного твердого вещества тем, что последнее не имеет правильного расположения атомов в трехмерной структуре. В общем, в зависимости от применяемых условий кристаллизации могут быть получены различные множественные кристаллические формы (полиморфизм) одного и того же соединения. Такие различные кристаллические формы имеют различные трехмерные структуры и имеют различные физико-химические свойства. Данное изобретение охватывает отдельные кристаллические формы и смеси двух или более указанных кристаллических форм. Кристаллические формы соединения (I) включают, например, кристалл, имеющий основные пики на расстояниях решетки d=19,53, 13,03, 9,75, 4,17, 4,00, 3,82, 3,68 и 3,41 Е, определенных рентгенографией порошковым методом с применением медных лучей K (кроме того, расстояние решетки d может быть рассчитано на основе уравнения 2dsin кристалл, имеющий основные пики на расстояниях решетки d=19,36, 12,87, 9,63, 4,70, 4,64, 4,28, 4,10, 3,92, 3,77 и 3,48 Е, определенных рентгенографией порошковым методом с применением медных лучей K кристалл, имеющий основные пики на расстояниях решетки d=19,62, 13,06, 9,82, 4,72, 4,63, 4,56, 4,15, 3,98, 3,93, 3,82, 3,45 и 3,40 Е, определенных рентгенографией порошковым методом с применением медных лучей K кристалл, имеющий основные пики на расстояниях решетки d=22,52, 5,17, 4,60, 4,28 и 3,87 Е, определенных рентгенографией порошковым методом с применением медных лучей K Когда кристаллические формы соединения (I) выдерживают таким образом, что они открыты для атмосферного воздействия или смешаны с водой или растворителем, они могут абсорбировать воду или растворитель с образованием гидрата или сольвата. Данное изобретение охватывает такие гидраты и сольваты. Соединение (I) может быть получено методом, аналогичным описанному в патенте Японии №2569251. Кристаллические формы соединения (I) могут быть получены из перенасыщенного раствора. Перенасыщенный раствор может быть получен растворением соединения (I) в подходящем растворителе, доведением рН указанного раствора, концентрацией указанного раствора, охлаждением указанного раствора, добавлением растворителя, в котором соединение (I) растворяется незначительно, к раствору соединения (I) в растворителе, в котором соединение (I) растворяется легко, или подобными действиями. Суспензию кристаллического или аморфного твердого вещества соединения (I) в подходящем растворителе превращают во взвесь и затем перемешивают для превращения чередующегося кристалла (опосредованная растворителем трансформация). Кроме того, осаждение кристаллов в реакционном сосуде происходит спонтанно или оно может быть инициировано или ускорено добавлением кристаллических затравок, механическим стимулированием, таким как применение ультразвуковых волн или натяжением внутри реакционного сосуда. Температура для кристаллизации соединения (I) или его фармакологически приемлемой соли обычно составляет от 0 до 60°С, предпочтительно от 5 до 45°С. Осажденные кристаллы могут быть собраны фильтрацией, центрифугированием или декантацией. Выделенные кристаллы могут быть промыты подходящим растворителем. Промывочный растворитель может включать, например, воду; спирт, такой как этанол, изопропанол; кетон, такой как ацетон; сложный эфир, такой как метилформиат, этилформиат, метилацетат, этилацетат; ароматический углеводород, такой как толуол, ксилол; нитрил, такой как ацетонитрил; простой эфир, такой как диэтиловый эфир, тетрагидрофуран или их смесь. Предпочтительно применяют метилацетат, который содержит воду или безводный. Выделенные кристаллы могут быть высушены при температуре от 10 до 100°С, предпочтительно от 30 до 50°С до тех пор, пока вес указанных кристаллов не станет постоянным, при необходимости, в присутствии осушителя, такого как силикагель или хлорид кальция и при пониженном давлении. Высушенные кристаллы могут абсорбировать воду в условиях относительной влажности от 20 до 90% и при температуре от 10 до 30°С, предпочтительно при относительной влажности от 50 до 80% и температуре от 20 до 30°С, до тех пор, пока вес указанных кристаллов не станет постоянным. Полученные таким образом кристаллы могут быть далее очищены перекристаллизацией или очисткой из суспензии. Перекристаллизацию проводят методами, известными специалистам в данной области техники, такими как (1) метод охлаждения: соединение (I) или его фармакологически приемлемую соль растворяют в горячем растворителе и затем полученный раствор охлаждают, (2) метод концентрирования: раствор соединения (I) или его фармакологически приемлемой соли концентрируют, (3) метод осаждения: растворитель, в котором соединение (I) или его фармакологически приемлемая соль растворяется незначительно, добавляют к раствору соединения (I) или его фармакологически приемлемой соли в растворителе, в котором соединение (I) или его фармакологически приемлемая соль