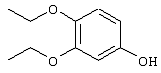
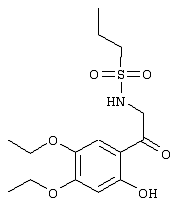
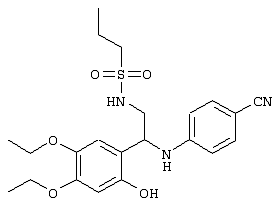
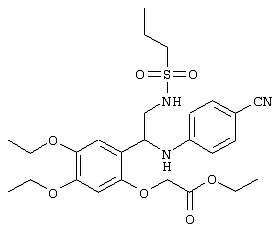
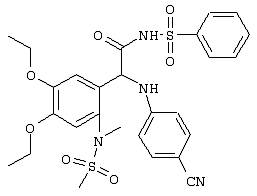
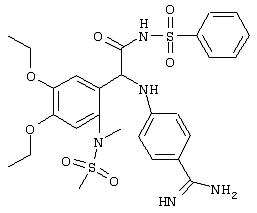
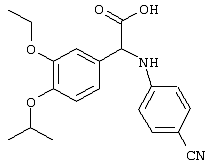
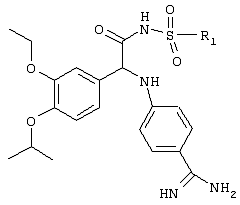
Патент на изобретение №2252935
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ
(57) Реферат:
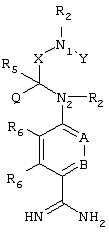
Изобретение относится к новому соединению, имеющему следующую структуру: где А и В независимо друг от друга обозначают CH или CR3, Х обозначает С=O или (CR4aR4b)m, где m равно 1 или 2; Y обозначает S(O)n-R1, где n равно 1 или 2, S(O)n-NR2R2, где n равно 1 или 2, S(O)n-OR2, где n равно 1 или 2; N1 и N2 обозначают атомы азота; Q и R1 независимо друг от друга обозначают (1) необязательно замещенный алкил, имеющий от 1 до 10 атомов углерода; (2) необязательно замещенный аралкил, содержащий арильный фрагмент, имеющий от 6 до 10 кольцевых атомов углерода, который связан с алкильным фрагментом, включающим от 1 до 10 атомов углерода; (3) необязательно замещенный аралкенил, содержащий арильный фрагмент, имеющий от 5 до 10 кольцевых атомов, который связан с алкенильным фрагментом, включающим от 1 до 10 атомов углерода; (4) необязательно замещенный арил, имеющий от 6 до 10 кольцевых атомов углерода; (5) необязательно замещенный гетероарил, имеющий от 5 до 10 кольцевых атомов, где кольцевые атомы выбирают из группы, включающей атомы углерода и гетероатомы, причем гетероатомы представляют собой серу; каждый R2 обозначает Н; R3 обозначает Н; R4a, R4b, R5 и R6 независимо друг от друга обозначают Н; R7 и R8 независимо друг от друга обозначают Н или С1-С6алкил; и его кислотно-аддитивные соли и соли присоединения оснований. Изобретение также относится к способу получения указанного соединения и к фармацевтической композиции, обладающей ингибирующим действием в отношении серинпротеазных ферментов, и к способам лечения на его основе. Технический результат – получение новых соединений и фармацевтической композиции на их основе в целях предупреждения тромбоза или лечения аномального тромбоза. 5 с. и 6 з.п. ф-лы, 7 табл.
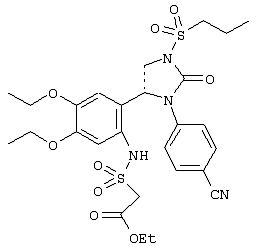
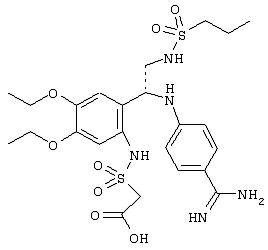
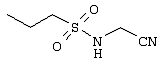
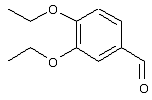
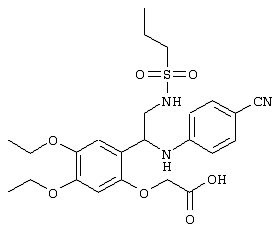
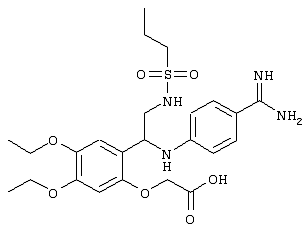
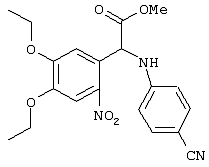
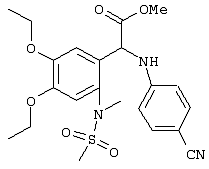
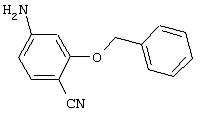
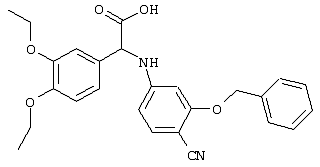
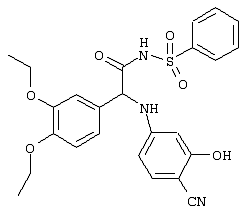
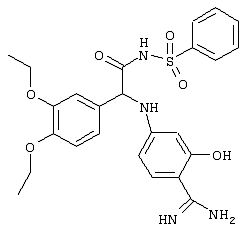
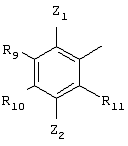
Обсуждение известного уровня техники Область техники изобретения Одним из объектов настоящего изобретения являются новые соединения, которые являются ингибиторами тканевого фактора (ТФ)/фактора VIIa, фактора VIIa, фактора Ха, тромбина и/или калликреина, а также композиции, содержащие эти соединения. Соединения могут применяться для ингибирования этих факторов и для лечения опосредуемых ими нарушений. Например, соединения могут применяться для предупреждения тромбоза или для лечения аномального тромбоза у млекопитающих путем ингибирования ТФ/фактора VIIa, фактора Ха, тромбина и/или калликреина. Предпосылки создания изобретения Нормальный гемеостаз является результатом сложного баланса между процессами инициации образования сгустка, формирования и растворения сгустка. Сложные взаимосвязи между клетками крови, специфическими протеинами плазмы и сосудистой поверхностью поддерживают текучесть крови, если не происходит повреждение и потеря крови. Начало многих серьезных болезней связано с аномальным гемеостазом. Например, образование локального тромба в результате разрушения атеросклеротической бляшки является основной причиной возникновения острого инфаркта миокарда и нестабильной стенокардии. Лечение окклюзивного коронарного тромба либо с помощью тромболитической терапии, либо с использованием чрескожной ангиопластики может сопровождаться острым повторным тромболитическим закрытием поврежденного сосуда. Кроме того, большой процент пациентов, подвергавшихся хирургическому вмешательству, особенно в области нижних конечностей, страдает от образования тромбов в венозной сосудистой системе, что приводит к уменьшению притока крови к поврежденной области. Следовательно, существует необходимость в безопасных и эффективных терапевтических антикоагулянтах, предназначенных для ограничения или предупреждения образования тромбов. Коагулирование крови представляет собой жизненно важный процесс для сохранения общей воды организма при повреждении ткани и является важным компонентом защитных механизмов хозяина. Коагулирование или образование сгустка включает последовательную активацию множества зимогенов в процессе, приводящем к образованию тромбина и превращению фибриногена в непроницаемый поперечно сшитый фибриновый сгусток. Образование тромбина является результатом каскада коагулирования крови, который хорошо изучен и подробно охарактеризован (см., например, Lawson J.H. и др., J. Biol. Chem. 269: 23357 (1994)). Реакции этого каскада коагулирования включают фазы инициации, амплификации и распространения. Кроме того, каскад подразделяют на внешний и внутренний пути. Внутренний путь включает факторы XII, XI и IX и приводит к образованию комплекса фактора IХа с его кофактором, т.е. фактором VIIIa. Этот комплекс превращает фактор Х в фактор Ха. Фактор Ха представляет собой фермент, который образует комплекс с его кофактором, т.е. фактором Va, и быстро превращает протромбин в тромбин. Тромбин превращает фибриноген в мономеры фибрина, которые полимеризуются с образованием сгустка. Внешний путь включает фактор VIIa и тканевый фактор, которые образуют комплекс (ТФ/фактор VIIa), и превращают фактор Х в фактор Ха. Так же как и во внутреннем пути, фактор Ха превращает протромбин в тромбин. Тромбин (фактор IIа), как указывалось выше, играет основную роль в каскаде коагулирования, превращая фибриноген в фибрин. Вследствие этого специалистами в области химического синтеза были предприняты многочисленные попытки разработать ингибиторы тромбина (см., например, патенты US 5656600, 5656645, 5670479, 5646165, 5658939, 5658930 и WO 97/30073). Кроме того, в качестве синтетических ингибиторов тромбина предложены такие соединения, как N-арилсульфинированные фенилаланинамиды. Известные ингибиторы фактора Ха включают бисамидиновые соединения (Katakura S., Biochem. Biophys. Res. Commun., 197: 965 (1993)) и соединения, основной которой является аргининовый скелет (WO 93/15756; WO 94/13693). Также в качестве ингибиторов фактора Ха описаны фенил- и нафтилсульфонамиды (WO 96/100022; WO 96/16940; WO 96/40679). ТФ/фактор VIIa представляет собой серинпротеазный комплекс, который принимает участие в коагулировании крови путем активации фактора Х и/или фактора IX. Фактор VIIa образуется из предшественника, т.е. фактора VII, который синтезируется в печени и секретируется в кровь, где он циркулирует в виде одноцепочечного гликопептида. Последовательность кДНК фактора VII охарактеризована (Hagen и др., Proc. Natl. Acad. Sci. U.S.A., 83: 2112-2416 (1986)). Известны многочисленные природные и синтетические ингибиторы комплекса ТФ/фактор VIIa, и они обладают различной активностью и избирательностью действия. Ингибитор пути тканевого фактора (TFPI; Broze, Thromb. Haemostas., 74: 90 (1995)) и антикоагулянтный пептид с2 нематод (NAPc2; Stanssens и др., Proc. Natl. Acad. Sci. U.S.A., 93: 2149 (1996)) связывают фактор Ха перед образованием четвертичного комплекса ингибитора с комплексом ТФ/фактор VIIa. Также известны небольшие протеины, являющиеся непосредственными ингибиторами (Dennis и др., J. Biol. Chem. 35: 22317 (1994)), и неактивные формы ТФ/фактора Vila. (Kirchhofer и др., Arteriosclerosis, Thrombosis and Vascular Biol., 15: 1098 (1995); Jang и др., Circulation, 92: 3041 (1995)). Кроме того, получены синтетические пептиды и растворимые формы мутантного ТФ, которые сохраняют сродство к связыванию, но обладают пониженной кофакторной активностью (Roenning Throb. Res. 82: 73 (1996); Kelley и др. Blood, 89: 3219 (1997)). В патенте US 5679639 описаны полипептиды и антитела, которые ингибируют активность серинпротеаз. В патенте US 5580560 описан мутантный фактор Vila, который обладает более продолжительным временем полужизни. В патентах US 5504067 и 5504064 описан укороченный ТФ, предназначенный для лечения кровотечения. Также установлено, что слитые протеины, включающие домен Куница (Kunitz)-тканевый фактор, являются бифункциональными антикоагулянтами (Lee и др., Biochemistry, 36: 5607-5611 (1997)). Комплекс ТФ/фактор VIIa считается перспективной мишенью для разработки ингибиторов, проявляющих избирательное действие в отношении кровотечения, связанного с хирургическим вмешательством, и предупреждения внутрисосудистых тромбозов (Harker и др., Thromb. Haemostas., 74:464 (1995)). Соединения, которые блокируют или ингибируют ферменты каскада коагулирования, могут найти терапевтическое применение для лечения или предупреждения тромбоза у млекопитающих, имеющих состояние, для которого характерен аномальный тромбоз. Например, для артериальной сосудистой сети органа образование аномального тромба вследствие деградации сформированной атеросклеротической бляшки является основной причиной острого инфаркта миокарда и нестабильной стенокардии. Лечение окклюзивного коронарного тромба с помощью тромболитической терапии или с использованием чрескожной внутрипросветной ангиопластики (РТСА) может сопровождаться повторным закрытием сосуда. Что касается венозной сосудистой сети органа, то у многих пациентов, подвергнутых хирургическому вмешательству, прежде всего в абдоминальной и низлежащих областях тела, известны