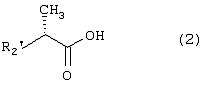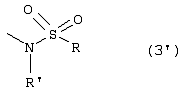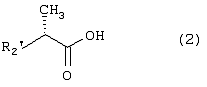Патент на изобретение №2255084
|
||||||||||||||||||||||||||
(54) N-(2-АРИЛПРОПИОНИЛ)СУЛЬФОНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
(57) Реферат:
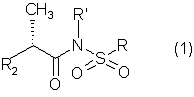
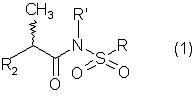
Изобретение относится к новым N-(2-арилпропионил)сульфонамидам формулы 1 где R2 означает фенил, тиофенил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, С1-С4-алкила, фенила, фенокси, бензила, бензоила, С1-С7-ацилокси, 2-тиеноила или 1-оксо-2-изоиндолила; R означает линейный или разветвленный C1-C16-алкил, трифторметил, циклогексил, о-толил, 3-пиридил, п-цианофенилметил, п-аминометилфенилметил, 3-циано-1-пропил, алкоксиэтиленовую группу СН3-(CH2)ni-(OCH2CH2)mi-, в которой ni и mi равно целому числу от 1 до 3, или группу P1P2N-CH2-CH2-, в которой P1 и Р2 представляют собой независимо Н, C1-С3-алкил, бензилоксикарбонил, R’ означает Н или линейный или разветвленный C1-С3-алкил, или их соли с сильными и средними основаниями. Соединения формулы 1 обладают ингибирующей активностью в отношении индуцированных интерлейкином-8 хемотаксиса и дегрануляции нейтрофилов и могут быть использованы в фармацевтической композиции для профилактики и лечения поражений ткани. 4 н. и 9 з.п. ф-лы, 2 фиг., 2 табл.
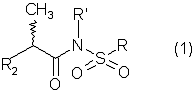
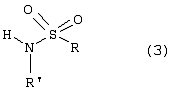
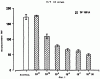
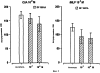
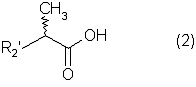
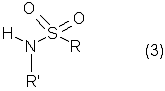
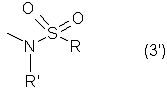
Настоящее изобретение относится к N-(2-арилпропионил)сульфонамидам и фармацевтическим препаратам на их основе, используемым для профилактики и лечения поражений тканей благодаря способствованию рекрутингу полиморфно-ядерных нейтрофилов (PMN лейкоцитов) в очаги воспаления. Хемотаксисом называется клеточная реакция, которая вызывает миграцию специфических клеток крови (макрофагов, гранулоцитов, полиморфно-ядерных нейтрофилов) по градиенту концентрации стимулирующего агента, которая следует за химическим стимулом (хемотаксином). Наиболее характерными хемотаксинами являются продукты расщепления комплемента С5а, некоторые N-формилпептиды, образующиеся при лизисе бактериальной поверхности, или синтетической природы, такой как формил-метионил-лейцил-фенилаланин (f-MLP), и главным образом ряд цитокинов, приписываемых интерлейкину ИЛ-8. Интерлейкин ИЛ-8 является эндогенным хемотаксическим фактором, который вырабатывается большинством содержащих ядро клеток (фибробластами, макрофагами, эндотелиальными и эпителиальными клетками) в ответ на обработку TNF-a (фактором некроза опухоли), интерлейкинами IL-la и IL-lfi и липополисахаридами стенок бактериальных клеток (LPS), также как и собственно нейтрофилами в случае, когда они подвергаются воздействию LPS и N-формилпептидов бактериальной природы (fMPL-подобные пептиды). Этот хемотаксический фактор [также известный как фактор активации нейтрофилов (NAF), хемотаксический фактор Т клеток, производимый моноцитами хемотаксический фактор нейтрофилов (MDNCF)] регулирует ряд ИЛ-8-подобных хемокинов [GROa, fi, V и NAP-2], которые связываются с ИЛ-8 рецептором (Chanq et al., J. Immunol., 148, 451, 1992). Нейтрофилы под воздействием хемотаксических факторов претерпевают изменение предрасполагающей к миграции и активации формы, которое заканчивается реакцией дегрануляции, поглощением кислорода и генерацией активных форм кислорода. Все эти события, которые происходят в течение нескольких секунд с момента воздействия хемотаксинов на нейтрофилы, биохимически характеризуются временным увеличением внутриклеточного уровня ионов Са2+, ионов Na+ и цAМФ с последующими изменениями мембранного потенциала. Увеличение вызванной хемотаксином внутриклеточной концентрации ионов Са2+ сопровождается активацией G белков, фосфолипаз С и A2 и каскада арахидоновой кислоты с образованием продуктов циклооксигеназы и липооксигеназы (смотри L.Наrvath в Regulation of neutrophil chemotaxis, Ann. Reports in Med. Chem., page 233-235, vol.24, 1992, и цитированную там литературу). Нейтрофилы являются ранней защитой от бактериальных инфекций из-за их способности к быстрой миграции из периферической крови через места соединения эндотелия и межклеточного вещества тканей в участки, где они оказывают свое специфическое действие, т.е. разрушают микроорганизмы, удаляют поврежденные клетки и стимулируют восстановление тканей (М.A.Goucerot-Podlcalo et al., Pathol. Biol. (Paris), 44, 36, 1996). В специфических патологических условиях, характеризуемых усилением рекрутинга нейтрофилов, благоприятное по сути восстанавливающее здоровье действие нейтрализуется усилением повреждения ткани, которое замедляет лечебный процесс и в наиболее серьезных случаях приводит к смерти пациента. Совсем недавно неожиданные убедительные данные подтвердили гипотезу о том, что поражения при ишемии и реперфузии и гипероксическое повреждение легких связаны с присутствием активированных нейтрофилов и что поражение клетки напрямую зависит от этой активации. Это доказано на экспериментальных моделях [N.Sekido et al., Nature, 365, 654, 1993, и Т.Matsumoto et al., Lab. Investig., 11, 119, 1997] и клинико-патологическими исследованиями у людей [А.Mazzone et al., Recent Prog. Med., 85, 397, 1994; Т.С.Fisher и H.J.Meiselmann, Thromb. Res., 74 (Suppl.1), S21-S34, 1994; G.Ricevuti et al., Atheroscl., 91, 1, 1991], исходя из которых повреждения клетки напрямую и строго связывают со степенью и распространением инфильтрата. PMN лейкоцитов, причем все чаще предполагается, что цитокин ИЛ-8 является наиболее специфическим и мощным активатором. При патогенезе таких состояний среди различных активирующих нейтрофилы хемотаксических факторов, таких как С5а, PAF, LTB4, которые опосредуют прямую миграцию этих нейтрофилов, ИЛ-8 может играть важную роль также в том, что его выработка мононуклеарными фагоцитами может опосредовать миграцию других клеток воспаления и путем выработки ИЛ-8 заново, и путем индуцирования его выработки близлежащими неиммунными клетками [А.Р.Metiko et al., J Clin. Invest, 90, 791 (1992)]. У пациентов, пораженных острой дыхательной недостаточностью (ARDS), увеличенное присутствие нейтрофилов в дыхательных путях и в легочных жидкостях (которое характеризует заболевание) в значительной мере коррелирует с высокой концентрацией цитокина. С другой стороны, концентрации NAP-1/ИЛ-8, присутствующие в легочных эдемных жидкостях этих пациентов, соответствуют оптимуму концентраций, требующихся для индукции хемотаксиса нейтрофилов (Е.J.Miller et al., Am. Rev. Respir. Dis., 146, 437, 1992); в заключение, явно существует строгая зависимость между смертностью и высокими концентрациями ИЛ-8 в альвеолярных жидкостях (A.Kurdowska et al., J. Immunol., 157, 2699, 1996). В этом контексте особенно значимыми являются результаты, полученные с использованием аНТН-ИЛ-8 антител, способных нейтрализовывать эффекты цитокинов на моделях острой дыхательной недостаточности и эндотоксиемического повреждения легких путем внутритрахеального введения малыми дозами инактивированного нагреванием Streptococcus pyogenes (ОК-432) (К.Yokoi et al., Lab. Invest., 16, 375, 1997). У пациентов, пострадавших от острого инфаркта миокарда, в течение 22 часов с момента его начала было показано значительное увеличение ИЛ-8 в сыворотке крови (13-1100 нг/л). Это увеличение может играть первостепенную роль в развитии повреждения миокарда благодаря высокому стимулирующему влиянию цитокина на нейтрофилы и роль последних в повреждении ткани (Y.Abe et al., Br.Heart J., 70, 132, 1993), которая в клиническом развитии случая ишемии явно зависит в большей степени от процесса реперфузии, чем от времени продолжительности ишемии. Недавно было доказано, что воздействие анти-ИЛ-8 антител уменьшает эдему головного мозга и распространение области повреждения на модели реперфузии при временной очаговой ишемии головного мозга кролика (Т.Matsumoto et al., Lab. Invest., 77, 119, 1997). Значительное увеличение ИЛ-8 в ткани головного мозга, наблюдаемое на этой модели, свидетельствует о том, что и локальная продукция ИЛ-8 в перфузированных тканях и его присутствие в активированных нейтрофилах в стенках сосудов обуславливают степень повреждения ткани. Считается, что рецепторное взаимодействие между нейтрофилом и хемотаксическим агентом имеет решающее значение для хемотаксиса; ИЛ-8 участвует в нем через два различных рецептора (CXCR1 и CXCR2), присутствующих на