растворяется легко. Очистка из суспензии включает сбор кристаллов, которые получают перемешиванием суспензии определенного соединения в подходящем растворителе. Растворитель, применяемый в методе очистки из суспензии соединения (I), включает, например, кетон, такой как ацетон, метилэтилкетон; сложный эфир, такой как метилацетат, этилацетат; нитрил, такой как ацетонитрил; галогенированный углеводород, такой как метиленхлорид, хлороформ; ароматический углеводород, такой как толуол, ксилол; спирт, такой как этанол, изопропанол; простой эфир, такой как диэтиловый эфир, тетрагидрофуран; амид, такой как N,N-диметилформамид; воду; алифатический углеводород, такой как гексан; простой эфир, такой как диизопропиловый эфир, диэтиловый эфир; или подобные, и их смеси. Предпочтительно, применяют кетон, такой как ацетон, метилэтилкетон; сложный эфир, такой как метилформиат, этилформиат, метилацетат, этилацетат; нитрил, такой как ацетонитрил; спирт, такой как этанол, изопропанол, и эти растворители, содержащие воду, и более предпочтительно, метилацетат, содержащий воду или безводный. Кристаллы, полученные перекристаллизацией и очисткой из суспензии, также выделяют методами, аналогичными таким же, как описанные выше. Если кристаллические формы соединения (I) применяют в качестве лекарственного средства, предпочтительно, в качестве агента для лечения или профилактики опухолей, указанные кристаллические формы могут вводиться сами или в виде смеси указанных кристаллических форм с подходящими фармакологически приемлемыми эксципиентом(ами) и/или разбавителем(ями). Композиции в соответствии с данным изобретением могут представлять собой единичные лекарственные формы, такие как таблетки, капсулы, гранулы, порошки, сиропы, инъекции, мази, растворы, суспензии, аэрозоли, пастилки или подобные формы для орального или парентерального введения. Фармацевтические композиции могут быть получены известными методами с применением добавок, таких как эксципиенты, связующие агенты, дезинтегранты, смазывающие агенты, стабилизирующие агенты, корригенты, суспендирующие агенты, разбавители и растворители. Примеры эксципиентов включают производные сахара, такие как лактоза, сахароза, глюкоза, маннит или сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, Примеры связующих агентов включают описанные выше эксципиенты; желатин; поливинилпирролидон; макроголь; или подобные. Примеры дезинтегрантов включают описанные выше эксципиенты, химически модифицированный крахмал или производные целлюлозы, такие как натрий кросс-кармеллоза, натрий карбоксиметилкрахмал, поперечно-сшитый поливинилпирролидон или подобные. Примеры смазывающих агентов включают тальк; стеариновую кислоту; производные стеарата металлов, такие как стеарат кальция, стеарат магния; коллоидную двуокись кремния; вигум; воск, такой как пчелиный воск или спермацет; борную кислоту; гликоль; производное карбоновой кислоты, такое как фумаровая кислота, адипиновая кислота; карбоксилат натрия, такой как бензоат натрия; сульфат, такой как сульфат натрия; лейцин; лаурилсульфат, такой как лаурилсульфат натрия или лаурилсульфат магния; производные кремниевой кислоты, такие как ангидрид кремниевой кислоты, гидрат кремниевой кислоты; производные крахмала, такие как описанные выше в качестве эксципиента; или подобные. Примеры стабилизирующего агента включают производные эфира пара-гидроксибензойной кислоты, такие как метилпарабен, пропилпарабен; производные спирта, такие как хлорбутанол, бензиловый спирт, фенетиловый спирт; хлорид бензалкония; производные фенола, такие как фенол, крезол; тимерозал; уксусный ангидрид; сорбиновая кислота; или подобные. Примеры корригентов включают подсластители, подкислители и вкусовые добавки или подобные, которые обычно применяют. Примеры растворителей включают воду, этанол, глицерин или подобные. Доза кристаллической формы соединения (I) зависит от таких факторов, как симптомы, вес тела и возраст пациента. Подходящая доза составляет от 0,1 мг (предпочтительно, 1 мг) в день до 100 мг (предпочтительно 50 мг) в день. Кристаллическая форма соединения формулы (I) может вводиться в виде либо разовой единичной дозы, либо, при желании, доза может быть разделена на удобные субъединицы, вводимые от одного до нескольких раз в течение дня, в зависимости от симптомов пациента. Предпочтительный вариант осуществления данного изобретения Данное изобретение далее описано с помощью примеров. Примеры тестирования и примеры препаративных форм Пример 1 Кристалл В (а) К 2′-циано-2′-деокси-N4-пальмитоил-1- (b) К 2′ -циано-2′-деокси-N4-пальмитоил-1- Пример 2 Кристалл С (а) К 2′-циано-2′-деокси-N4-пальмитоил-1- (b) К 2′-циaнo-2′-дeoкcи-N4-пaльмитoил-1- Пример 3 Кристалл С(I) 1) Высушенные кристаллы С выдерживают в течение примерно 20 мин в атмосфере, увлажненной до влажности более чем 45% с получением желаемых кристаллов С(I). 