случаи образования тромба, который уменьшает ток крови и может привести к легочной эмболии. Синдром диссеминированного внутрисосудистого коагулирования как в венозной, так и в артериальной системах обычно возникает при септическом шоке, некоторых вирусных инфекциях и раке и может привести к быстрому и широко распространенному образованию тромбов и к нарушению функции органа (недостаточности). РТСА и реканализация являются наиболее предпочтительными процедурами при лечении окклюзии сосудов. Однако артериальный тромбоз, являющийся результатом этих процедур, остается основной причиной нарушения функции органа. Для наиболее широко распространенного в настоящее время антикоагулянта гепарина обнаружено, что он не является полностью эффективным в отношении лечения и предупреждения острого артериального тромбоза или ретромбоза. Синтез и разработка небольших молекул ингибиторов, в которых за основу принимается известная трехмерная структура протеинов, является основным современным путем создания лекарственных средств. Создан целый ряд ингибиторов тромбина, которые имеют хирудиновый тип строения (Stubbs и Bode, Current Opinion in Structural Biology, 4: 823-832 (1994)). Описаны новые синтетические ингибиторы тромбина, а также ингибиторы фактора Ха и ТФ/фактора VIIa (см., например, Annual Reports in Medicinal Chemistry, Academic Press, San Diego, CA, 1995-1997). В патенте US 5589173 описано применение антагониста тканевого фактора и тромболитического агента для лечения инфаркта миокарда. В патенте US 5399487 описаны нафталинсульфонамиды, которые могут применяться для определения активности протеолитического фермента или в качестве ингибиторов фермента. В настоящее время продолжает существовать необходимость в разработке соединений, которые являются эффективными ингибиторами ферментов каскада коагулирования и которые обладают повышенной ингибирующей активностью и/или избирательным действием в отношении определенных ферментов каскада. Краткое изложение сущности изобретения Таким образом, одним из объектов настоящего изобретения являются новые соединения, которые ингибируют факторы/ферменты каскада коагулирования и которые могут применяться для предупреждения или лечения образования тромба в артериальных или венозных сосудах. Эти соединения могут применяться в качестве ингибиторов факторов коагулирования и в качестве антикоагулянтов в целом. Согласно одному из вариантов осуществления изобретения объектом изобретения являются ингибиторы, которые избирательно ингибируют фактор VIIa, ТФ/фактор VIIa по сравнению с фактором Ха, тромбином или калликреином. Согласно этому варианту осуществления соединения предпочтительно ингибируют ТФ/фактор VIIa примерно на порядок (10Х) лучше, более предпочтительно примерно на два порядка (100Х) лучше, еще более предпочтительно примерно на три порядка (1000Х) лучше, чем фактор Ха, тромбин и/или калликреин. Согласно еще одному варианту осуществления объектом изобретения являются соединения, которые специфично ингибируют фактор Ха по сравнении с фактором VIIa, ТФ/фактором VIIa, тромбином или калликреином. Согласно этому варианту осуществления соединения предпочтительно ингибируют Ха примерно на порядок (10Х) лучше, более предпочтительно примерно на два порядка (100Х) лучше, еще более предпочтительно примерно на три порядка (1000Х) лучше, чем ТФ/фактор VIIa, тромбин и/или калликреин. Согласно еще одному варианту осуществления объектом изобретения являются соединения, которые специфично ингибируют тромбин по сравнению с фактором VIIa, ТФ/фактором VIIa, Ха или калликреином. Согласно этому варианту осуществления соединения предпочтительно ингибируют тромбин примерно на порядок (10Х) лучше, более предпочтительно примерно на два порядка (100Х) лучше, еще более предпочтительно примерно на три порядка (1000Х) лучше, чем ТФ/фактор VIIa, фактор Ха и/или калликреин. Еще одним объектом изобретения является способ ингибирования активности ТФ/фактора VIIa, Ха или тромбина путем приведения этих ферментов в контакт с эффективным для ингибирования количеством новых ингибиторов по настоящему изобретению или композиции, содержащей эти соединения. Еще одним объектом изобретения является способ лечения заболевания, опосредуемого ТФ/фактором VIIa, Ха или тромбином, предусматривающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества одного из соединений по изобретению или композиции, содержащей это соединение. И еще одним объектом изобретения является способ предупреждения тромбоза или лечения аномального тромбоза, предусматривающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества одного из соединений по изобретению или композиции, содержащей это соединение и носитель или эксципиент. Эти и другие объекты станут очевидными после изучения нижеприведенного описания, в котором представлены соединения по настоящему изобретению, имеющие указанную ниже структуру: где А и В независимо друг от друга обозначают СН, СR3 или N; Х обозначает С=O или (CR4aR4b)m, где m равно 1 или 2; Y обозначает S(O)n-R1, где n равно 1 или 2, S(O)n-NR2R2, где n равно 1 или 2, S(O)n-OR2, где n равно 1 или 2, C(O)R1, C(S)R1, C(O)-OR1, C(O)-NR2R2; N1 и N2 обозначают атомы азота; Q и R1 независимо друг от друга обозначают (1) необязательно замещенный алкил, имеющий от 1 до примерно 10 атомов углерода; (2) необязательно замещенный аралкил, содержащий арильный фрагмент, имеющий от 6 до примерно 10 кольцевых атомов углерода, который связан с алкильным фрагментом, включающим от 1 до примерно 10 атомов углерода; (3) необязательно замещенный гетероаралкил, содержащий гетероарильный фрагмент, имеющий от 5 до примерно 10 кольцевых атомов, который связан с алкильным фрагментом, включающим от 1 до примерно 10 атомов углерода; (4) необязательно замещенный карбоциклоалкил, содержащий карбоциклильный фрагмент, имеющий от 3 до примерно 10 кольцевых атомов углерода, который связан с алкильным фрагментом, включающим от 1 до примерно 10 атомов углерода; (5) необязательно замещенный гетероциклоалкил, содержащий гетероциклильный фрагмент, имеющий от 3 до примерно 10 кольцевых атомов, который связан с алкильным фрагментом, включающим от 1 до примерно 10 атомов углерода; (6) необязательно замещенный алкенил, имеющий от 2 до примерно 10 атомов углерода; (7) необязательно замещенный аралкенил, содержащий арильный фрагмент, имеющий от 5 до примерно 10 кольцевых атомов, который связан с алкенильным фрагментом, включающим от 2 до примерно 10 атомов углерода; (8) необязательно замещенный гетероаралкенил, содержащий гетероарильный фрагмент, имеющий от 5 до примерно 10 кольцевых атомов, который связан с алкенильным фрагментом, включающим от 2 до примерно 10 атомов углерода; (9) необязательно замещенный карбоциклоалкенил, содержащий карбоциклильный фрагмент, имеющий от 3 до примерно 10 кольцевых атомов углерода, который связан с алкенильным фрагментом, включающим от 2 до примерно 10 атомов углерода; (10) необязательно замещенный гетероциклоалкенил, содержащий гетероциклильный фрагмент, имеющий от 3 до примерно 10 кольцевых атомрв, который связан с алкенильным фрагментом, включающим от 2 до примерно 10 атомов углерода; (11) необязательно замещенный арил, имеющий от 6 до примерно 10 кольцевых атомов углерода; (12) необязательно замещенный гетероарил, имеющий от 5 до примерно 10 кольцевых атомов, где кольцевые атомы выбирают из атомов углерода и гетероатомов, причем гетероатомы представляют собой азот, кислород или серу; (13) необязательно замещенный карбокциклил, имеющий от 3 до примерно 10 атомов углерода; (14) необязательно замещенный гетероциклил, имеющий от 3 до примерно 10 кольцевых атомов, где кольцевые атомы выбирают из атомов углерода и гетероатомов, причем гетероатомы представляют собой азот, кислород или серу; R2 каждый независимо друг от друга обозначает Н, алкил, замещенный алкил, C(O)R7 или C(NH)R7 или N2R2 и N2R2 вместе образуют группу N1-CO-N2; R3 обозначает Н, С1-С6алкил, С1-С6алкокси, галоген или ОН; R4a и R5 независимо друг от друга обозначают группу, выбранную из ряда, включающего Н, незамещенный или замещенный алкил, незамещенный или незамещенный алкоксиалкил, незамещенный или замещенный галоалкил, незамещенный или замещенный арил, алкил-OR7, алкил-NR7R8, алкил-ОС(O)ОR7, алкил-С(О)OR7, алкил-С(O)R7, OC(O)R7, C(O)OR7, C(O)R7, и группы, в которых алкил, R7 или R8 замещены 1-3 заместителями, выбранными из ряда, включающего F, Cl, Br, I, OR7, SR7, NR7R8, OC(OR7), C(O)OR7, С(O)R7, C(O)NR7R8, NHC(NH)NH2, РО3, незамещенный или замещенный индолил или незамещенный или замещенный имидазолил; R4b обозначает Н, алкил или замещенный алкил; R6 каждый независимо друг от друга выбирают из ряда, включающего Н, C1-С6алкил, С1-С6алкил-ОR7, C1-C6-алкил-NR7R8, С1-С6 галоалкил, галоген, циано, OR7, SR7, NR7R8, C(O)OR7, C(O)R7 и OC(O)R7; R7 и R8 независимо друг от друга обозначают Н или С1-С6алкил; и их кислотно-аддитивные соли и соли присоединения оснований и пролекарства. Кроме того, объектами изобретения являются композиции, содержащие указанные соединения, и описанные ниже способы. Подробное описание предпочтительных вариантов осуществления Определения Понятие “заболевание, опосредуемое фактором VIIa, ТФ/фактором VIIa, фактором Ха, тромбином или калликреином” обозначает болезнь или физиологическое состояние, включающее коагулирование крови, при котором ингибирование одного или нескольких из этих ферментов снижает или устраняет по меньшей мере один из физиологических симптомов болезни или состояния. Понятие “тромбоз” обозначает развитие или образование сгустка крови или тромба в кровеносном сосуде млекопитающего или в синтетическом сосуде, таком как пластиковая или стеклянная трубка или сосуд. Тромб, который отделен от своего исходного места и обнаружен в другом месте, называют тромботическим эмболом. Понятие “аномальный тромбоз” обозначает тромбоз, встречающийся у млекопитающего, отличающегося от здорового млекопитающего. Понятие “алкил” в контексте настоящего описания индивидуально или в качестве составляющей другого понятия обозначает разветвленную или неразветвленную насыщенную алифатическую углеводородную группу, имеющую указанное количество атомов углерода, или, если количество атомов специально не указано, имеющую до 12 атомов углерода включительно. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 2-метилбутил, 2,2-диметилпропил, н-гексил, 2-метилпентил, 2,2,-диметилбутил, н-гептил, 3-гептил, 2-метилгексил и т.п. Понятия “(низш.) алкил”, “С1-С6алкил” и “алкил, имеющий 1-6 атомов углерода” являются синонимами и используются взаимозаменяемо. Предпочтительные “C1-С6алкильные” группы обозначают метил, этил, 1-пропил, изопропил, 1-бутил или втор-бутил. Понятия “замещенный алкил” или “замещенный Сn-Сmалкил”, где m и n представляют собой целые числа, указывающие количество атомов углерода в алкильной группе, обозначают алкильные группы, как они определены выше, которые замещены 1, 2 или 3 атомами галогена (F, Cl, Вr, I), трифторметилом, гидрокси-, незамещенной и замещенной С1-С7алкокси-, защищенной гидрокси-, амино- (включая алкил- и диалкиламино), защищенной амино-, незамещенной и замещенной С1-С7ацилоксигруппой, незамещенным и замещенным С3-С7гетероциклилом, незамещенной и замещенной фенокси-, нитро-, карбокси-, защищенной карбокси-, незащищенной и защищенной карбоксиалкоксигруппой, незамещенным и замещенным ацилом, карбамоилом, карбамоилокси-, циано-, метилсульфониламино-, незамещенной и замещенной бензилоксигруппой, незамещенным и замещенным C3-С6карбоциклилом или С1-С4алкоксигруппой. Замещенные алкильные группы могут быть замещены одним (предпочтительно), двумя или тремя одинаковыми или различными заместителями. Примеры указанных выше замещенных алкильных групп включают (но не ограничиваясь ими): цианметил, нитрометил, гидроксиметил, тритилоксиметил, пропионилоксиметил, аминометил, карбоксиметил, карбоксиэтил, трифторэтил, трифторпропил, карбоксипропил, 2-аминопропил, алкоксикарбонилметил, аллилоксикарбониламинометил, карбамоилоксиметил, метоксиметил, этоксиметил, трет-бутоксиметил, ацетоксиметил, хлорметил, бромметил, йодметил, трифторметил, 6-гидроксигексил, 2,4-дихлор(н-бутил), 2-амино (изопропил), 2-карбамоилоксиэтил и т.п. Алкильная группа также может быть замещена карбоциклической группой. Примеры включают циклопропилметил, циклобутилметил, циклопентилметил и циклогексилметил, а также соответствующие -этильные, -пропильные, -бутильные, -пентильные, -гексильные группы и т.д. Примеры предпочтительных групп из указанного ряда включают замещенную метильныю группу, например метильную группу, замещенную такими же заместителями, которые указаны для замещенной “Сn-Сmалкильной” группы. Примеры замещенных метильных групп включают такие группы, как гидроксиметил, защищенный гидроксиметил (например, тетрагидропиранилоксиметил), ацетоксиметил, карбамоилоксиметил, трифторметил, хлорметил, карбоксиметил, бромметил и йодметил. Понятие “алкокси” обозначает группы, имеющие указанное количество атомов углерода, такие как метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, втор-бутокси-, трет-бутокси-группы и т.п. Понятие “замещенная, алкоксигруппа” обозначает указанные алкоксигруппы, замещенные такими же заместителями, которые приведены для замещенной “Сn-Сmалкильной” группы, например 2,2,2-трифторэтокси, 2,2,2-трифторпропокси и т.д. Понятие “ацилокси” в контексте настоящего описания обозначает карбоацилоксигруппы, имеющие указанное количество атомов углерода, такие как формилокси, ацетокси, пропионилокси, бутирилокси, пентаноилокси, гексаноилокси, гептаноилокси и т.п. Понятие “замещенная ацилоксигруппа” обозначает указанные ацилоксигруппы, замещенные такими же заместителями, которые указаны для замещенной “Сn-Сmалкильной” группы. Понятия “алкилкарбонил”, “алканоил” и “ацил” в контексте настоящего описания используются взаимозаменяемо и обозначают группы, имеющие указанное количество атомов углерода, такие как формил, ацетил, пропионил, бутирил, пентаноил, гексаноил, гептаноил, бензоил и т.п. Понятия “карбоциклил”, “карбоциклический” и “карбоцикло” индивидуально или в качестве фрагмента сложной группы, такой как карбоциклоалкильная группа, обозначают моно-, би-, или трициклическое алифатическое кольцо, имеющее 3-14 атомов углерода, предпочтительно 3-7 атомов углерода. Предпочтительные карбоциклические группы включают циклопропил, циклобутил, циклопентил и циклогексил. Понятия “замещенный карбоциклил” и “замещенная карбоциклогруппа” обозначает, что эти группы замещены такими же заместителями, которые указаны для замещенной “Сn-Сmалкильной” группы. “Карбоциклоалкильная группа” обозначает карбоциклогруппу, как она определена выше, ковалентно связанную с алкильной группой, как она определена выше. Понятие “алкенил” обозначает разветвленную или неразветвленную углеводородную группу, имеющую указанное количество атомов углерода, которая содержит одну или несколько двойных связей углерод-углерод, каждая двойная связь независимо друг от друга образует цис-, транс- или негеометрический изомер. Понятие “замещенный алкенил” обозначает эти алкенильные группы, замещенные такими же заместителями, которые указаны для замещенной “Сn-Сmалкильной” группы. Понятие “алкинил” обозначает разветвленную или неразветвленную углеводородную группу, имеющую указанное количество атомов углерода, которая содержит одну или несколько тройных связей углерод-углерод. Понятие “замещенный алкинил” обозначает эти алкинильные группы, замещенные такими же заместителями, которые указаны для замещенной “Сn-Сmалкильной” группы. Понятия “алкилтио-” и “замещенная С1-С12алкилтиогруппа” обозначает С1-С12алкильную и замещенную С1-С12алкильную группы соответственно, которые присоединены к атому серы, который в свою очередь является местом присоединения алкилтио- или замещенной алкилтиогруппы к указанной группе или заместителю. Понятие “арил” в контексте настоящего описания индивидуально или в качестве составляющей другого понятия обозначает гомоциклическую ароматическую группу, несконденсированную или сконденсированную, которая имеет указанное количество атомов углерода, или, если количество атомов специально не указано, имеет вплоть до 14 атомов углерода. Предпочтительные арильные группы включают фенил, нафтил, бифенил, фенантренил, нафтаценил и т.п. (см., например, Lang’s Handbook of Chemistry (ред. Dean J.A), 13-oe изд., таблицы 7-2 [1985]). Понятие “замещенный фенил” или “замещенный арил” обозначает фенильную группу или арильную группу, замещенную 1, 2, 3, 4 или 5, предпочтительно 1-2, 1-3 или 1-4 заместителями, выбранными из ряда, включающего галоген (F, Сl, Вr, I), гидрокси-, защищенную гидрокси-, циано-, нитрогруппу, алкил (предпочтительно С1-С6алкил), алкокси-(предпочтительно С1-С6алкокси-), бензилокси-, карбокси-, защищенную карбоксигруппу, карбоксиметил, защищенный карбоксиметил, гидроксиметил, защищенный гидроксиметил, аминометил, защищенный аминометил, трифторметил, алкилсульфониламино-, арилсульфониламино-, гетероциклилсульфониламиногруппу, гетероциклил, арил или другие указанные группы. Одна из метиновых (СН) и/или метиленовых (СН2) групп в этих заместителях в свою очередь может быть замещена группой, аналогичной перечисленным выше. Примеры “замещенного фенила” включают (но не ограничиваясь ими) моно- или ди(гало)фенильную группу, такую как 4-хлорфенил, 2,6-дихлорфенил, 2,5-дихлорфенил, 3,4-дихлорфенил, 3-хлорфенил, 3-бромфенил, 4-бромфенил, 3,4-дибромфенил, 3-хлор-4-фторфенил, 2-фторфенил и т.п.; моно- или ди (гидрокси) фенильную группу, такую как 4-гидроксифенил, 3-гидроксифенил, 2,4-дигидроксифенил, его защищенное гидроксипроизводное и т.п.; нитрофенильную группу, такую как 3-или 4-нитрофенил; цианфенильную группу, например, 4-цианфенил; моно- или ди((низш.) алкил) фенильную группу, такую как 4-метилфенил, 2,4-диметилфенил, 2-метилфенил: 4-(изопропил) фенил, 4-этилфенил, 3-(н-пропил)фенил и т.п.; моно- или ди(алкокси)фенильную группу, например, 3,4-диметоксифенил, 3,4-диэтоксифенил, 3-этокси-4-изопропоксифенил, 3-этокси-втор-бутоксифенил, 3-метокси-4-бензилоксифенил, 3-метокси-4-(1-хлорметил)бензилоксифенил, 3-этоксифенил, 4-(изопропокси)фенил, 4-(трет-бутокси)фенил, 3-этокси-4-метоксифенил и т.п.; 3-или 4-трифторметилфенил; моно- или дикарбоксифенил или (защищенный карбокси)фенильную группу, такую как 4-карбоксифенил; моно- или ди(гидроксиметил) фенил или (защищенный гидроксиметил) фенил, такой как 3-(защищенный гидроксиметил) фенил или 3,4-ди(гидроксиметил)фенил; моно- или ди(аминометил)фенил или (защищенный аминометил) фенил, такой как 2-(аминометил) фенил или 2,4-(защищенный аминометил) фенил; или моно- или ди(N-(метилсульфониламино)фенил, такой как 3-(N-метилсульфониламино)фенил. Кроме того, понятие “замещенный фенил” обозначает дизамещенные фенильные группы, в которых заместители отличаются друг от друга, например, 3-метил-4-гидроксифенил, 3-хлор-4-гидроксифенил, 2-метокси-4-бромфенил, 4-этил-2-гидроксифенил, 3-гидрокси-4-нитрофенил, 2-гидрокси-4-хлорфенил и т.п., а также тризамещенные фенильные группы, в которых 1, 2 или 3 заместителя являются различными, например, 3-метокси-4-бензилокси-6-метилсульфониламино, 3-метокси-4-бензилокси-6-фенилсульфониламино, и тетразамещенные фенильные группы, в которых заместители являются различными, такие как 3-метокси-4-бензилокси-5-метил-6-фенилсульфонила-мино. Предпочтительные замещенные фенильные группы включают 3-метоксифенил, 3-этоксифенил, 4-бензилоксифенил, 4-метоксифенил, 3-этокси-4-бензилоксифенил, 3,4-диэтоксифенил, 3-метокси-4-бензилоксифенил, 3-, метокси-4-(1-хлорметил)бензилоксифенил, 3-метокси-4-(1-хлорметил) бензилокси-6-метилсульфониламинофенил. Кроме того, понятие “замещенный фенил” обозначает фенильные группы, включающие сконденсированные с ними арильную, фенильную или гетероарильную группу. Сконденсированное кольцо также может быть замещено любым количеством, предпочтительно 1, 2 или 3 заместителями, указанными выше для “замещенных алкильных” групп. Понятие “аралкил” обозначает 1, 2 или 3 арильные группы, имеющие указанное количество атомов углерода, присоединенные к алкильной группе, имеющей указанное количество атомов углерода, включая (но не ограничиваясь ими): бензил, нафтилметил, фенэтил, бензгидрил (дифенилметил), тритил и т.