поверхности нейтрофилов человека и некоторых Т клетках (L. Xu et al., J.Leukocyte Biol., 57, 335, 1995). Это может быть доказано путем использования N-(2-гидрокси-4-нитрофенил)-N’-(2-бромфенил)мочевины, которая избирательно ингибирует связывание ИЛ-8 с мембранным рецептором нейтрофилов CXCR2 с IC50 22 нм (J.R.White et al., J. Biol. Chem., 273, 10095, 1998). Следствием является блокировка хемотаксиса нейтрофилов человека, стимулированных ИЛ-8 (1 нМ) и GROa (10 нМ) со сходными значениями IC50 (20-60 нМ) [но не в случае хемотаксиса, стимулированного компонентом комплемента С5а в диапазоне концентраций от 50 до 330 нМ], а не задержка мобилизации внутриклеточных ионов Ca2+, индуцированная ИЛ-8 или оптимальными концентрациями LIB4, у тех же самых нейтрофилов человека. Использование фенилмочевин в лечении интерлейкин-8(ИЛ-8)-опосредованных патологий было недавно заявлено в изобретении WO 98/07418 (28.02.1998). В ходе исследований, направленных на выявление вклада, который отдельные энантиомеры (S) и (R) кетопрофена дают в противовоспалительной активности рацемата, и их роли в модуляции цитокинов (Р.Ghezzi et al., J. Exp. Pharm. Ther., 1998 в печати), к удивлению было обнаружено, что соли отдельных энантиомеров с хиральными или нехиральными органическими основаниями в зависимости от дозы ингибировали увеличение внутриклеточной концентрации ионов Ca2+ ([Ca2+]i), индуцированного ИЛ-8 у PMN лейкоцитов (заявка на выдачу патента Италии №MI 98A000146 (28.01.1998), принадлежащая настоящему заявителю). Одно конкурентное исследование с ионами La (лантана) доказало, что эффекты ингибирования этими солями ответа нейтрофилов на ИЛ-8 являлись ни следствием рецепторного взаимодействия, ни ослабленной экспрессии некоторого количества рецепторов ИЛ-8, а результатом избирательного блокирования действия катионов Ca2+ в количестве, достаточном для предупреждения хода событий, типичного для вызванной цитокином активации нейтрофила: хемотаксиса и дегрануляции нейтрофилов (с высвобождением эластазы, катепсина и других ферментов). Более того, отдельные энантиомеры ингибируют хемотаксис и увеличение [Ca2+]i, индуцированное С5а и fMLP, хотя и менее эффективно. Основным отличием между двумя энантиомерами является более низкая эффективность (по крайней мере в 10-100 раз) энантиомера (R) в качестве ингибитора СО ферментов. Поэтому по сравнению с энантиомером (S) энантиомер (R) обладает более слабой ингибирующей активностью по отношению к синтезу простагландинов, которые, в свою очередь, оказывают ингибирующее и контролирующее действие по отношению к высвобождению цитокинов, которые, подобно TNF-a, способствуют усилению величины провоспалительных эффектов, типичных для нейтрофилов. Как следствие, энантиомер (S) является менее терапевтически эффективным в лечении нейтрофил-зависимых патологий и воспалительных состояний, таких как псориаз, идиопатический фиброз легких, острая дыхательная недостаточность, реперфузионные повреждения и гломерулонефрит. Сейчас было обнаружено, что N-ацилсульфонамиды 2-арилпропионовых кислот являются эффективными ингибиторами хемотаксиса и дегрануляции нейтрофилов, индуцированных ИЛ-8. Сходные свойства также были доказаны для R(2)-[(4-изобутил)фенил]пропионамида (описанного в W.H.Pirke and J.Me Cune, J.Chromatography, 469, 67, 1989). Поэтому изобретение также относится к их использованию в лечении нейтрофил-зависимых патологий. Более конкретно настоящее изобретение относятся к новым N-(2-арилпропионил)сульфонамидам общей формулы (1) в которой R2 является арильной группой, R является линейной или разветвленной С1-С16-алкильной группой, трифторметильной, циклогексильной, орто-толильной, 3-пиридильной 2-пиридилэтильной, пара-цианфенилметильной, пара-аминофенилметильной, 3-циан-1-пропильной, 4-аминобутильной группой, алкоксиэтилен СН3-(CH2)ni-(OCH2CH2)mi-группой, в которой ni равно 0 или 1 и mi равно целому числу от 1 до 3, или P1P2N-CH2-CH2-группой, в которой P1 и P2 независимо являются водородом, C1-С3-алкильной, бензилоксикарбонильной, R’ является водородом или линейной или разветвленной C1-С3-алкильной группой, предпочтительно водородом. “Арильная группа” предпочтительно означает фенильную группу, необязательно замещенную 1-3 заместителями, которые являются одинаковыми или различными, выбранными из атомов галогенов, С1-С4-алкильной, С1-С4-алкоксильной, гидроксильной, С1-С7-ацилоксильной, цианогруппы, нитрогруппы, аминогруппы, C1-С3-ациламино, галоген-С1-С3-алкильной, галоген-С1-С3-алкоксигруппы, бензоильной группы или арильного радикала известных противовоспалительных 2-арилпропионовых кислот, таких как ибупрофен, кетопрофен, напроксен, супрофен, карпрофен, пирпрофен, фенопрофен. Предпочтительными арильными остатками 2-арилпропионовой кислоты являются 4-изобутилфенил, 3-бензоилфенил, 5-бензоил-2-ацетоксифенил, 3-феноксифенил, 5-бензоилтиен-2-ил, 4-тиеноилфенил, 1-оксо-2-изоиндоленилфенил, 3-хлор-4-(2,5-дигидро-1Н-пиррол-1-ил)фенил, 6-метокси- в котором А является бензильной, бензоильной или N-гидроксибензилиминовой, 1-гидроксифенил-1-метильной группой, В является водородом, гидроксигруппой, C1-С3-ацилоксигруппой или аминогруппой. R2 предпочтительно является арильным остатком известной противовоспалительной 2-арилпропионовой кислоты, как описано выше. Особенно предпочтительными значениями PR2 являются 4-(2-метилпропил)фенил, 3-феноксифенил, 2-[4-(1-оксо-2-изоиндолинил)фенил], 5-бензоилтиен-2-ил, 4-тиеноилфенил. C1-С3-ацилоксигруппой предпочтительно является ацетильная группа; линейной или разветвленной С1-С16-алкильной группой предпочтительно является метильная, гексильная, додецильная или гексадецильная группа; C1-С3-алкильной группой предпочтительно является метильная группа. Особенно предпочтительными соединениями по изобретению формулы (1) являются такие, в которых метильная группа имеет конфигурацию R. Соединения по изобретению могут быть получены с использованием методов, таких как взаимодействие в инертном растворителе эквимолекулярных количеств кислоты формулы (2) в которой R2’ имеет те же значения, что и R2, или является группой, которая может быть преобразована в R2 путем удаления любых защитных групп, с эквимолекулярными количествами сульфонамида формулы (3) в которой R и R’ имеют описанные выше значения, в присутствии эквимолекулярного количества и/или незначительного избытка конденсирующего агента, например карбодиимида (такого как дициклогексилкарбодиимид), растворимого карбодиимида (такого как гидрохлорид N-(3-деметиламинопропил)-N’-этилкарбодиимида) или 1,1’-карбонилдиимидазола и каунтер-основания, выбранного из группы, включающей триэтиламин, 4-(N,N-диметиламино)пиридин, 1,8-диазабицикло[5.4.0]ундец-7-ен и 1,5-диазабицикло[4.3.0]нон-5-ен. Альтернативно, подобный эффективный процесс заключается во взаимодействии сульфонамид-аниона формулы (3’) в которой R имеет описанные выше значения, в инертном растворителе с подходящей реактивной формой кислот формулы (2); хлориды кислот формулы (2) являются наиболее предпочтительными реакционноспособными формами в процессе приготовления хиральных N-ацилсульфонамидов по изобретению, во избежание трудностей, связанных с теми частичными рацемизациями, которые могут иметь место во время взаимодействия сульфонилхлорида формулы (4) с анионом амида формулы (5) В том случае, когда R2’ в соединениях, полученных в соответствии с описанными выше методами, представляет собой бензоильную группу, указанная бензоильная группа могла бы приводить к различным соединениям формулы (1) посредством известных реакций оксимов или посредством восстановления карбонильной группы через спирт до углеводорода (бензила) при поглощении одного или двух эквивалентов водорода в присутствии подходящего катализатора. N-Ацилсульфонамидогруппа соединений формулы (1) является группой, достаточно кислотной для образования солей прибавления сильных и средних оснований, таких как L-лизин, L-аргинин, трометамин или хиральные амины, такие как эфедрин, цинхонин, цинхонидин и т.д., тем самым обеспечивая оптическое разделение указанных сульфонамидов на их отдельные энантиомеры (R) и (S). Различные хроматографические методы, необязательно на хиральных носителях (смотри, например, W.H.Pirkle and J.McCune, цитированные выше), такие как ТСХ или различные способы колоночной хроматографии, могут быть использованы для очистки и разделения энантиомеров и диастереомеров, оптическая чистота которых оценивается с использованием R-(-)-2-[(4’-изобутил)фенил]пропионамида в качестве эталона. Хлорангидриды алкилсульфоновой, арилсульфоновой или гетероарилсульфоновой кислот, а также их амиды являются известными коммерчески доступными и удобными при получении соединений. Поэтому, например, после преобразования полиоксиэтиленового спирта формулы СН3-(CH2)ni-(OCH2CH2)ni-OH (в которой ni и mi имеют описанные выше значения) в соответствующие тозилаты или галогениды (X=Сl, Вr или I), взаимодействие последних с избытком тиомочевины дает соответствующие соли изотиоурония: СН3-CH2)ni-(OCH2CH2)min-S-(C=NH)-NH2· HX, которые напрямую преобразуются в соответствующие сульфонилхлориды: Н3-(CH2)ni-(OCH2CH2)min-SO2Cl путем хлорирования при комнатной температуре, а затем в соответствующие сульфонамиды (J.