2) К высушенным кристаллам С добавляют воду в количестве, которое соответствует около 33% мас. применяемых кристаллов, и полученную смесь перемешивают в течение 3 мин с получением желаемых кристаллов С(I). Пример 4 Кристалл D (a) К 2′-циано-2′-деокси-N4-пальмитоил-1- (b) К 2′-циано-2′-деокси-N4-пальмитоил-1- Пример тестирования 1. Тестирование устойчивости При тестировании устойчивости кристаллы В, С и D в соответствии с данным изобретением, полученные в примерах 1, 2 и 4, соответственно, и аморфный порошок (аморфный А) соединения общей формулы (I), описанного в примере 1 (1d) патента Японии №2569251, применяют в качестве контроля. Эти соединения раздельно помещают в закрытые пробками сосуды и хранят при температуре 60°С в атмосфере азота в течение 17 дней, и содержимое этих соединений проверяют на 5, 10 и 17 дни после начала хранения. Содержание данных соединений определяют количественно высокоэффективной жидкостной хроматографией (ВЭЖХ), и содержание оставшихся соединений (%) рассчитывают, исходя из содержания соединения (I), определенного в каждый момент отбора образца, исходя из начального содержания (100%), определенного непосредственно перед началом хранения. Условия проведения ВЭЖХ следующие: Колонка: L-колонка ODS (4,6 мм Ч 250 мм) (Chemicals Inspection and Testing Institute) Подвижная фаза: ацетонитрил:вода:уксусная кислота = 750:250:1 Скорость потока: 1,0 мл/мин Длина волны определения: 249 нм Температура колонки: 40°С
Исходя из результатов, представленных в таблице 1, устойчивость аморфного порошка (аморфный А) соединения (I) при температуре 60°С в атмосфере азота очень низкая, и остаточное содержание понижается до 62,5% после хранения в течение 17 дней. Наоборот, остаточное количество кристаллов В и С, полученных в примерах 1 и 2, соответственно, при тех же условиях хранения составляет 100% для каждого соединения и остаточное количество кристаллов D, полученных в примере 4, составляет 98,4%, что говорит о том, что устойчивость кристаллов в соответствии с данным изобретением очень высокая. Пример препаративной формы 1. Раствор 1 Получают раствор, содержащий соединение, полученное в примере 1 (10% (мас./мас.)), хлорид бензалкония (0,04% (мас./мас.)), фенетиловый спирт (0,04% (мас./мас.)) и очищенную воду (89,56% (мас./мас.)). Пример препаративной формы 2. Раствор 2 Получают раствор, содержащий соединение, полученное в примере 1 (10% (мас./мас.)), хлорид бензалкония (0,04% (мас./мас.)), пропиленгликоль (30% (мас./мас.)) и очищенную воду (39,96% (мас./мас.)). Пример препаративной формы 3. Порошок Получают порошок, содержащий соединение, полученное в примере 1 (40% (мас./мас.)) и лактозу (60% (мас./мас.)). Пример препаративной формы 4. Аэрозоль Получают аэрозоль, содержащий соединение, полученное в примере 1 (10% (мас./мас.)), лецитин (0,5% (мас./мас.)), фулон 11 (34,5% (мас./мас.)) и фулон 12 (55% (мас./мас.)). Возможность промышленного применения Кристаллические формы в соответствии с данным изобретением имеют значительно лучшую устойчивость при хранении и простоту применения по сравнению с аморфным порошком соединения (I). Указанные кристаллические формы демонстрируют превосходное метаболическое распределение, такое как оральная абсорбция и подобные, и, поэтому, являются полезными лекарственными средствами (предпочтительно, в качестве агентов для лечения или профилактики опухолей). Описание чертежей На фигуре 1 представлена порошковая рентгенограмма кристаллического продукта, полученного в примере 1, полученная при облучении кристаллического продукта медными K На фигуре 2 представлена порошковая рентгенограмма кристаллического продукта, полученного в примере 2, полученная при облучении кристаллического продукта медными K На фигуре 3 представлена порошковая рентгенограмма кристаллического продукта, полученного в примере 3, полученная при облучении кристаллического продукта медными K На фигуре 4 представлена порошковая рентгенограмма кристаллического продукта, полученного в примере 4, полученная при облучении кристаллического продукта медными K Кроме того, на данных фигурах вертикальные оси каждой порошковой рентгенограммы показывают интенсивность дифракции в импульс/секунду (сПз) и горизонтальные оси показывают угол дифракции как значение 2 Приложение Получение кристаллического 2′-циано-2′-деокси-N4-пальмитоил-1- 2′-циано-2′-деокси-N4-пальмитоил-1- Получение кристаллического 2′-циано-2′-деокси-N4-пальмитоил-1- 2′-циано-2′-деокси-N4-пальмитоил-1- Получение кристаллического 2′-циано-2′-деокси-N4-пальмитоил-1- 2′-циано-2′-деокси-N4-пальмитоил-1- Получение кристаллического 2′-циано-2′-деокси-N4-пальмитоил-1- 2′-циано-2′-деокси-N4-пальмитоил-1- Получение кристаллического 2′-циано-2′-деокси-N4-пальмитоил-1- 2′-циано-2′-деокси-N4-пальмитоил-1-
Способ тестирования такой же, как в примере тестирования 1 настоящей заявки.