п. Предпочтительной аралкильной группой является бензильная группа. Понятие “замещенный аралкил” обозначает алкильную группу, предпочтительно С1-С8алкильную группу, замещенную на любом из атомов углерода арильной группой, предпочтительно С6-С10арильной группой, связанной с алкильной группой в любом положении арильного кольца, и замещенную на алкильном фрагменте 1, 2 или 3 группами, выбранными из ряда, включающего галоген (Р, Сl, Вr, I), гидрокси-, защищенную гидрокси-, амино-, защищенную амино-, С1-С6ацилокси-, нитро-, карбокси-, защищенную карбоксигруппу, карбамоил, карбамоилокси-, циано-, С1-С6алкилтио-, N-(метилсульфониламино)- или С1-C4алкоксигруппу. Необязательно арильная группа может быть замещена 1, 2, 3, 4 или 5 группами, выбранными из ряда, включающего галоген, гидрокси-, защищенную гидрокси-, нитрогруппу, С1-С6алкил, С1-С6алкокси-, карбокси-, защищенную карбоксигруппу, карбоксиметил, защищенный карбоксиметил, гидроксиметил, защищенный гидроксиметил, аминометил, защищенный аминометил или N-(метилсульфониламино) группу. Как указывалось ранее, если либо C1-С8алкильный фрагмент, либо арильный фрагмент или оба являются дизамещенными, то заместители могут быть одинаковыми или различными. Эта группа также может представлять собой замещенный аралкильный фрагмент замещенной арилоксигруппы. Примеры “замещенного аралкила” и этой группы, когда она входит в, состав “замещенной аралкоксигруппы”, включают такие группы, как 2-фенил-1-хлорэтил, 1-фенил-1-хлорметил, 1-фенил-1-бромметил, 2-(4-метоксифенил)этил, 2,6-дигидрокси-4-фенил(н-гексил), 5-циан-3-метокси-2-фенил(н-пентил), 3-(2,6-диметилфенил)-н-пропил, 4-хлор-3-аминобензил, 6-(4-метоксифенил)-3-карбокси(н-гексил), 5-(4-аминометилфенил)-3-(аминометил)(н-пентил) и т.п. Понятие “карбоксизащитная группа” в контексте настоящего описания обозначает одно из эфирных производных группы карбоновой кислоты, обычно применяемое для блокирования или защиты группы карбоновой кислоты в случае, когда реакции осуществляют на других функциональных группах соединения. Примеры таких карбоксизащитных групп включают такие фрагменты, как 4-нитробензил, 4-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4’-диметоксибензгидрил, 2,2’,4,4’-тетраметоксибензгидрил, алкил, такой как метил, этил, изопрорпил, трет-бутил или трет-амил, тритил, 4-метокситритил, 4,4’-диметокситритил, 4,4’,4’’-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, бета-(триметилсилил)этил, бета-(ди(н-бутил) метилсилил) этил, пара-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилилметил)проп-1-ен-3-ил и т.п. Виды применяемых карбоксизащитных групп не имеют решающего значения в случае, когда дериватизированная карбоновая кислота является стабильной в условиях последующей(их) реакции(ий) в других положениях молекулы, и они могут быть удалены в соответствующий момент без нарушения остальной части молекулы. В частности, важно не приводить молекулу с защищенной карбоксигруппой в контакт с сильными нуклеофильными основаниями или использовать восстановительные условия, в которых применяют высокоактивные металлические катализаторы, такие как никель Ренея. (Такие жесткие условия удаления, которые также необходимо избегать при удалении аминозащитных групп и карбоксизащитных групп, описаны ниже). Предпочтительными карбоксизащитными группами являются аллильная и пара-нитробензильная группы. Аналогичные карбоксизащитные группы, которые, применяются в области химии цефалоспоринов, пенициллинов и пептидов, также могут использоваться для защиты заместителей карбоксильной группы. Дополнительные примеры этих групп можно обнаружить у Е. Haslam, “Protective Groups in Organic Chemistry”, ред. J.G.W. McOmie, Plenum Press, New York, N.Y., 1943, глава 5 и у T.W. Green, “Protective Groups in Organic Synthesis”, John Wiley and Sons, New York, NY, 1981, глава 5. Понятие “защищенная карбоксигруппа” относится к карбоксигруппе, замещенной одной из перечисленных выше карбоксизащитных групп. Понятие “амидозащитная группа” в контексте настоящего описания относится к любой группе, обычно применяемой в области химии пептидов для защиты атомов азота пептида от нежелательных побочных реакций. Такие группы включают пара-метоксифенил, 3,4-диметоксибензил, бензил, O-нитробензил, ди (пара-метоксифенил) метил, трифенилметил, (пара-метоксифенил) дифенилметил, дифенил-4-пиридилметил, мета-2-(пиколил)-N’-оксид, 5-дибензосуберил, триметилсилил, трет-бутилдиметилсилил и т.п. Дополнительные описания этих защитных групп можно обнаружить в “Protective Groups in Organic Synthesis”, Theodora W. Greene, 1981, John Wiley and Sons, New York. Понятия “гетероциклическая группа”, “гетероциклический”, “гетероциклил” или “гетероцикло” индивидуально или в качестве составной части сложной группы, такой как гетероциклоалкильная группа, используют взаимозаменяемо, и они относятся к моно-, би- или трициклическому насыщенному или неароматическому ненасыщенному кольцу, имеющему указанное количество атомов, как правило, от 3 до примерно 10 кольцевых атомов, где кольцевые атомы представляют собой углерод и 1, 2, 3 или 4 атома азота, серы или кислорода. Как правило, 5-членное кольцо содержит 0-2 двойных связи, а 6-или 7-членное кольцо имеет 0-3 двойные связи, и гетероатомы, представляющие собой азот или серу, необязательно могут быть окислены, и любой гетероатом, представляющий собой азот, необязательно может быть кватернизован. Примеры включают пирролидинил, оксиранил, оксетанил, тетрагидрофуранил, 2,3-дигидрофуранил, 2Н-пиранил, тетрагидропиранил, тииранил, тиетанил, тетрагидротиетанил, азиридинил, азетидинил, 1-метил-2-пирролил, пиперидинил и 3,4,5,6-тетрагидропиперидинил. “Гетероциклоалкильная” или ” гетероциклоалкенильная” группа представляет собой гетероциклогруппу, как она определена выше, ковалентно связанную с алкильной или алкенильной группой, как она определена выше. Если не указано иное, “гетероарил” индивидуально или в качестве фрагмента сложной группы, такой как гетероаралкильная группа, обозначает моно-, би- или трициклическую ароматическую кольцевую систему, имеющую указанное количество атомов, где по меньшей мере одно кольцо представляет собой 5-, 6-или 7-членное кольцо, содержащее от 1 до 4 гетероатомов, выбранных из группы, включающей азот, кислород и серу, и предпочтительно по меньший один гетероатом представляет собой азот (см. Lang’s Handbook of Chemistry, выше). Под определение подпадают любые бициклические группы, в которых любое из перечисленных выше гетероарильных колец сконденсировано с бензольным кольцом. Предпочтительными являются гетероарилы, в которых гетероатом представляет собой азот или кислород. Примерами гетероарильных (замещенных или незамещенных) групп, подпадающих под понятие “гетероарил” являются следующие кольцевые системы: тиенил, фурил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, тетразолил, тиатриазолил, оксатриазолил, пиридил, пиримидил, пиразинил, пиридазинил, тиазинил, оксазинил, триазинил, тиадиазинил, оксадиазинил, дитиазинил, диоксазинил, оксатиазинил, тетразинил, тиатриазинил, оксатриазинил, дитиадиазинил, имидазолинил, дигидропиримидил, тетрагидропиримидил, тетразоло[1,5-b] пиридазинил и пуринил, а также сконденсированные с бензольным кольцом производные, например, бензоксазолил, бензофурил, бензотиазолил, бензотиадиазолил, бензотриазолил, бензоимидазолил и индолил. Согласно настоящему изобретению также могут применяться 5-членные кольцевые системы, содержащие атом серы или кислорода и 1-3 атома азота. Примеры таких предпочтительных групп включают тиазолил, в частности тиазол-2-ил и N-оксид тиазол-2-ила, тиадиазолил, в частности 1,3,4-тиадиазол-5-ил и 1,2,4-тиадиазол-5-ил, оксазолил, предпочтительно оксазол-2-ил, и оксадиазолил, такой как 1,3,4-оксадиазол-5-ил и 1,2,4-оксадиазол-5-ил. Примеры систем, входящих еще в одну предпочтительную группу 5-членных кольцевых систем с 2-4 атомами азота, включают имидазолил, предпочтительно имидазол-2-ил; триазолил, предпочтительно 1,3,4-триазол-5-ил; 1,2,3-триазол-5-ил, 1,2,4-триазол-5-ил, и тетразолил, предпочтительно 1H-тетразол-5-ил. Примерами систем, входящих в предпочтительную группу сконденсированных с бензольным кольцом производных, являются бензоксазол-2-ил, бензотиазол-2-ил и бензимидазол-2-ил. Также приемлемыми конкретными примерами перечисленных выше гетероциклических кольцевых систем являются 6-членные кольцевые системы, содержащие 1-3 атома азота. Их примеры включают пиридил, такой как пирид-2-ил, пирид-3-ил и пирид-4-ил; пиримидил, предпочтительно пиримид-2-ил и пиримид-4-ил; триазинил, предпочтительно 1,3,4-триазин-2-ил и 1,3,5-триазин-4-ил; пиридазинил, в частности пиридазин-3-ил, и пиразинил. Предпочтительную группу составляют N-оксиды пиридина и N-оксиды пиридазина и пиридил, пиримид-2-ил, пиримид-4-ил, пиридазинил и 1,3,4-триазин-2-ил. Заместители для необязательно замещенных гетероциклических кольцевых систем и дополнительные примеры описанных выше 5- и 6-членных кольцевых систем можно обнаружить у W.Druckheimer и др., патент US 4278793. Особенно предпочтительная группа “гетероарила” включает: 1,3-тиазол-2-ил, 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ил, натриевую соль 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ила, 1,2,4-тиадиазол-5-ил, 3-метил-1,2,4-тиадиазол-5-ил, 1,3,4-триазол-5-ил,. 