М.Sprague and Т.В.Johnson, J. Am. Chem. Soc., 59, 1837, 1937). 2-Алкиламиноэтансульфонамиды формулы P1P2-N-CH2-CH2-SO2NH2, в которой P1 и Р2 являются C1-С3-алкильной группой или, как описано выше, образуют кольцо с атомом азота, являются известными соединениями или могут быть получены с использованием известных методов. Для синтеза амидов таурина (смотри, например. Miller et al., J. Am. Chem. Soc., 62, 2099, 1940, и Н.Mcllwain, J. Chem. Soc., 75, 1941, в качестве основного метода получения 2-диалкиламиноэтансульфонамидов, в A. Le Berre and С.Porte, Bull. Soc. Chim. (France), 11-602, 1978 описано добавление вторичных аминов (диметиламина, пиперидина или морфолина) к этиленсульфонамиду [CH2=CH-SO2NH2], синтез которого описан в A.S.Matlack, J. Org. Chem., 23, 729, 1958, и в цитируемых в ней ссылках. Известно, что сульфонамиды формулы (3) и сульфонилхлориды формулы (4) являются соединениями, которые могут быть получены с использованием хорошо известных методов. 2-Арилпропионовые кислоты формулы (2) являются хорошо известными соединениями, используемыми в качестве обезболивающих и противовоспалительных лекарств. Известно несколько процедур для их получения как в качестве отдельных энантиомеров, так и в качестве рацематов. Описано множество эффективных методов для оптического разделения рацематов. Энантиоселективный синтез обычно относится к (S) энантиомерам арилпропионовых кислот, но они также могут быть модифицированы с получением (R) энантиомеров путем соответствующего подбора реагентов (хиральных помощников), например, для использования алкиларилкетонов в качестве субстратов для Более детально, ариловые кислоты формулы (2), в которых R2 является 3-бензоил-2-гидроксифениловой или 3-бензоил-2-аминофениловой группой, являются известными соединениями. Способ их получения описан в заявке на выдачу патента Италии 1283649 (23.04.1998), принадлежащей настоящему заявителю. Конкретными примерами соединений изобретения являются: R(-)-N-2-[(4-изобутилфенил)пропионил]метансульфонамид и его соль L(+)-лизина; R(-)-N-2-[(3-бензоилфенил)пропионил]метансульфонамид; R(-)-N-(2-[3-(1 R(-)-N-метил,N-2-[(4-изобутилфенил)пропионил]метансульфонамид; R(-)-N-[2-(3-бензилфенил)пропионил]метансульфонамид; (±)N-[2-(5’-бензоил-2’-ацетоксифенил)пропионил]метансульфонамид; гидрохлорид R(-)-N-2-[(4-изобутилфенил)пропионил]-2-аминоэтилсульфонамида; R(-)-N-[2-(4-изобутилфенил)пропионил]-2-фталимидоэтансульфонамид; амид R(-)-N-[2-(3-бензоилфенил)пропионил],N’-карбобензилокситаурина; амид R(-)-N-[2-(3-феноксифенил)пропионил],N’-карбобензилокситаурина; амид R(-)-N-[2-[4-(1-оксо-2-изоиндолинилфенил]пропионил],N’-карбобензилокситаурина; N-[2-(5-бензоилтиен-2-ил)пропионил]-(2-карбобензилоксиамино)этансульфонамид; N-[2-(4-тиеноилфенил)пропионил]-(2-карбобензилоксиамино)этансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-(2-карбобензилоксиамино)этансульфонамид; R(-)-N-[2-(3-бензоилфенил)пропионил]-(2-амино)этансульфонамид; R(-)-N-[2-(3-феноксифенил)пропионил]-(2-амино)этансульфонамид; N-[2-(5-бензоилтиен-2-ил)пропионил]-(2-амино)этансульфонамид; N-[2-(4-тиеноилфенил)пропионил]-(2-амино)этансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]циклогексилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]додецилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-пара-цианофенилметансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3-циано-1-пропансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-2-метоксиэтансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6-диоксагептилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6-диоксаоктилсульфонамид; R(-)-N-[2-(4-изобутилфенил)лропионил]-3,6,9-триоксадецилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6,9-триоксаундецилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]циклогексилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]додецилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-пара-цианофенилметансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3-циано-1-пропансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-2-метоксиэтансульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6-диоксагептилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6-диоксаоктилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6,9-триоксадецилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6,9-триоксаундецилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-2-диметиламиноэтилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-2-(пиперидин-1-ил)этилсульфонамид; R(-)-N-[2-(4-изобутилфенил)пропионил]-2-(морфолин-4-ил)этилсульфонамид. Соединения по изобретению изучали, используя PMN клетки, полученные седиментацией на декстране гепаринизированной крови от здоровых добровольцев; мононуклеарные клетки удаляли посредством Ficoll/Hypaque, тогда как эритроциты удаляли обработкой гипотоническими растворами. Жизнеспособность PMN лейкоцитов подсчитывали путем эксклюзии Turk и трипановым синим, тогда как доля содержащих ядро РМ-клеток в цитоцентрифугате оценивалась путем окрашивания Diff Quinck (W.J.Ming et al., J. Imnunol., 138, 1469, 1987). В каждом из ниже описанных тестов in vitro PMN инкубировали с соединениями по изобретению в течение 10 минут при температуре 37° С. В тестах на хемотаксис и в тестах по определению уровня ионов Ca2+ в цитозоле рекомбинантный ИЛ-8 человека (Рерrо Tech.) использовали в качестве стимулирующего агента: лиофилизированный белок растворяли в HBSS (сбалансированном солевом растворе Хенкса) в концентрации 100 нг/мл и использовали после растворения в HBSS в концентрации 10 нг/мл в тестах на хемотаксис и в концентрации 25-50 нг/мл при определении клеточных модификаций [Ca2+]i. В тестах на хемотаксис использовали (в соответствии W.Falket et al., J. Immunol. Metods, 33, 239, 1980) PVP фильтры с 5 мм пористостью и плексигласовую микрокамеру, способную выполнять 48 повторений. Микрокамера состоит из куска плексигласа, содержащего 48 ячеек емкостью 25 [мл каждая, и снабжена крышкой, которая на своей обратной стороне содержит 48 пор, расположенных таким образом, что когда крышка закрыта и прикручена к подлежащей части, в микрокамере создаются компартменты емкостью 50 мкл. Равные концентрации тестируемых соединений помещали в верхние ячейки, содержащие суспензию PMN, а в нижние ячейки, которые содержали носитель, необязательно добавляли ИЛ-8 (или другой стимулирующий агент). В качестве примера, R(-)-N-2-[(4-изобутилфенил)пропионил]метансульфонамид (DF 1681), в зависимости от дозы ингибировал хемотаксис, индуцированный ИЛ-8 (10 нг/мл), в диапазоне концентраций от 10-6 до 10-11 М. Представленные как среднее значение + S.D. трех независимых опытов результаты, выраженные в виде количества мигрировавших PMN, представлены на фиг. 1 в виде гистограмм. (S)(+)-N-2-[(4-Изобутилфенил)пропионил]метансульфонамид и R(-)-N-2-[(4-изобутилфенил)пропионил]метансульфонамид имели значения IC50 в диапазоне от 10-7 до 10-8 М. Представленные на фиг. 2 результаты подтверждают тот факт, что соединения по изобретению избирательно ингибируют ИЛ-8-индуцированный хемотаксис вместо хемотаксиса, индуцированного С5а (10-9 М) или f-MLP (10-8 М), по крайней мере для диапазона концентраций DF 1681 от 10-6 до 10-7 М [представленные как среднее значение + S.D. трех независимых тестов результаты выражены в виде количества мигрировавших PMN]. Изменения [Са2+]i в цитозоле определяли на экспериментальной модели, описанной C.Bizzarri et al. (Blood, 86, 2388, 1995), с использованием предметных стекол, содержащих прилипшие PMN с добавленным 1 В качестве примера, результаты, полученные с R(-)-N-2-[3-(бензоилфенил)пропионил]метансульфонамидом (DF 1661) и R(-)-N-2-[(4-изобутилфенил)пропионил]метансульфонамидом (DF 1681) с использованием PMNs лейкоцитов от трех разных доноров, представлены в таблице 1. Эти лейкоциты были признаны ответившими на ИЛ-8 (50 нг/мл) тогда, когда в ответ на стимул [Ca2+]i, была выше, чем контрольный уровень, на 34% (нормализованный к 100).