Формула изобретения
1. Кристаллическая форма соединения формулы (I) или его гидрат где указанная кристаллическая форма имеет основные пики на расстояниях решетки 19,53, 13,03, 9,75, 4,17, 4,00, 3,82, 3,68 и 3,41 ангстрема, определенных рентгенографией порошковым методом с применением медных лучей K 2. Кристаллическая форма соединения формулы (I) где указанная кристаллическая форма имеет основные пики на расстояниях решетки 19,36, 12,87, 9,63, 4,70, 4,64, 4,28, 4,10, 3,92, 3,77 и 3,48 ангстрема, определенных рентгенографией порошковым методом с применением медных лучей K 3. Кристаллическая форма соединения формулы (I) где указанная кристаллическая форма имеет основные пики на расстояниях решетки 19,62, 13,06, 9,82, 4,72, 4,63, 4,56, 4,15, 3,98, 3,93, 3,82, 3,45 и 3,40 ангстрем, определенных рентгенографией порошковым методом с применением медных лучей K 4. Кристаллическая форма соединения формулы (I) где указанная кристаллическая форма имеет пики на расстояниях решетки 22,52, 5,17, 4,60, 4,28 и 3,87 ангстрем, определенных рентгенографией порошковым методом с применением медных лучей K 5. Фармацевтическая композиция, обладающая противоопухолевым действием, содержащая кристаллическую форму по любому из пп.1–4 в качестве активного ингредиента. 6. Фармацевтическая композиция по п.5 для профилактики или лечения опухолей. 7. Применение кристаллической формы по любому из пп.1–4 при изготовлении лекарственного средства для профилактики или лечения опухолей у теплокровных животных. 8. Применение по п.7, где указанным теплокровным животным является человек. 9.Способ профилактики или лечения опухолевых заболеваний, включающий введение теплокровному животному, нуждающемуся в такой профилактике или лечении, фармакологически эффективного количества кристаллической формы по любому из пп.1–4. 10. Способ по п.9, в котором теплокровным животным является человек.
РИСУНКИ
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 (длина волны
(длина волны  =1,54 Е),
=1,54 Е),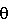 =n
=n -D-арабинофуранозилцитозину (30 г), который является соединением, описанным в примере 1 (Id) патента Японии № 2569251, добавляют метилацетат, содержащий воду в количестве 2,5% об. (300 мл), и полученную смесь нагревают до температуры приблизительно 55°С с получением прозрачного раствора. Далее раствор охлаждают до температуры 5°С со скоростью приблизительно 0,5°С в минуту. При охлаждении до около 45°С в процессе охлаждения из раствора выделяются пластинчатые кристаллы. После дальнейшего перемешивания при температуре 5°С в течение 20 мин выделенные кристаллы собирают фильтрацией и промывают метилацетатом, содержащим воду в количестве 2,5% об. (30 мл) с получением желаемых кристаллов В (28,78 г, чистота 97,9%) с 96,0% [N/N] выходом.
-D-арабинофуранозилцитозину (30 г), который является соединением, описанным в примере 1 (Id) патента Японии № 2569251, добавляют метилацетат, содержащий воду в количестве 2,5% об. (300 мл), и полученную смесь нагревают до температуры приблизительно 55°С с получением прозрачного раствора. Далее раствор охлаждают до температуры 5°С со скоростью приблизительно 0,5°С в минуту. При охлаждении до около 45°С в процессе охлаждения из раствора выделяются пластинчатые кристаллы. После дальнейшего перемешивания при температуре 5°С в течение 20 мин выделенные кристаллы собирают фильтрацией и промывают метилацетатом, содержащим воду в количестве 2,5% об. (30 мл) с получением желаемых кристаллов В (28,78 г, чистота 97,9%) с 96,0% [N/N] выходом.