2-метил-1,3,4-триазол-5-ил, 2-гидрокси-1,3,4-триазол-5-ил, натриевую соль 2-карбокси-4-метил-1,3,4-триазол-5-ила, 2-карбокси-4-метил-1,3,4-триазол-5-ил, 1,3-оксазол-2-ил, 1,3,4-оксадиазол-5-ил, 2-метил-1,3,4-оксадиазол-5-ил, 2-(гидроксиметил)-1,3,4-оксадиазол-5-ил, 1,2,4-оксадиазол-5-ил, 1,3,4-тиадиазол-5-ил, 2-тиол-1,3,4-тиадиазол-5-ил, 2-(метилтио)-1,3,4-тиадиазол-5-ил, 2-амино-1,3,4-тиадиазол-5-ил, 1Н-тетразол-5-ил, 1-метил-1Н-тетразол-5-ил, 1-(1-(диметиламино)эт-2-ил)-1H-тетразол-5-ил, 1-(карбоксиметил)-1Н-тетразол-1-5-ил, натриевую соль 1-(карбоксиметил)-1Н-тетразол-5-ила, 1-(метилсульфоновая кислота)-1Н-тетразол-5-ил, натриевую соль 1-(метилсульфоновая кислота)-1H-тетразол-5-ила, 2-метил-1Н-тетразол-5-ил, 1,2,3-триазол-5-ил, 1-метил-1,2,3-триазол-5-ил, 2-метил-1,2,3-триазол-5-ил, 4-метил-1,2,3-триазол-5-ил, N-оксид пирид-2-ила, 6-метокси-2-(N-оксид)пириаз-3-ил, 6-гидроксипиридаз-3-ил, 1-метилпирид-2-ил, 1-метилпирид-4-ил, 2-гидроксипиримид-4-ил, 1,4,5,6-тетрагидро-5,6-диоксо-4-метиластриазин-3-ил, 1,4,5,6-тетрагидро-4-(формилметил)-5,6-диоксоастриазин-3-ил, 2,5-дигидро-5-оксо-6-гидроксиастриазин-3-ил, натриевую соль 2,5-дигидро-5-оксо-6-гидроксиастриазин-3-ила, натриевую соль 2,5-дигидро-5-оксо-6-гидрокси-2-метиластриазин-3-ила, 2,5-дигидро-5-оксо-6-гидрокси-2-метиластриазин-3-ил, 2,5-дигидро-5-оксо-6-метокси-2-метиластриазин-3-ил, 2,5-дигидро-5-оксоастриазин-3-ил, 2,5-дигидро-5-оксо-2-метиластриазин-3-ил, 2,5-дигидро-5-оксо-2,6-диметиластриазин-3-ил, тетразоло[1,5-b]пиридазин-6-ил и 8-аминотетразоло [1,5-b] пиридазин-6-ил. Альтернативная группа “гетероарила” включает: 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ил, натриевую соль 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ила, 1,3,4-триазол-5-ил, 2-метил-1,3,4-триазол-5-ил, 1H-тетразол-5-ил, 1-метил-1Н-тетразол-5-ил, 1-(1-(диметиламино)эт-2-ил)-1Н-тетразол-5-ил, 1-(карбоксиметил)-1Н-тетразол-5-ил, натриевую соль 1-(карбоксиметил)-1Н-тетразол-5-ила, 1-(метилсульфоновая кислота)-1Н-тетразол-5-ил, натриевую соль 1-(метилсульфоновая кислота)-1H-тетразол-5-ила, 1,2,3-триазол-5-ил, 1,4,5,6-тетрагидро-5,6-диоксо-4-метиластриазин-3-ил, 1,4,5,6-тетрагидро-4-(2-формилметил)-5,6-диоксоастриазин-3-ил, натриевую соль 2,5-дигидро-5-оксо-6-гидрокси-2-метиластриазин-3-ила, 2,5-дигидро-5-оксо-6-гидрокси-2-метиластриазин-3-ил, тетразоло[1,5-b] пиридазин-6-ил и 8-аминотетразоло [1,5-b] пиридазин-6-ил. “Гетероаралкильная или “гетероаралкенильная” группа обозначает гетероарильную группу, как она определена выше, ковалентно связанную с алкильной группой или алкенильной группой, как они определены выше. “Фармацевтически приемлемые соли” включают как кислотно-аддитивные соли, так и соли присоединения оснований. “Фармацевтически приемлемая кислотно-аддтивная соль” обозначает соли, которые сохраняют биологическую эффективность и свойства свободных оснований и не являются нежелательными с биологической или любой иной точки зрения, и образованы с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота и т.п., и органическими кислотами, выбранными из ряда, включающего такие классы органических кислот, как алифатические, циклоалифатические, ароматические, аралифатические, гетероциклические, карбоновые и сульфоновые кислоты, например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, глюконовую кислоту, молочную кислоту, пировиноградную кислоту, щавелевую кислоту, яблочную кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, аспарагиновую кислоту, аскорбиновую кислоту, глутаминовую кислоту, антраниловую кислоту, бензойную кислоту, циннамовую кислоту, миндальную кислоту, эмбоновую кислоту, фенилуксусную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, пара-толуолсульфоновую кислоту, салициловую кислоту и т.п. “Фармацевтически приемлемые соли присоединения оснований” включают соли, образованные из неорганических оснований, такие как соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.п. Особенно предпочтительными являются соли аммония, калия, натрия, кальция и магния. Соли, образованные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в естественных условиях замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, поламинные смолы и т.п.Особенно предпочтительными органическими нетоксичными основаниями являются изопропиламин, диэтиламин, этанооламин, триметамин, дициклогексиламин” холин и кофеин. Понятие “пролекарство” в контексте настоящего описания обозначает производное исходной молекулы лекарства, которое усиливает требуемые фармацевтические характеристики или свойства (например, транспорт, биологическую доступность, фармакодинамические характеристики и т.д.) и которое должно быть подвергнуто внутри организма биотрансформации, спонтанной или ферменативной, для высвобождения активного исходного лекарства. Варианты осуществления Изобретение в целом относится к соединениям, имеющим указанную ниже структуру В этой структуре R2, R5, R6,, А, В, N1, N2, Q, X и Y имеют указанные выше значения. При этом алкил предпочтительно обозначает незамещенный или замещенный С1-С6алкил; алкенил предпочтительно обозначает незамещенный или замещенный С2-С6алкенил; алкинил предпочтительно обозначает незамещенный или замещенный С2-С6алкинил; арил предпочтительно обозначает незамещенный или замещенный нафтил или фенил; более предпочтительно фенил; аралкил предпочтительно обозначает незамещенный или замещенный бензил. Переменная m предпочтительно равна 1. Группа Y предпочтительно обозначает S(O)n-R1, где n равно 1 или 2, или группу S(O)n-NR2R2, где n равно 1 или 2, более предпочтительно S(O)n-R1. Согласно одному из предпочтительных вариантов осуществления R1, например, когда Y обозначает S(O)n-R1, выбирают из ряда, включающего С1-С6алкил, С2-С6алкенил, С2-С6алкинил, С3-С6циклоалкил, фенил, нафтил, бензил и гетероарил, имеющий 5-6 кольцевых атомов, выбранных из атомов углерода и 1-2 гетероатомов, где гетероатомы представляют собой N, S или О, и R1 необязательно замещен 1-3 заместителями, выбранными из ряда, включающего галоген, нитро, С1-С6алкил, NR7R8, OR7, SR7, С1-С6алкил-C(O)OR7, C1-C6алкил-C(O)R7, С1-С6алкил-С(O)R7, С1-С6алкил-ОR7, С1-С6галоалкил, C1-C6алкил-NR7R8, С(О)OR7, ОС(O)R7, C(O)NR7R8, OC(O)NR7R8, NHC(O)R7 и NHC(O)NR7R8, где R7 и R8 независимо друг от друга обозначают Н или С1-С6алкил. В этом варианте осуществления все остальные переменные R2, R5, R6, А, В, Q, X и Y независимо друг от друга могут быть выбраны из любых групп, удовлетворяющих соответствующим приведенным выше определениям. Согласно второму предпочтительному варианту осуществления Q обозначает фенил, необязательно замещенный 1-5, предпочтительно 2-4, более предпочтительно 2-3 заместителями, выбранными из ряда, включающего галоген, нитро, С1-С6алкил, С2-С6алкенил, С2-С6алкинил, NR7R8, OR7, SR7, С1-С6алкил-С(O)OR7, ОС1-С6алкил-С(O)OR7, С1-С6алкил-ОR7, ОС1-С6алкил-OR7, С1-С6алкил-NR7R8, ОС1-С6алкил-NR7R8, С1-С6алкил-C(O)NR7R8, OC1-C6алкил-C(O)NR7R8, С1-С6алкил-С(O)R7, ОС1-С6алкил-C(O)R7, С1-С6 галоалкил, O-аралкил (например, бензилокси), С(O)ОR7, C(O)NR7R8, OC(O)NR7R8, NHC(O)R7, NHC(O)NR7R8, NR7S(O)R1, NR7S(O)nR7, S(O)nR7, S(O)nNR7, где R7 и R8 независимо друг от друга обозначают Н или С1-С6алкил. В этом варианте осуществления все остальные переменные R2, R5, R6, А, В, Q, X и Y (и R1) независимо друг от друга могут быть выбраны из любых групп, удовлетворяющих соответствующим приведенным выше определениям. Каждый алкильный, алкенильный и алкинильный фрагмент также может быть замещен, как указано выше. Согласно третьему предпочтительному варианту осуществления Q имеет структуру где R9 обозначает Н, С1-С6алкил, С2-С6алкенил, С2-С6алкинил, С1-С6алкокси, гидрокси, NR7R8, SR7 или ОR7, где R7 и R8 независимо друг от друга обозначают Н или замещенный или незамещенный С1-С6алкил; R10, R11 и Z2 каждый независимо друг от друга выбирают из ряда, включающего Н, галоген, нитро, циано, С1-С6алкил, С6-С10арил, NR7R8, OR7, SR7, C1-С6алкил-С(O)R7, С1-С6алкил-С(O)NR7R8, С1-С6алкил-С(O)ОR7, C1-С6алкил-ОС(O)R7, С1-С7алкил-ОR7, ОС1-С6алкил-С(O)R7, OC1-С6алкил-С(О)R7, ОС1-С6алкил-ОС(O)R7, ОС1-С6алкил-OR7, OC1-С6алкил-С(O)NR7R8, С1-С6галоалкил, OR12, С1-С6алкил-R12, ОС1-С6алкил-R12, C(O)OR7, C(O)OR12, C(O)NR7R8, OC(O)NR7R8, NR7C(O)R7, NR7C(O)R12, NR7C(O)-NR7R8, NR7C(O)R7, NR7C(O)OR12, NR7C(O)n-R1, NR7S(O)n-R7 и NR7S(O)n-R12, где R7 и R8 независимо друг от друга обозначают Н или незамещенный или замещенный C1-С6алкил, R12 обозначает незамещенный или замещенный С6-С10арил или гетероциклил, как он определен выше, и n равно 1 или 2; Z1 обозначает Н, С1-С6алкил, С1-С6алкокси, галоген или нитро. В этом варианте осуществления все остальные переменные R2, R5, R6, А, В, X и Y независимо друг от друга могут быть выбраны из любых групп, удовлетворяющих приведенным выше определениям. Каждый алкильный, алкенильный и алкинильный фрагмент также может быть замещен, как указано выше. Согласно различным объектам изобретения Z1 и Z2 могут обозначать водород; Z1, Z2 и R11 могут обозначать водород; или Z1, R10 и R11 могут обозначать водород; а остальные кольцевые заместители иметь указанные выше значения. Согласно следующему варианту осуществления заместители в 4 и 5 положениях или в 5 и 6 положениях кольца, когда Q обозначает замещенный фенил, могут быть связаны друг с другом с образованием незамещенного или замещенного карбоциклического или гетероциклического кольца. Примеры таких соединений приведены ниже, где символ предпочтительно обозначает 5-членное или 6-членное карбоциклическое или гетероциклическое кольцо, сконденсированное с фенильным кольцом в указанных ниже положениях. Примеры приемлемых 5-членных или 6-членных карбоциклических или гетероциклических колец, которые могут быть сконденсированы с фенильным кольцом, включают приведенные ниже кольцевые системы, в которых R6 имеет указанные выше значения. Согласно еще одному предпочтительному варианту осуществления Y обозначает S(O)n-R1, где n равно 1 или 