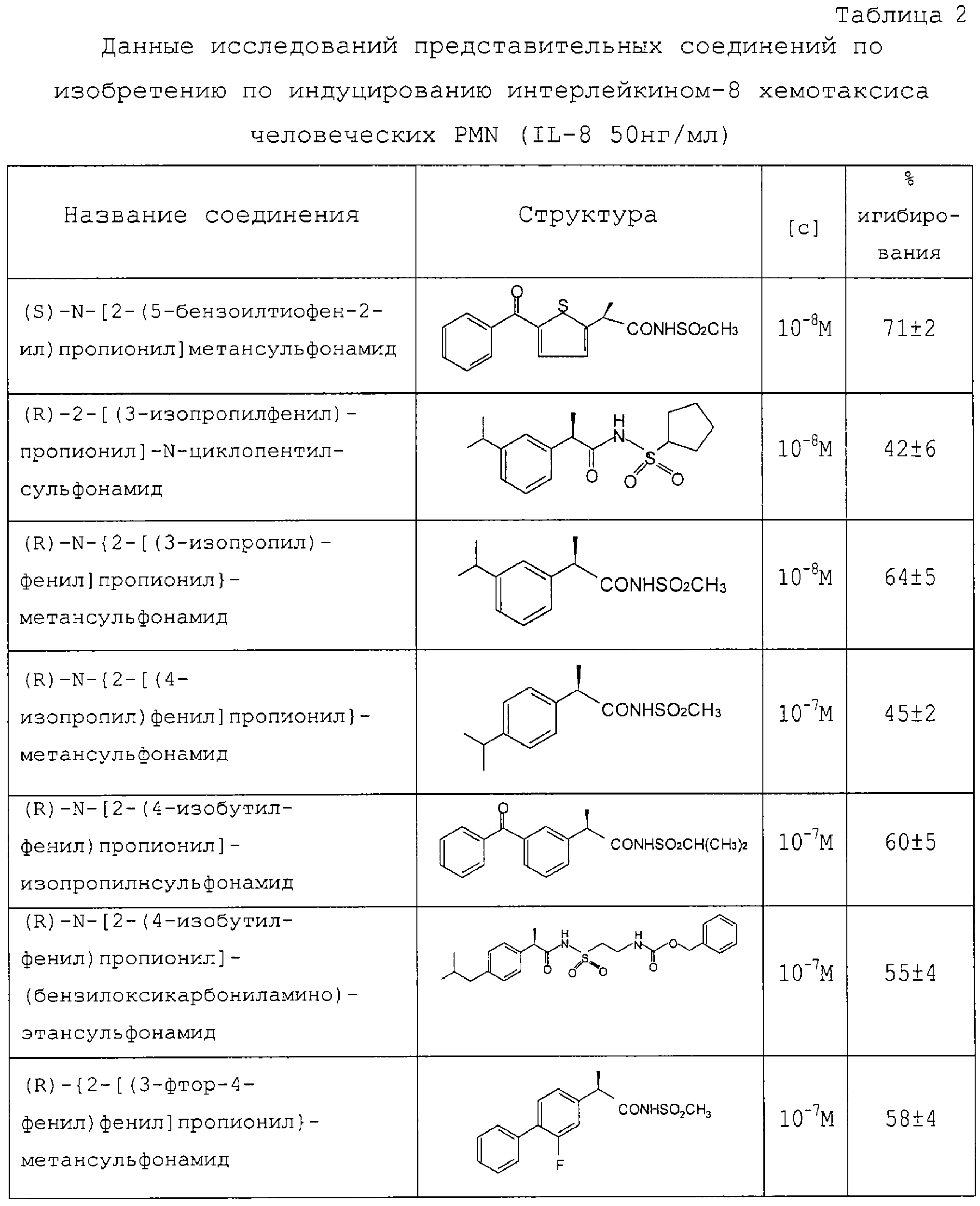
Ответы, выраженные в % по сравнению с контрольными значениями [Са2+]i, равны среднему значению ответивших клеток и включают среднюю ошибку измерения (SEM); n равно числу повторов. В ходе этих тестов процент неответивших клеток различался в различных группах: 30% в группе, на которую воздействовали только ИЛ-8, 40% в группе, на которую предварительно воздействовали DF 1661, и 70% в группе, на которую предварительно воздействовали DF 1681. N-2-Арилпропионилсульфонамиды по изобретению формулы (1) характеризуются способностью ингибировать in vitro хемотаксис PMN лейкоцитов человека, стимулированный интерлейкином 8, см. таблицу 2. Ацилсульфонамиды изобретения в зависимости от дозы ингибируют хемотаксис PMN лейкоцитов человека, со значением IC50 (дозы, ингибирующей эффект на 50%) в диапазоне концентраций от 10-7 до 10-9 М с замечательным уровнем избирательности и специфичности к ИЛ-8-индуцированному хемотаксису. Концентрации выше на один или два порядка требуются для ингибирования in vitro хемотаксиса, индуцированного другими хемотаксическими факторами: С5а, формилпептидами бактериальной и синтетической природы (f-LMP). Специфичность соединений изобретения доказывается тем фактом, что они ингибируют стимулированное ИЛ-8 увеличение в PMN человека внутриклеточной концентрации [Ca2+]i, чье увеличение связывают с активацией PMNL человека [J.H.Liu et al., J.Infect. Dis., 166, 1089 (1992)]. Независимо от абсолютной структуры метильной группы соединения по изобретению значительно не влияют на продукцию СО и простагландинов. Фактически на LPS-стимулированных макрофагах мышей (1 мг/мл) показано, что соединения по изобретению (в диапазоне концентраций от 10-5 до 10-7 М) ингибируют продукцию PGE2, обычно менее чем это статистически значимо, и никогда не выше чем 10-15% от значения контроля. Это не относящееся к делу ингибирование синтеза PGE2 является полезным, так как соединения по изобретению, в отличие от некоторых 2-арилпропионовых кислот, в случае макрофагов мышей (LPS-стимулированных) не являются подходящими стимулами для усиления синтеза TNF Принимая во внимание эти экспериментальные данные и то, что интерлейкин 8 (ИЛ-8) и его аналоги являются вовлеченными в процесс инфильтрации нейтрофилами при патологиях, таких как псориаз (B.J.Nickoloff et аl., Am.J.Pathol., 138, 129, 1991), ревматоидный артрит (M.Selz et al., J.Clin. Invest. 87, 463, 1991), язвенный колит (Y.R.Mahkla et al., Clin. Sci., 82, 273, 1992), респираторный дистресс-синдром взрослых (ARDS) и идиопатический фиброз (Р.С.Саrrе et al., J. Clin. Invest., 88, 1802, 1991, и E.J.Miller et al., Am. Rev. Respir. Dis. цитированный выше), гломерулонефрит (Т.Wada et al., J. Exp. Med., 180, 1135, 1994), соединения по изобретению могут быть использованы при лечении этих патологий. Для этих терапевтических целей соединения по изобретению удобно представлены в фармацевтических композициях с использованием общепринятых методик и наполнителей, таких как описанные в “Remington’s Pharmaceutical Sciences Handbook” Mack Publishing Co., New York, 18th Ed,, 1990. Композиции по изобретению могут вводиться внутримышечно, внутривенно, в виде шариков, в дерматологических препаратах (кремах, лосьонах, спреях и мазях), а также перорально в виде капсул, таблеток, сиропов, композиций с контролируемым высвобождением и тому подобное. Средняя дневная доза зависит от различных факторов, таких как тяжесть болезни и состояния пациента (возраст, пол и вес). Доза как правило варьируется от 1 или нескольких миллиграмм до 1500 мг соединений ежедневно, необязательно разделенных на части для многократного введения. Более высокие дозировки также могут вводиться ввиду низкой токсичности соединений по изобретению, даже для длительного курса лечения. Следующие примеры подробнее иллюстрируют изобретение. ПОЛУЧЕНИЯ Способ, используемый для получения алкилсульфонамидов, арилалкилсульфонамидов, гетероалкилсульфонамидов и алкоксиполиоксиэтиленсульфонамидов формулы СН3-(СН3)ni-(OCH2-CH2)mi-SO2NH2, в которой ni равно нулю или 1 и mi равно целому числу от 1 до 3, осуществляют, как описано J.M.Sprague and Т.В.Jonson J.A.C.S., 59,. 1837, 1937, и Е.Miller et al., ibidem, 62, 2099 (1940). Так, например, гидрохлорид 2-этоксиэтилтиомочевины получают путем взаимодействия 2-этоксиэтилхлорида с небольшим молярным избытком тиомочевины в спирте при перегонке с обратным холодильником. При барботировании Сl2 в растворе соли, охлажденном до примерно 25° С, выделяется маслянистая желтая масса, которую растворяют в этиловом эфире и сушат над сульфатом натрия. Растворитель выпаривают с получением 2-этоксиэтилсульфонилхлорида, который медленно добавляют к раствору гидроксида аммония с получением 2-этоксиэтансульфонамида, который, если требуется, кристаллизуют из разбавленного спирта или очищают методом колоночной хроматографии. С использованием описанного выше процесса приготавливают следующие соединения: 2-метоксиэтансульфонамид; 2-(2-метоксиэтокоси)этансульфонамид или 3,6-диоксагептилсульфонамид; (2-этоксиэтокси)этансульфонамид или 3,6-диоксаоктилсульфонамид; 3,6,9-триоксадецилсульфонамид; 3,6,9-триоксаундецилсульфонамид. ПРИМЕР 1 ОСНОВНЫЕ МЕТОДЫ ПРИГОТОВЛЕНИЯ N-АЦИЛСУЛЬФОНДМИДА А) путем взаимодействия хлорангидрида кислоты формулы (2) с анионом сульфонамида формулы (3) R(-)-N-[2-(4-изобутил)пропионил]метансульфонамида а) Получение хлорангидрида кислоты формулы (2) Суспензию R(-)-2-(4-изобутил)пропионовой кислоты (R-ибупрофен, 4 г, 0019 моль) в тионилхлориде (7,4 мл) перегоняли с обратным холодильником в течение 4 часов, затем оставляли охлаждаться при комнатной температуре. Избыток тионилхлорида выпаривали в вакууме. Остатки тионилхлорида удаляли двукратным промыванием оставшейся массы несколькими каплями сухого диоксана и выпаривали растворитель в вакууме. 