2, предпочтительно 2. В этом варианте R1 может иметь указанные выше значения и каждая из остальных переменных независимо друг от друга может иметь любое из указанных выше значений. Также предпочтительными являются соединения, в которых Q обозначает замещенный фенил и R10 выбирают из ряда, включающего С1-С6алкил, C1-С6алкокси, С1-С6аминоалкил, C1-С6галоалкил, С1-С6гидроксиалкил, фенил, фенокси, бензил, бензилокси, а также фенокси- и бензилоксигруппу, замещенную С1-С6алкилом, С1-С6алкоксигруппой, галогеном, С1-С6галоалкилом, С1-С6гидроксиалкилом, С1-С6аминоалкилом, ОС(O)-С1-С6алкилом, С(O)O-С1-С6алкилом и С(O)ОН, где каждая из остальных переменных независимо друг от друга может быть выбрана из любого указанного выше значения. Также представляют интерес соединения, в которых R11 обозначает NR7С1-С6алкил-С(O)NR7R8, NR7S(O)nR7 или NR7S(O)nR12, n равно 1 или 2 и/или в которых Z1=Z2=H и/или в которых R10 обозначает OR7, OR12, ОС7-С10аралкил, ОС1-С6алкил-ОR7 или ОС1-С6алкил-OR12, где R7 и R12 являются незамещенными или замещены, как определено выше. Приемлемые замещенные R7 и R12 включают эти группы, которые замещены, как указано выше, например, имеющие 1 или 2 С1-С6алкокси-, С1-С6алкокси-С1-С6алкоксигрупп, атомов галогена, С1-С6галоалкильных, С1-С6гидроксиалкильных, С1-С6аминоалкильных, ОС(O)-С1-С6алкильных, С(O)O-С1-С6алкильных, С1-С6алкилС(O)ОR7-, С1-С6алкилOС(O)ОR7-или С(O)ОН-групп. В этих соединениях каждая из остальных переменных независимо друг от друга может иметь любое указанное выше значение. Также представляют интерес те соединения, в которых Y обозначает S(O)n-R1, где n равно 1 или 2, т.е. представляющие собой дисульфонамидные соединения. Согласно другому варианту осуществления А и В независимо друг от друга обозначают СН или CR3, где R3 обозначает Н, С1-С6алкил или ОН, и где остальные переменные независимо друг от друга могут иметь любое указанное выше значение. Согласно еще одному варианту осуществления R6 обозначает Н или R3 обозначает СН3, где остальные переменные независимо друг от друга могут иметь любое указанное выше значение. И согласно еще одному предпочтительному варианту осуществления Х обозначает карбонильную группу (С=O), где остальные переменные независимо друг от друга могут иметь любое указанное выше значение. В этом варианте осуществления предпочтительно m равно 1. В таблице 1, ниже, представлены примеры некоторых предпочтительных групп в различных положениях определенных соединений по изобретению. В этой таблице представлена группа конкретных соединений, которые получают путем выбора всех уникальных комбинаций заместителей, взятых по одному из каждой колонки для каждой переменной, и объединения этих групп со структурой, представленной в верхней части таблицы 1. Методы получения Соединения по настоящему изобретению могут быть получены с помощью методов, основанных на применении стандартных химических методик, которые описаны и на которые сделаны ссылки в известных руководствах (например, у March J. “Advanced Organic Chemistry” McGraw-Hill, New York, 1997; Collman J.P., Hegedus L.S., Norton J.R., Finke R.G. “Principles and Applications of Organotransition Metall Chemistry”, University Science, Mill Valley, 1987; Larock R.C., “Comprehensive Organic Transformations”, Verlag, New York, 1989). Основной промежуточный продукт для синтеза соединений по изобретению имеет приведенную ниже формулу В этой формуле А, В, R2, R4a, R4b, R5, R6, m и Q имеют значения, в том числе предпочтительные значения, которые указаны выше. Это соединение может быть получено с помощью нескольких альтернативных путей синтеза. После получения цианогруппа может быть превращена в амидиногруппу (C(NH)NH2), например, с помощью известных процессов, таких как реакция Пиннера. Циансодержащее соединение, имеющее указанную выше формулу, может быть подвергнуто взаимодействию с гидроксиламином, предпочтительно в спиртовом растворителе, с последующим восстановлением в присутствии Ni Ренея, предпочтительно в спиртовом растворителе, или может быть сначала подвергнуто взаимодействию с этанольным раствором НСl, а затем со спиртовым раствором аммиака с получением соответствующих амидиносоединений. В альтернативном варианте требуемое амидиносоединение может быть получено с помощью модифицированной реакции Пиннера с использованием пиридина/диэтиламина (1/1)/сероводорода с последующим добавлением метилйодида/ацетонитрила, а затем ацетата аммония/этанола. Одним из путей синтеза соединений, имеющих указанную выше формулу, является реакция конденсации с использованием соответствующим образом замещенных предшественников, как указано на схеме ниже Эту реакцию конденсации осуществляют в присутствии катализатора, предпочтительно такого катализатора, как кислота Льюиса, и алкилового спирта (ROH), предпочтительно (низш.)алкилового спирта, такого как метанол, этанол, изопропанол и т.д., с последующим гидролизом промежуточного продукта, предпочтительно с избытком воды, как правило, с использованием примерно до 10 эквивалентов воды. Приемлемые кислоты Льюиса включают эфират, BF3, АlСl3 и т.д. W-NC обозначает изонитрил, в котором W может представлять собой любую приемлемую углеводородную группу, как правило, алкильную, карбоциклоалкильную или аралкильную группу, предпочтительно имеющую не более примерно 12 атомов углерода. Особенно предпочтительным изонитрилом является бензилизонитрил. Продукт, представляющий собой сложный эфир, может быть очищен стандартными методами, включая жидкостную хроматографию высокого давления (ЖХВД), хроматографию на колонках, перекристаллизацию и т.д. Восстановление образовавшегося сложного эфира с получением спирта может быть осуществлено с использованием любого известного восстановителя ([Н]), который предпочтительно должен восстановить сложный эфир раньше, чем нитрил. Приемлемые восстановители и методы хорошо известны в данной области (см., например, Modern Synthetic Reactions. H.O. House, W.A. Benjami Inc., 2-ое изд., 1977). Пригодным восстановителем является борогидрид лития. Затем спирт может быть превращен в амин с помощью известных химических реакций. Приемлемые условия включают сначала взаимодействие спирта с водородистым азидом, DEAD и трифенилфосфином (РРh3), а затем с РРh3 и водой, или сначала с фталимидом, DEAD и РРh3, а затем с гидразином. Эти реакции приведены ниже на схеме. В альтернативном варианте сложный эфир может быть подвергнут взаимодействию с реагентом, имеющим нуклеофильный атом углерода, для введения приемлемых R4a-групп.Такие реагенты могут включать активированный углерод метилена, как, например, метилен, соседний с одним или несколькими сильными группами, являющимися акцепторами электронов, такими как нитро (NO2), карбоалкокси (COOR4а) и т.д., реагенты Гриньяра (R4аMgHal, где Hal обозначает галоген) и т.д., и затем превращены в спирт или амин. Превращение аминной функциональной группы в сульфонамид и превращение нитрильной функциональной группы в амидин может быть осуществлено в любом требуемом порядке. Предпочтительная реакционная схема приведена ниже. Эти превращения осуществляют с использованием известных химических реакций, методов очистки и разделения. Амин может быть превращен в сульфонамид путем взаимодействия с соответствующим образом замещенным сульфонилхлоридом (ClSO2R1) в присутствии основания. Нитрил может быть подвергнут взаимодействию с гидроксиламином в спиртовом растворителе с последующим восстановлением, например, с помощью никеля Ренея и водорода, или путем взаимодействия со смесью HCl/спирт, а затем аммиак/спирт. Пример приемлемой последовательности реакций приведен ниже. На этой схеме а – обозначает ВF3ОЕt2/ЕtOН, б – обозначает LiВН4/ДМЭ (диметиловый эфир), в – обозначает фталимид, DIAD (диизопропилазодикарбоксилат)/РРh3/ТГФ, г – обозначает H2NNH2/EtOH, д – обозначает R1SO2Cl, e – обозначает H2/Pt/C/EtOH и ж – обозначает R7SO2Cl/NEt3, NH2OH-HCl/NEt3, H2/Ra-Ni/MeOH. Аналогичная схема синтеза, которая может применяться для получения соответствующих соединений, в которых Х обозначает карбонил, приведена ниже. Соединения, в которых m равно 2, могут быть получены с помощью процесса, приведенного ниже на схеме, где спирт является гомологом спирта, указанного на вышеприведенной схеме, и он может быть превращен в амин (и в другие требуемые соединения) аналогичным образом. На приведенной ниже схеме (а) обозначает основание и (б) обозначает восстановитель, такой как LiBH4. Соединения, в которых Y обозначает C(O)-R1; C(O)-OR1; С(О)-NR1R2, получают согласно описанному выше процессу, используя соответствующий ацилгалогенид (предпочтительно ацилхлорид), алкилгалоформиат (предпочтительно хлорформиат) или изоцианат, как показано ниже на схеме: Пример приемлемой последовательности реакций приведен ниже Полученные в результате указанных выше реакций конденсации сложные эфиры также могут функционировать в качестве промежуточных продуктов для синтеза соединений, в которых Х обозначает карбонильную группу. Превращение сложного эфира в карбоновую кислоту может быть легко осуществлено омылением с помощью гидроксида щелочного металла, такого как гидроксид лития, натрия или калия. Сочетание сульфонамида с кислотой осуществляют сначала путем активации карбоксилата для его сочетания с использованием, например, карбонилдиимидазола или других обычных активирующих агентов, которые применяются для синтеза пептидов. Вторую часть реакции сочетания осуществляют путем смешения алкил- или арилсульфонамида с сильным основанием, например, ДБУ (1,8-диазабицикло[5.4.0]ундец-7-ен) или гидридом натрия, предпочтительно в безводном растворителе, таком как углеводородный или эфирный растворитель, например, тетрагидрофуран. Нитрил превращают в амидин с помощью ранее описанных методов. В этом наиболее предпочтительном варианте осуществления Q обозначает замещенный фенил, заместителями которого являются указанные выше Z1, Z2 и R9-R11. Еще один метод получения промежуточных продуктов, которые могут применяться для получения соединений по изобретению, приведен ниже, и он включает синтез иминных производных из легко доступных альдегидов и кетонов с последующим добавлением нуклеофильного реагента, содержащего нуклеофильный атом углерода, обозначенного как “Nu”. “Nu” может представлять собой такой фрагмент как CHR4aNO2, CHR4aCOOR, CH(NO2)(COOR) и т.