4,66 г (0,019 моль) R(-)-2-(4-изобутил)пропионилхлорида получали в виде желтого масла, которое растворяли в нескольких миллилитрах безводного тетрагидрофурана (ТГФ). b) получение аниона сульфонамида Метансульфонамид (2,3 г, 0,0243 моль) добавляли к суспензии трет-бутоксида калия (2,73 г, 0,244 моль) в безводном ТГФ (28 мл); смесь перемешивали в течение 30 минут при комнатной температуре. После этого добавляли R(-)-2-(4-изобутил)пропионилхлорид (4,66 г, 0,019 моль) при перемешивании и оставляли реакционную смесь перемешиваться в течение ночи при комнатной температуре. Выделенные неорганические соли отфильтровывали, растворитель выпаривали в вакууме и маслянистый остаток распределяли между CH2Cl2 (30 мл) и насыщенным раствором фосфата мононатрия. Органическую фазу промывали водой (2× 10 мл) и водные фазы экстрагировали CH2Cl2 (2× 10 мл). Объединенные органические экстракты сушили над Na2SO4 и выпаривали растворитель, затем к раствору маслянистого остатка в безводном метаноле (10 мл) добавляли две микрокапсулы концентрированной серной кислоты для того, чтобы этерифицировать в метиловый эфир любые следы непрореагировавшей R(-)-2-(4-изобутил)пропионовой кислоты. Смесь выдерживали в течение ночи при комнатной температуре, растворитель осторожно выпаривали в вакууме, остаток распределяли между водой (10 мл) и метиленхлоридом (25 мл). Водные фазы отбрасывали и органическую фазу экстрагировали насыщенным раствором NаНСО3 (2× 20 мл). Основные фазы объединяли, подкисляли концентрированной НСl и экстрагировали CH2Cl2 (3× 15 мл). После обычного промывания до нейтральных значений рН объединенные органические экстракты сушили над Na2SO4 и растворитель выпаривали в вакууме с получением 1,86 г (0,0066 моль) R(-)-N-[2-(4-изобутил)пропионил]метансульфонамида; т.пл. 103-105° C (разл.); [ В) прямой конденсацией кислоты формулы (2) с сульфонамидом формулы (3) в присутствии конденсирующего агента N,N-Диметиламинопиридин (2,363 г, 0,0194 моль), гидрохлорид N-(3-диметиламинопропил)-N’-этилкарбодиимида (3,721 г, 0,0194 моль) и метансульфонамид (1,845 г, 0,0194 моль) добавляли последовательно к раствору R-(-)-ибупрофена (4 г, 0,0194 моль) в безводном CH2Cl2 (30 мл); смесь оставляли перемешиваться в течение ночи. Растворитель выпаривали, остаток омыливали этилацетатом (40 мл) и комбинированные органические фазы промывали водой (15 мл) и 20% водным раствором лимонной кислоты (2× 10 мл) и сушили над Na2SO4, затем растворитель выпаривали с получением 2,2 г (0,0076 моль) R(-)-N-[2-(4-изобутилпропионил]метансульфонамида в виде белого твердого вещества, т.пл. 103-105° С (разл.); [ ПРИМЕР 2 С помощью процедур, описанных в примере 1, приготавливали следующие соединения. S(+)-N-[2-(4-изобутил)пропионил]метансульфонамид: т.пл. 109-111° С (разл.); [ R(-)-N-[2-(3-бензоилфенил)пропионил]метансульфонамид: бледно-желтоое масло; [ S(+)-N-[2-(3-бензоилфенил)пропионил]метансульфонамид: бледно-желтое масло; [ S(+)-N-[2-(2-фтор-4-бифенилил)пропионил]метансульфонамид: т.пл. 130-132° С; [ R(-)-N-[2-(2-фтор-4-бифенилил)пропионил]метансульфонамид: т.пл. 106-108° С; [ ПРИМЕР 3 R(-)-N-Метил,N-[2-(4-изобутил)пропионил]метансульфонамид N-ацилсульфонамиды являются достаточно кислыми веществами, дающими после реакции с диазоалканами, N-ацил-N-алкилсульфонамиды вследствие процесса, описанного здесь и далее. Раствор R(-)-N-[2-(4-изобутил)пропионил]метансульфонамида (0,3 г, 0,0001 моль) в сухом этиловом эфире, охлажденный на ледяной бане (Т=4° С), добавляли по каплям к 0,67 н. раствору диазометана в этиловом эфире до появления устойчивого желтого цвета. Избыток диазометана разрушали добавлением к охлажденному раствору нескольких капель ледяной уксусной кислоты. Раствор выдерживали при комнатной температуре и выпаривали растворитель. Остаток вновь растворяли в толуоле и выпаривали в вакууме с целью удаления остатков уксусной кислоты. Маслянистый остаток очищали методом колоночной хроматографии (н-гексан/СН2Сl2, 7:3) с получением R(-)-N-метил-[2-(4-изобутил)пропионил]метансульфонамида (0,24 г, 0,00076 моль) в виде бесцветного масла, [ Это же соединение может быть приготовлено в соответствии с процедурой примера 1, используя эквимолярные количества N-метилметансульфонамида вместо метансульфонамида. ПРИМЕР 4 R(-)-N-[2([3-(1 К раствору 0,5 г N-(R(-)-2-[3-(бензоилфенил)пропионил])метансульфонамида в этаноле добавляли 0,05 мл триэтиламина и каталитическое количество 5% палладия на активном углероде и выдерживали в атмосфере водорода до момента поглощения одного молярного эквивалента газа. Катализатор отфильтровывали через целит и упаривали элюат досуха. Остаток распределяли между этиловым эфиром и 5% водным раствором NаН2РО4 и органическая фаза давала 0,4 г R(-)-N-[2([3-(1 ПРИМЕР 5 R(-)-N-[2[3-(бензилфенил)пропионил]метансульфонамид К раствору 0,5 г N-(R(-)-2-[3-(бензоилфенил)пропионил])метансульфонамида в этаноле добавляли 0,05 мл триэтиламина и каталитическое количество 5% палладия на активном углероде и выдерживали в атмосфере водорода до того момента поглощения по крайней мере двух молярных эквивалентов газа. Катализатор отфильтровывали через целит и упаривали элюат досуха. Остаток распределяли между этиловым эфиром и 5% водным раствором NаН2РО4 и органическая фаза давала 0,4 г R(-)-N-2[3-бифенил)пропионил]метансульфонамида. ПРИМЕР 6 (±)N-2-[(5’-Бeнзoил-2’-aцeтoкcифeнил)пpoпиoнил]мeтaнсульфонамид К раствору 6 г (+)4-ацетокси-3-(1’-метил-2’-пропен-1-ил)бензофенона в CH2Cl2 (125 мл) с добавленным эквивалентным объемом воды добавляли подряд при сильном перемешивании ледяную уксусную кислоту (12 мл), Aliquat 336 (0, 37 г) и, маленькими порциями, перманганат калия до общих 9,5 г (0,060 моль). Смесь выдерживали при перемешивании в течение 20 часов при комнатной температуре до полного исчезновения начального продукта. Реакционную смесь обеспечивали добавлением водного раствора метабисульфита натрия (7,2 г в H2O, 15 мл), затем добавляли CH2Cl2 (10 мл) и разделяли фазы. Органическую фазу промывали насыщенным солевым раствором (2× 25 мл) и сушили над Na2SO4, растворитель выпаривали под вакуумом с получением 6,2 г (±)2-(5’-беноил-2’-ацетоксифенил)пропионовой кислоты хорошей чистоты в виде маслянистой жидкости. HPLC Н2O/СН3СN t=0 60 40, t=12 0 100, t=15 0 100. Bondapak C18 20 см, 1=254 нм, комнатная температура, 5,5 минут), ТСХ (СН2Сl2/СН3ОН 9:1) Rf=0,2; 1H-ЯМР (CDCl3): Используя это соединение в процессе примера 1, получали (+)N-2-[(5’-бензоил-2’-ацетоксифенил)пропионил]метансульфонамид. ПРИМЕР 7 L(±)-лизиновая соль R(-)-N-[2-(4-изобутил)пропионил]метансульфонамида К раствору L(+)-лизина (129 мг; 0,88 ммоль) в воде (1,3 мл добавляли раствор R(-)-N-[2-(4-изобутилпропионил]метансульфонамида (250 мг; 0,88 ммоль) в 1 мл метанола. Растворитель выпаривали и остаточную массу доводили этиловым эфиром (5 мл) и перемешивали в течение ночи при комнатной температуре. Кристаллин, высоко гигроскопичный выделенный материал, быстро фильтровали в атмосфере азота, промывали на фильтре безводным этиловым эфиром и сушили под вакуумом при 50° С в течение 2 часов с получением 360 мг L(+)-лизиновой соли R(-)-N-[2-(4-изобутил)пропионил]метансульфонамида в виде бледно-желтого порошка. [ ПРИМЕР 8 R(-)-2-[(4’-Изобутил)фенил]пропионамид Используя в процедуре примера 1 28% водный раствор NH4OH вместо сульфонамидо-аниона, в соответствии с описанным здесь процессом, приготавливали амиды кислот формулы (2). Раствор 1 г R(-)-2[4-изобутилфенил]пропионилхлорида в сухом ацетонитриле (1,5 мл) добавляли по каплям в охлажденный до 0-5° С 28% раствор NH4ОН (3 мл) с такой скоростью, чтобы температура реакционной смеси не превышала +5° С. Смесь оставляли перемешиваться на 1 час при комнатной температуре, растворители выпаривали в условиях пониженного давления с получением остатка, который растворяли в этилацетате (5 мл). Раствор охлаждали до 0-4° С с целью отделить белый кристаллический преципитат (1,218 г; 5,93 ммоль) R(-)-2-[(4’-изобутил)фенил]пропионамида, который отфильтровывали и сушили под вакуумом, т.пл. 125-127° С; [ ПРИМЕР 9 Гидрохлорид R(-)N-[2-(4-изобутил)пропионил]-(2-амино)этилсульфонамида Следуя методу, описанному Winterbottom R. et al., J. Am. Chem. Soc., 69, 1393-1401 (1947), 21,4 г фталевого ангидрида (0,145 моль) добавляли к суспензии таурина (17 г, 0,137 моль) и ацетата калия (14,2 г, 0,145 моль) в ледяной уксусной кислоте (48 мл), нагреваемой с обратным холодильником. Нагревание продолжали с целью полного растворения реагентов (2,5 часа); после охлаждения до 0-5° С образующийся преципитат, выделенный путем фильтрации, промывали ледяной уксусной кислотой и абсолютным спиртом, сушили на воздухе и под вакуумом (50° С) с получением 31,2 г калийной соли 2-фталимидоэтансульфоновой кислоты [т.пл. >300° С; 1H-ЯМР (D2O) 28% раствор аммиака (15,5 мл) в ацетонитриле (15 мл) добавляли по каплям к раствору 2-фталимидоэтансульфонилхлорида в охлажденном до 0-4° С ацетонитриле (30 мл), смесь размешивали в течение 30 секунд при 0-4° С, растворитель выпаривали и к остаточной массе добавляли горячую воду с целью отделить преципитат 2-фталимидоэтансульфонамида (0,8 г) [т.пл.=206-209° С; 1ЯMP (ДМСО-d6): 0,75 г (0,00295 моль) соединения добавляли в условиях инертного газа к суспензии трет-бутоксида калия (0,331 г, 0,00295 моль) в безводном THF (7 мл); смесь перемешивали в течение 1 часа, затем к ней добавляли раствор R(-)2-(4-изобутилфенил)пропионилхлорида [свежеприготовленного путем реакции 0,47 г кислоты (0,00227 моль) с тионилхлоридом] в безводном THF (2 мл). Смесь выдерживали при перемешивании при комнатной температуре в течение 24 часов; твердый остаток отфильтровывали, раствор упаривали досуха и остаток разделяли между водой и этилацетатом. Комбинированные органические фазы промывали водой (2× 25 мл) и насыщенным солевым раствором (25 мл) и сушили над. Na2SO4 как обычно, растворитель выпаривали под вакуумом с получением маслянистого остатка, который очищали методом флэш-хроматографии (элюент CH2Cl2/CH3OH 98:2) с выходом прозрачного масла, 0,6 г N-[R(-)2-(4-изобутилфенил)пропионил]-2-фталимидоэтансульфонамида. 1H-ЯМР (СDСl3): Раствор соединения (0,5 г, 1,12 ммоль) в этаноле (4 мл) с добавленным 85% гидратом. гидразина (0,4 мл) перегоняли с обратным холодильником в течение 1 часа. После выпаривания растворителя под вакуумом, растворения водой и подкисления 2 н. соляной кислотой фталилгидразид выделяли путем фильтрации. Элюат упаривали досуха с получением 0,332 г гидрохлорида R(-)-N-[2-(4-изобутилфенил)пропионил]-(2-амино)этансульфонамида, также называемого гидрохлоридом R(-)N-[2-(4-изобутилфенил)пропионил]тауринсульфонамида. ПРИМЕР 10 Используя в способе по примеру 9 N’-карбобензилокситауринамид, полученный в соответствии с Н.Mcllwain, J. Chem. Soc, 75, 1941, и хлорид 2-арилпропионовой кислоты, выбранной из группы, включающей R(-)2-(3-бензоилфенил)пропионовую, R(-)2-(3-феноксифенил)пропионовую, R(-)2-[4-(1-оксо-2-изоиндолинил)фенил]пропионовую, 2-(5-бензоилтиен-2-ил)пропионовую, 2-(4-тиеноилфенил)пропионовую и R(-)-2-(4-изобутилфенил)пропионовую кислоты, получали следующие соединения соответственно: R(-)-N-[2-(3-бензоилфенил)пропионил]-(2-карбобензилокси)этансульфонамид 1H-ЯМР (CDCl3): 1H-ЯМР (CDCl3): R(-)-N-[2-[4-(l-оксо-2-изоиндолинил)фенил)пропионил]-(2-карбобензилокси)этансульфонамид 1H-ЯМР (СDСl3): N-[2-(5-бензоилтиен-2-ил)пропионил]-(2-карбобензилокси)этансульфонамид 1H-ЯМР (СDСl3): N-{2-[4-(2-тиеноил)фенил]пропионил}-(2-карбобензилокси)этансульфонамид 1H-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-(2-карбобензилокси)этансульфонамид 1Н-ЯМР (ДМСО-d6): ПРИМЕР 11 Суспензию 2 г R(-)-N-[2-(4-изобутилфенил)пропионил]-(2-карбобензилокси)этансульфонамида и 0,1 г палладиевой черни в смеси воды (20 мл), метанола (20 мл) и уксусной кислоты (6 мл) перемешивали в атмосфере водорода; выделение диоксида углерода останавливали через полчаса. Суспензию фильтровали с целью удаления палладия, растворитель выпаривали в вакууме. Остаток помещали в вакуум в присутствие NaOH. На раствор этого остатка в этаноле (5 мл) воздействовали насыщенным раствором НСl в этаноле для выделения гидрохлорида R(-)-N-[2-(4-изобутилфенил)пропионил]-(2-амино)этансульфонамида, который собирали фильтрацией. Используя в подобном способе карбобензилокситауринимиды по примеру 10, получали следующие соединения в виде гидрохлоридов: R(-)-N-[2-(3-бензоилфенил)пропионил]-(2-амино)этансульфонамид 1H-ЯМР (СDСl3): R(-)-N-[2-(3-феноксифенил)пропионил]-(2-амино)этансульфонамид; R(-)-N-[2-[4-(1-оксо-2-изоиндолинил)фенил]пропионил]-(2-амино)этансульфонамид 1H-ЯМР (CDCl3): N-[2-(5-бензоилтиен-2-ил)пропионил]-(2-амино)этансульфонамид 1H-ЯМР (CDCl3): N-{2-[4-(2-тиеноил)фенил]пропионил}-(2-амино)этансульфонамид 1H-ЯМР (CDCl3): ПРИМЕР 12 Используя в способе по примеру 1 сульфонамид, выбранный из группы, включающей циклогексилсульфонамид, гексилсульфонамид, додецилсульфонамид или гексадецилсульфонамид, парацианофенилметансульфонамид, 3-циано-1-пропансульфонамид, 2-метоксиэтансульфонамид, 3,6-диоксагептилсульфонамид, 3,6-диоксаоктилсульфонамид, 3,6,9-триоксадецилсульфонамид и 3,6,9-триоксаундецилсульфонамид, получали следующие соединения: R(-)-N-[2-(4-изобутилфенил)пропионил]циклогексилсульфонамид 1H-ЯМР (CDCl3): R(-)-N-[2-(4-изобутилфенил)пропионил]гексилсульфонамид 1H-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]додецилсульфонамид 1H-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]гексадецилсульфонамид 1Н-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-пара-цианофенилметансульфонамид 1Н-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-3-циано-1-пропансульфонамид 1Н-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-2-метоксиэтансульфонамид 1Н-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6-диоксагептилсульфонамид 1Н-ЯМР (CDCl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6-диоксаоктилсульфонамид 1Н-ЯМР (CDCl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6,9-триоксадецилсульфонамид 1Н-ЯМР (CDCl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-3,6,9-триоксаундецилсульфонамид 