д., которые получают с помощью хорошо известных реакций Гриньяра, реакций, в которых основание применяют для удаления протона из атома углерода, соседнего с группой, являющейся акцептором электрона (СО, COO, NO2) и т.д. “Nu” может быть превращен в такую группу, как CHR4aNH2 или СНR4aСН2ОН или CHR4aNH2CH2OH с помощью приведенных ниже известных реакций восстановления. В этих промежуточных продуктах аминогруппа может быть дополнительно сульфирована или ацилирована каким-либо иным образом, как описано выше. Ниже приведен пример приемлемой последовательности реакций. Для получения указанных выше спиртовых промежуточных продуктов может применяться другой процесс синтеза. Как показано на приведенной ниже схеме, реакция первичного стирольного производного с перацидом, как правило, позволяет получить смесь продуктов, содержащих не обозначающие водород заместители R4a и/или R5, как показано ниже, которые без разделения могут быть превращены в спирт взаимодействием с циананилином или соответствующим цианпиридином. Затем спирт может применяться для получения соединений по изобретению согласно описанному выше методу. Если требуется получить соответствующие соединения, в которых А и В обозначают азот, применяемые в описанных выше реакциях анилин или замещенный анилин заменяют на соответствующие аминопиридины или замещенные аминопиридины. Соединения, в которых азот сульфонамида несет заместитель, могут быть получены с помощью общепринятого алкилирования атома азота с использованием известных реакций, например, путем алкилирования с помощью диалкилсульфата, алкилгалогенида и т.д. согласно известным процессам. В предпочтительном варианте осуществления Q обозначает замещенный арил и более предпочтительно замещенную фенильную группу и имеет приведенную ниже структуру. В этой структурной формуле Z1, Z2, R9-R11 имеют указанные выше общие значения и значения, приведенные в предпочтительных вариантах осуществления. Соединения по этому варианту осуществления получают согласно вышеприведенной схеме I, используя соответствующим образом замещенный бензальдегид, имеющий строение Q-CHO (R5 обозначает Н). Эти замещенные бензальдегиды могут быть получены от производителей или могут быть легко получены из известных бензальдегидов с помощью хорошо известных методов синтеза. Согласно одному из вариантов осуществления Q замещен нитрогруппой. Предпочтительно нитрогруппа располагается на радикале R11 (где Z1, Z2, R9 и R10 имеют указанные выше общие значения и. значения, приведенные в предпочтительных вариантах осуществления), причем нитрогруппа может быть дополнительно восстановлена с получением аминогруппы с помощью приемлемого восстановителя. Как правило, цианаминное производное или циансульфонамидное производное, приведенные на схеме 3, могут быть подвергнуты взаимодействию с восстановителем, который предпочтительно должен восстанавливать нитрогруппу на R11, а не цианогруппу. Могут применяться любые восстановители, обладающие этими свойствами, например, водород и катализатор Pt/C. Анилин, образовавшийся в результате восстановления, затем может быть подвергнут взаимодействию с сульфонилхлоридом (ClSO2W, где W имеет указанные выше значения) с получением дисульфонамидного соединения. Синтез производных циклической мочевины, в которых N1-R2 и N2-R2 вместе образуют характерную для мочевины связь, т.е. N1-C(O)-N2, позволяет получить дополнительные соединения по изобретению, а процессы их получения представляют собой дополнительный вариант способа получения энантиомерно чистых соединений по изобретению. Производные циклической мочевины могут применяться, например, для получения диалкоксибиссульфонамидов и других соединений по изобретению, как показано ниже на схеме. В альтернативном варианте азотная кислота на приведенной ниже схеме может быть заменена серной кислотой с получением производных сульфоновой кислоты, которые дополнительно могут быть превращены в сульфонамиды и сульфоны с помощью известных реакций. Другие соединения по изобретению, включая гетероциклические соединения, могут быть легко получены с использованием обычных исходных продуктов, которые могут применяться согласно процессам, приведенным на указанных выше схемах синтеза. Например, используя в качестве исходных продуктов простые нитро- и гидроксизамещенные альдегиды, после описанной выше реакции конденсации получают соответствующие сложные эфиры, которые могут быть непосредственно превращены в циклические производные уретана или оксазола, которые затем могут быть подвергнуты ранее описанной обработке с получением соединений по изобретению. Эти реакции схематично приведены ниже для колец, сконденсированных в положении 5 и положении 6. Соединения, в которых кольцо слито в 4 положении и 5 положении с фенильным кольцом, получают аналогичными методами, используя в качестве исходных продуктов соответствующим образом замещенный альдегид, как показано ниже. Другие слитые гетероциклические соединения получают, используя общепринятые реакции химического синтеза и соответствующим образом замещенные исходные продукты, которые хорошо известны в области химического синтеза, в результате чего получают дополнительные соединения по изобретению. Например, слитые фурановые кольцевые системы могут быть получены из соответствующих гало- и гидроксизамещенных альдегидов, как показано ниже. Под объем изобретения также подпадают пролекарства описанных выше соединений. Приемлемые пролекарства включают известные аминозащитные и карбоксизащитные группы, которые отщепляются в физиологических условиях, например, в результате гидролиза, с образованием исходного соединения. Предпочтительным классом пролекарств являются соединения, в которых атом азота на амино-, амидино-, аминоалкиленамино-, иминоалкиленамино- гуанидиногруппе замещен гидроксигруппой (ОН), алкилкарбонильной (-CO-W) группой, алкоксикарбонильной (-CO-OW) группой, ацилоксиалкилалкоксикарбонильной (-CO-O-W-O-CO-W) группой, где W обозначает одновалентную или двухвалентную группу, указанную выше, или группу формулы -С(O)-O-СР1Р2-галоалкил, где Р1 и Р2 имеют одинаковые или различные значения и обозначают Н, (низш.)алкил, (низш.)алкокси, циано, гало(низш.)алкил или арил. Предпочтительно атом азота представляет собой один из атомов азота амидиногруппы соединений по изобретению. Эти соединения в виде пролекарства получают взаимодействием описанных выше соединений по изобретению с активированным ацильным производным для связывания атома азота соединения по изобретению с карбонилом активированного ацильного производного. Приемлемые активированные карбонильные производные содержат легко отщепляемую уходящую группу, связанную с углеродом карбонила, и включают ацилгалогениды, ациламины, соли ацилпиридиния, ацилалкоксиды, в частности ацилфеноксиды, такие как пара-нитрофеноксиацил, динитрофеноксиацил, фторфеноксиацил и дефторфеноксиацил. Реакции, как правило, являются экзотермическими и их осуществляют в инертных растворителях при пониженных температурах, таких как от -78, до примерно 50°С. Реакции также обычно осуществляют в присутствии неорганического основания, такого как карбонат калия или бикарбонат натрия, или органического основания, такого как амин, включая пиридин, триэтиламин и т.д. Один из путей получения пролекарств описан в WO 98/46576, опубликованной 22 октября 1998 г. С помощью описанных выше методов синтеза могут быть получены следующие репрезентативные соединения по изобретению (m равно 1), приведенные в таблице 2. В каждом из представленных в таблице радикалов Х может обозначать карбонил или (CR4aR4b)m, где m pавно 1 или 2; и бензамидиновое кольцо может нести в качестве заместителя галоген, гидрокси или алкил. Соединения по изобретению содержат один или несколько асимметричных атомов углерода. Следовательно, соединения могут существовать в виде диастереомеров, энантиомеров или их смесей. Описанные выше пути синтеза могут применяться с использованием в качестве исходных или промежуточных продуктов рацематов, диастереомеров или энантиомеров. Диастереомерные соединения могут быть разделены методами хроматографии или кристаллизации. Аналогично этому энантиомерные смеси могут быть разделены с использованием этих же или других методов, известных в данной области. Каждый из асимметричных атомов углерода может находиться в R-или S-конфигурации, и обе эти конфигурации подпадают под объем изобретения. Применение Как указано выше, полученные и отобранные согласно описанным выше анализам соединения по изобретению являются ингибиторами серинпротеаз, например, фактора VIIa, ТФ/фактора VIIa, фактора Ха, калликреина и/или тромбина. Эти соединения обладают способностью ингибировать каталитическую активность этих ферментов и вследствие этого ингибировать каскад коагулирования и предупреждать или ограничивать коагулирование и/или образование тромбов или эмболов в кровеносных сосудах и/или повышать время коагулирования крови. Таким образом, соединения по настоящему изобретению ингибируют способность ТФ/фактора VIIa превращать фактор Х в фактор Ха, ингибируют способность фактора Ха превращать протромбин в тромбин (фактор IIа); и/или способность тромбина превращать фибриноген в мономеры фибрина. Избирательное действие соединений по изобретению с качестве ингибиторов этих ферментов может быть определено на основе значений Кi, как описано ниже в примерах. Данные об избирательной активности некоторых репрезентативных соединений приведены ниже в таблицах 3-6.