1Н-ЯМР (СDСl3): При желании в тех случаях, когда раствор одного из цианосульфонамидов, выбранных из R(-)-N-[2-(4-изобутилфенил)пропионил]-пара-цианофенилметансульфонамида и R(-)-N-[2-(4-изобутилфенил)пропионил]-3-циано-1-пропансульфонамида, гидрировали в этаноле, содержащем газообразный НСl, в присутствии оксида платины, получали гидрохлориды следующих сульфонамидов: R(-)-N-[2-(4-изобутилфенил)пропионил]-пара-аминометил-фенилметансульфонамида 1Н-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-4-амино-1-бутансульфонамида 1Н-ЯМР (СDСl3): ПРИМЕР 13 Используя в способе по примеру 1 сульфонамид, выбранной из группы, включающей 2-диметиламиноэтилсульфонамид, 2-(пиперидин-1-ил)этилсульфонамид и 2-(морфолин-4-ил)этилсульфонамид, получали: R(-)-N-[2-(4-изобутилфенил)пропионил]-2-диметиламиноэтилсульфонамид 1Н-ЯМР (CDCl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-2-(пиперидин-1-ил)этилсульфонамид 1Н-ЯМР (CDCl3); R(-)-N-[2-(4-изобутилфенил)пропионил]-2-(морфолин-4-ил)этилсульфонамид 1Н-ЯМР (СDСl3): ПРИМЕР 14 Используя в способе по примеру 1 R(-)-2-(3-бензоилфенил)пропионилхлорид и сульфонамид, выбранный из группы, включающей трифторметилсульфонамид, орто-толилсульфонамид, 3-пиридилсульфонамид и 4-пиридилэтансульфонамид, получали следующие соединения. R(-)-N-[2-(3-бензоилфенил)пропионил]трифторметилсульфонамид 1Н-ЯМР (ДМСО-d6): R(-)-N-[2-(3-бензоилфенил)пропионил]-орто-толилсульфонамид 1Н-ЯМР (CDCl3): R(-)-N-[2-(3-бензоилфенил)пропионил]-(3-пиридил)сульфонамид 1Н-ЯМР (CDCl3): R(-)-N-[2-(3-бензоилфенил)пропионил]-2(4-пиридил)этансульфонамид 1Н-ЯМР (CDCl3): ПРИМЕР 15 К раствору 0,35 г R(-)-N-[2-(3-бензоилфенил)пропионил]трифторметилсульфонамида в 5 мл пиридина добавляли 0,11 г гидрохлорида гидроксиламина. Смесь оставляли взаимодействовать при комнатной температуре в течение 12 часов, затем раствор вывливали по каплям в 40 мл 2 н. H2SО4. Образующийся осадок экстрагировали этилацетатом (3× 8 мл). Органические фазы объединяли, промывали водой до нейтральных значений рН, сушили над сульфатом натрия и упаривали досуха с получением 0,36 г оксима R(-)-(Z,Е)-N-[2-(3-бензоилфенил)пропионил]трифторметилсульфонамида. Используя в описанном выше способе R(-)-N-[2-(3-бензоилфенил)пропионил]метансульфонамид, получали оксим R(-)-(Z,E)-N-[2-(3-бензоилфенил)пропионил]метансульфонамида. ПРИМЕР 16 R(-)2-(4’-Изобутилфенилпропионил-(2″-N-никотиноиламино)этансульфонамид Раствор никотиновой кислоты (0,565 г, 4,6 ммоль) в тионилхлориде (3,07 мл, 42,3 ммоль) кипятили с обратным холодильником в течение 3 часов и охлаждали до комнатной температуре с получением, после сушки в вакууме, никотиноилхлорида в виде бледно-желтого твердого вещества (0,65 г). Смесь полученного никотиноилхлорида (0,64 г, 4,52 ммоль) в N,N-диметилформамиде (ДМФ, 1 мл) добавляли по каплям к раствору R(-)2-(4’-изобутил)фенилпропионил(2’’-амино)этансульфонамида (0,6 г, 1,92 ммоль) и триэтиламина (1 мл) в ДМФ (10 мл). Полученную смесь перемешивали в течение 24 часов при комнатной температуре. ДМФ выпаривали в вакууме; неочищенный остаток очищали методом колоночной хроматографии (элюент: СНСl3/СН3ОН/циклогексан/NН4OН 60:14:24:2) с получением R(-)2-(4’-изобутил)фенилпропионил-(2’’-N-никотиноиламино)этансульфонамида, [ Используя в описанном выше способе изоникотиновую и пиколиновую кислоты вместо никотиновой кислоты получали следующие соединения. R(-)-N-[2-(4-изобутилфенил)пропионил]-(2’’-N-изоникотиноиламино)этансульфонамид 1H-ЯМР (СDСl3): R(-)-N-[2-(4-изобутилфенил)пропионил]-(2’’-N-пиколиноиламино)этансульфонамид 1H-ЯМР (СDСl3): ПРИМЕР 17 R(-)2-(4’-Изобутил)фенилпропионил-(2’’-N-карбоксикарбониламино)этансульфонамид Раствор этилхлороксоацетата (0,62 г, 4,52 ммоль) в ДМФ (1 мл) добавляли по каплям к перемешиваемому раствору R(-)2-(4’-изобутил)фенилпропионил-(2’’-амино)этансульфонамида (0,6 г, 1,92 ммоль) и триэтиламина (1 мл) в ДМФ (10 мл). Полученную смесь перемешивали в течение 24 часов при комнатной температуре. ДМФ выпаривали в вакууме, неочищенный остаток разбавляли водой и экстрагировали диэтиловым эфиром (3× 10 мл). Объединенные органические экстракты промывали водой (2× 15 мл) и насыщенным солевым раствором (20 мл), сушили над Na2SO4 и выпаривали в вакууме с получением R(-)2-(4′-изобутилфенилпропионил-(2”-N-этилоксалиламино)этансульфонамида в виде бесцветного масла (0,51 г, 1,25 ммоль). К раствору указанного выше R(-)2-(4’-изобутил)фенилпропионил-(2’’-N-этилoкcaлилaминo)этансульфонамида (0,5 г, 1,22 ммоль) в диоксане (1,55 мл) добавляли 1 н. NaOH (1,55 мл) и перемешивали в течение ночи при комнатной температуре. Диоксан удаляли в вакууме, затем растворимый в воде остаток разбавляли водой; добавляли 4 н. Н2SO4 до значения рН=2; водную фазу экстрагировали дихлорметаном (4× 15 мл); объединенные органические экстракты промывали водой (2× 15 мл) и насыщенным солевым раствором (15 мл), сушили над Nа2SO4 и выпаривали в вакууме с получением R(-)2-(4’-изобутил)фенилпропионил-(2’’-N-карбоксикарбониламино)этансульфонамида в виде бесцветного масла (0,426 г, 1,1 ммоль); ТСХ (CHCl3/CH3OH/H2O 65:25:4) Rf=0,5; [ ПРИМЕР 18 R(-)2-(4’-Изобутил)фенилпропионил-(2’’-N-бензилоксикарбониламино)этансульфонамид Таурин (1 г, 8 ммоль) растворяли в 2 н. NaOH (4,3 мл) и после охлаждения на ледяной бане одновременно добавляли 4 н. NaOH (2,14 мл) и раствор бензилоксикарбонилхлорида (3,27 мл, 8 ммоль) в толуоле (3 мл). После добавления смесь оставляли перемешиваться при температуре 0-5° С в течение 1 часа. Реакцию прекращали добавлением диэтилового эфира. Смесь обрабатывали и фазы разделяли. Водную фазу охлаждали до 0-5° С и добавляли 37% НСl до значения рН=2. Кислотную фазу экстрагировали этилацетатом (3× 10 мл) и объединенные органические экстракты промывали водой (2× 15 мл) и насыщенным солевым раствором (15 мл), сушили над Na2SO4 и выпаривали в вакууме с получением 2-(N-бензилоксикарбониламино)этансульфоновой кислоты в виде неочищенного продукта, очищаемого растиранием в порошок в диэтиловом эфире с получением чистого продукта в виде твердого белого вещества (1,46 г, 5,64 ммоль, выход 70,5%). 2-(N-Бензилоксикарбониламино)этансульфоновую кислоту (0,6 г, 2,31 ммоль) суспендировали в сухом толуоле (6 мл) и добавляли PCl5 (0,65 г, 3,11 ммоль). Смесь кипятили с обратным холодильником в течение 2 часов до полного растворения реагентов. После охлаждения при комнатной температуре растворитель выпаривали в вакууме с получением неочищенного 2-(N-бензилоксикарбониламино)этансульфонилхлорида, достаточно чистого для следующей стадии. Неочищенный 2-(N-бензилоксикарбониламино)этансульфонилхлорид растворяли в ацетонитриле (10 мл). Полученный раствор охлаждали на ледяной бане и добавляли в него по каплям раствор 28% NH4OH (5 мл) и ацетонитрила (5 мл) в соотношении 1:1. После перемешивания в течение 30 секунд растворители выпаривали в вакууме и неочищенный остаток перекристаллизовывали в метаноле с получением 2-(N-бензилоксикарбониламино)этансульфонамида в виде белого порошка (0,51 г, 1,96 ммоль). R(-)Ибупрофен (0,32 г, 1,55 ммоль) растворяли в сухом дихлорметане (7,5 мл) и к нему добавляли N,N-диметиламинопиридин (0,19 г, 1,55 ммоль) и дициклогексилкарбодиимид (0,32 г, 1,7 ммоль); полученную смесь оставляли перемешиваться в течение 30 секунд. Затем добавляли 2-(N-бензилоксикарбониламино) этансульфонамид (0,4 г, 1,55 ммоль) и смесь оставляли перемешиваться в течение ночи. Выпавший осадок N,N-дициклогексилмочевины отфильтровывали и фильтрат выпаривали в вакууме с получением неочищенного остатка, разбавленного ацетонитрилом, вторую порцию N,N-дициклогексилмочевины отфильтровывали и фильтрат после выпаривания растворителя разбавляли дихлорметаном. Органическую фазу промывали 2 н. НСl (2× 10 мл), насыщенным солевым раствором (15 мл), сушили над Na2SO4 и выпаривали в вакууме с получением R(-)2-(4’-изобутил)фенилпропионил-(2’’-N-бензилоксикарбамоиламино)этансульфонамида в виде белого порошка (0,44 г, 0,98 ммоль); т.пл.=107-109° С; ТСХ (СН2Сl2/СН3ОН 98:2) Rf=0,2; [
Формула изобретения
1. N-(2-Арилпропионил)сульфонамиды формулы 1 где R2 представляет собой фенил или тиофенил, необязательно замещенный от 1 до 3 заместителями, независимо выбранными из галогена, С1-С4-алкила, фенила, фенокси, бензила, бензоила, С1-С7-ацилокси, 2-тиеноила или 1 -оксо-2-изоиндолила; R представляет собой линейный или разветвленный С1-С16-алкил, трифторметил, циклогексил, о-толил, 3-пиридил, п-цианофенилметил, п-аминометилфенилметил, 3-циано-1-пропил, алкоксиэтиленовую группу СН3-(CH2)ni-(OCH2CH2)mi-, в которой ni и mi независимо равны целому числу от 1 до 3, или группу P1P2N-CH2-CH2-, в которой P1 и Р2 представляют собой независимо Н, C1-С3-алкил, бензилоксикарбонил, R’ представляет собой Н или линейный или разветвленный C1-С3-алкил, или их соли с сильными и средними основаниями. 2. Соединения по п.1, где R’ представляет собой водород. 3. Соединения по п.1 или 2, в которых R2 выбран из 4-изобутилфенила, 3-бензоилфенила, 5-бензоил-2-ацетоксифенила, 3-феноксифенила, 1-оксо-2-изоиндолинилфенила или остатка формулы в котором А представляет собой бензил или бензоил, В представляет собой водород или C1-С3-ацилокси. 4. Соединения по любому из вышеуказанных пунктов, в которых асимметрический атом углерода, замещенный метилом, имеет абсолютную конфигурацию R. 5. Соединение по п.1, представляющее собой R(-)-N-2-[(4-изобутилфенил)пропионил]метансульфонамид или его соль с L(+)-лизином. 6. Соединение по п.1, представляющее собой R(-)-N-2-[3-(бензоилфенил)пропионил]метансульфонамид. 7. Соединение по любому из пп.1-6 для использования в качестве лекарственного средства для профилактики или лечения поражений ткани, связанных с хемотаксисом и дегрануляцией нейтрофилов, индуцированных интерлейкином-8. 8. Соединение по любому из пп.1-6 для получения лекарственного средства, обладающего ингибирующей активностью в отношении индуцированных интерлейкином-8 хемотаксиса и дегрануляции нейтрофилов. 9. Соединение по п.8 для получения лекарственного средства для лечения псориаза, ревматоидного артрита, язвенного колита, острой дыхательной недостаточности, идиопатического фиброза, глорумеронефрита. 10. Соединение по п.8 или 9, представляющие собой R(-)-N-2-[(4-изобутилфенил)пропионил]метансульфонамид или его соль с L(+)-лизином. 11. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении индуцированных интерлейкином-8 хемотаксиса и дегрануляции нейтрофилов, содержащая в качестве активного ингредиента соединение по любому из пп.1-6 в смеси с подходящим носителем. 12. Способ получения соединения по любому из пп.1-6, включающий взаимодействие в инертном растворителе эквимолярных количеств кислоты формулы (2) где R2’ имеет те же значения, что и группа R2, указанная в п.1, с эквимолярным количеством сульфонамида формулы (3): где R и R’ имеют указанные в п.1 значения, в присутствии конденсирующего агента. 13. Способ получения соединения по любому из пп.1-6, включающий взаимодействие сульфонамиданиона формулы (3’) где R и R’ имеют значения, указанные в п.1, в инертном растворителе с хлорангидридом кислоты формулы (2) где R2‘ имеет значения, указанные в п.12.
РИСУНКИ
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
(73) Патентообладатель(и):
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 11.08.2006 № РД0011189
Извещение опубликовано: 20.09.2006 БИ: 26/2006
|
||||||||||||||||||||||||||
 -,
-,  – или
– или  -пиридокарбонил, карбоксикарбонил или карбалкоксикарбонил, или P1 и P2, вместе с атомом азота, к которому они присоединены, образуют морфолино;
-пиридокарбонил, карбоксикарбонил или карбалкоксикарбонил, или P1 и P2, вместе с атомом азота, к которому они присоединены, образуют морфолино;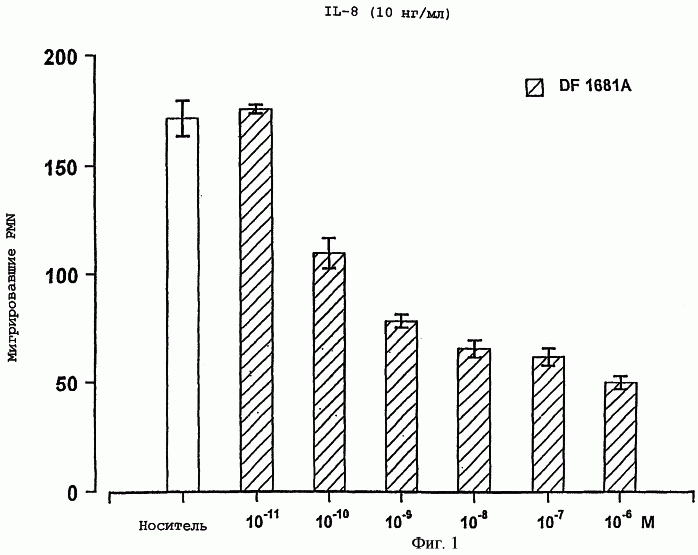
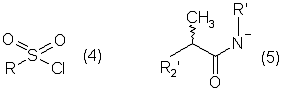
 -гидрокси-1
-гидрокси-1 М Fura-2AM для оценки изменений [Ca2+]i в реальном времени. В свою очередь PMN цитоцентрифугаты ресуспендировали в среде RPMI 1640 с 5% FCS (фетальной бычьей сывороткой) в концентрации 3× 106/мл и затем помещали на круглые стеклянные предметные стекла диаметром 25 мм, которые помещали в инкубатор на 30 минут при температуре 37° С. После того как трехкратными последовательными отмываниями BSS (сбалансированным солевым раствором) удаляли неприлипшие клетки, все прилипшие клетки в дальнейшем инкубировали максимум 4 часа перед добавлением Fura-2-АМ.
М Fura-2AM для оценки изменений [Ca2+]i в реальном времени. В свою очередь PMN цитоцентрифугаты ресуспендировали в среде RPMI 1640 с 5% FCS (фетальной бычьей сывороткой) в концентрации 3× 106/мл и затем помещали на круглые стеклянные предметные стекла диаметром 25 мм, которые помещали в инкубатор на 30 минут при температуре 37° С. После того как трехкратными последовательными отмываниями BSS (сбалансированным солевым раствором) удаляли неприлипшие клетки, все прилипшие клетки в дальнейшем инкубировали максимум 4 часа перед добавлением Fura-2-АМ. 7,3 (д, 2Н, J=8 Гц); 7,09 (д, 2Н, J=7 Гц); 3,42 (кв, 1H, J=8 Гц); 2,8 (с, 3Н); 2,45 (д, 2Н, J=7 Гц); 1,55 (м, 1H); 1,3 (д, 3Н, J=8 Гц); 0,95 (д, 6Н, J=7 Гц).
7,3 (д, 2Н, J=8 Гц); 7,09 (д, 2Н, J=7 Гц); 3,42 (кв, 1H, J=8 Гц); 2,8 (с, 3Н); 2,45 (д, 2Н, J=7 Гц); 1,55 (м, 1H); 1,3 (д, 3Н, J=8 Гц); 0,95 (д, 6Н, J=7 Гц).