Активность соединений по изобретению в качестве антикоагулянтов может быть определена с помощью приведенных ниже анализов. Анализы по определению протромбинового времени (ПВ) и активированного парциального тромбопластинового времени (АПТВ) свертывания могут быть проведены с использованием объединенной нормальной плазмы (человека или различных видов животных) после добавления в плазму возрастающих концентраций ингибиторов. Время свертывания определяют с использованием автоматического анализатора коагулирования типа ACL 300 (фирма Coulter Corp., Майами, штат Флорида) и поступающих в продажу реагентов следующим образом. ПВ-анализ: Водные растворы различных концентраций ингибитора добавляют в объединенную нормальную плазму в соотношении 1 часть ингибитора на 9 частей плазмы. Затем эти смеси вносят в чашки для анализа образцов. К реагентам в чашки добавляют Innovin® (фирма Dade International Inc, Майами, штат Флорида), представляющий собой смесь человеческого обезжиренного тканевого фактора и ионов Са+ +. Точные объемы образца и Innovin® автоматически переносят в ячейки акрилового ротора, предварительно уравновешенного при 37°С. После 2-минутного периода инкубации инициируют коагулирование путем смешения друг с другом двух компонентов с помощью центрифугирования. Коагулирование оценивают оптически и время свертывания регистрируют в секундах. Согласно Janson с соавторами (Jansori T.L. и др., Haemostasis, 14: 440-444, 1984) обезжиренный человеческий тканевый фактор является сильным инициатором коагулирования у всех изученных видов. В этой системе время свертывания в контрольных образцах плазмы (плазма плюс разбавитель для ингибитора), как правило, составляет 8-10 с. Строят график зависимости времени свертывания от концентрации ингибитора и для каждого ингибитора определяют концентрацию, при которой ПВ в 2 раза выше, чем в контрольной плазме. АПТВ-анализ: Ингибитор и плазму смешивают и переносят в чашки для образцов устройства типа ACL 300 согласно описанному выше процессу. К реагентам в чашках 1 и 2 соответственно добавляют Actin FS® и СаСl2 (фирма Dade International Inc, Майами, штат Флорида). Точные объемы образца и активатора (Actin FS®) автоматически переносят в ячейки акрилового ротора, предварительно уравновешенного при 37°С, и смешивают с помощью центрифугирования. После 2-минутного периода активации инициируют коагулирование, добавляя СаСl2. Осуществляют мониторинг коагулирования и оценивают данные согласно методу, описанному для ПВ-анализа. АПТВ для контрольных образцов плазмы, как правило, составляет 12-32 с в зависимости от того, плазма каких видов используется для анализа. Репрезентативные результаты ПВ-и АПТВ-анализов приведены ниже в таблице 7.
Соединения по изобретению могут применяться в качестве диагностических реагентов in vitro для ингибирования коагулирования в пробирках для взятия крови. Применение снабженных стопором пробироок с вакуумом внутри в качестве средства для взятия крови хорошо известно (см. Kasten B.L., “Specimen Collection”, Laboratory Test Handbook, 2-ое изд., Lexi-Comp. Inc. Cleveland, стр.16-17, ред-ры Jacobs и др., 1990). Такие вакуумные пробирки могут не содержать ингибирующих коагулирование добавок, в этом случае они применяются для выделения сыворотки из крови млекопитающих. Они также могут включать ингибирующие коагулирование добавки, такие как гепариновые соли, цитратные соли или оксалатные соли, и в этом случае они могут применяться для выделения сыворотки из крови млекопитающих. Соединения по изобретению могут быть внесены в пробирки для сбора крови и функционировать в качестве ингибиторов ТФ/фактора VIIa, фактора Ха, тромбина и/или калликреина и препятствовать коагулированию крови млекопитающего, поступающей в пробирки. При использовании в пробирках для сбора крови соединения по настоящему изобретению могут применяться индивидуально, в виде смесей или в сочетании с другими соединениями-ингибиторами коагулирования, которые известны в данной области. Количество соединения по изобретению должно представлять собой количество, достаточное для предупреждения или ингибирования образования сгустка при поступлении крови в пробирку. Эти соединения могут быть внесены в пробирки аналогично тому, как вносятся известные соединения, ингибирующие образование сгустка, такие как гепариновые соли. Жидкость, как правило, лиофилизируют с помощью известных методов. Обычно пробирки должны содержать от примерно 2 до примерно 10 мл крови млекопитающего и соединения добавляют в количестве, достаточном для предупреждения коагулирования этого количества крови. Приемлемая концентрация составляет 10-1000 нМ. Эти соединения также ингибируют образования эмбол и тромбов в кровеносной системе млекопитающих и вследствие этого могут применяться in vivo. Установлено, что тромбоэмболические нарушения непосредственно связаны с чувствительностью млекопитающего к образованию эмболов и тромбов. Например, образование тромба в венозном сосуде приводит к тромбофлебиту, который, как правило, лечат покоем и введением антикоагулянотов. Другие состояния, которые могут лечиться с использованием антикоагулянтов по изобретению, включают тромболимфангиит, тромбосинусит, тромбэндокардит, тромбангиит и тромбартериит. Млекопитающие, которых подвергают медицинским процедурам, таким как ангиопластика и тромболитическая терапия, особенно чувствительны к образованию тромбов. Соединения по настоящему изобретению могут применяться для ингибирования образования тромбов после ангиопластики. Они также могут применяться в сочетании с антитромболитическими агентами, такими как тканевый активатор плазминогена и его производные (патенты US 4752603; 4766075; 4777043; ЕР 199574; ЕР 238304; ЕР 228862; ЕР 297860; РСТ WO 89/04368; РСТ WO 89/00197), стрептокиназа и ее производные или урокиназа и ее производные, для предупреждения артериальной реокклюзии после тромболитической терапии. При введении в сочетании с вышеперечисленными тромболитическими агентами соединения по настоящему изобретению могут вводиться до, одновременно или последовательно с антитромболитическим агентом. Млекопитающие, которых подвергают почечному диализу, оксигенации крови, катетеризации сердца и аналогичным медицинским процедурам, а также млекопитающие, которым введены определенные протезные приспособления, также чувствительны к тромбоэмболическим нарушениям. Физиологические условия по известным или неустановленным причинам также могут приводить к тромбоэмболическим нарушениям. Таким образом, соединения, представленные в настоящем описании, могут применяться для лечения тромбоэмболических нарушений у млекопитающих. Соединения, представленные в настоящем описании, также могут применяться в качестве вспомогательных средств при антикоагуляционной терапии, например, в сочетании с аспирином, гепарином или варфарином и другими антикоагулянтами. Различные нарушения коагулирования, описанные выше, лечат с помощью соединений по изобретению таким образом, чтобы предотвратить кровотечение в результате такого нарушения. Применение соединений, представленных в настоящем описании, при таких и аналогичных нарушениях очевидно специалистам в данной области. Соединения по изобретению также могут применяться в качестве промежуточных продуктов в целом или в качестве предшественников ингибиторов связанной с коагулированием серинпротеазы и вследствие этого помимо лечения сердечно-сосудистого заболевания эти соединения могут применяться при метастатическом заболевании или любом заболевании, при котором показано ингибирование коагулирования. Как правило, ингибиторы, применяемые согласно способу по изобретению, приготавливают путем смешения при температуре окружающей среды и при пригодном значении рН и с требуемой степенью чистоты с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов в применяемых дозах и концентрациях. Значение рН композиции в основном зависит от конкретного примененного соединения и его концентрации, но предпочтительно находится в диапазоне от примерно 3 до примерно 8. Приемлемым вариантом осуществления является композиция в ацетатном буфере, рН 5. Соединения-ингибиторы для применения по настоящему изобретению предпочтительно являются стерильными. Соединение обычно должно храниться в виде твердой композиции, хотя также пригодны лиофилизированные композиции или водные растворы. Композиция по изобретению может быть приготовлена, разделена на дозы и введена согласно принятой медицинской практике. Факторы, которые следует при этом учитывать, включают конкретное нарушение, подлежащее лечению, конкретное млекопитающее, подлежащее лечению, клиническое состояние конкретного пациента, причину болезни, область доставки агента, метод введения, схему применения и другие факторы, известные практикующим врачам. “Терапевтически эффективное количество” соединения, подлежащего введению, должно соответствовать этим требованиям и оно представляет собой наименьшее количество, необходимое для предотвращения, облегчения симптомов или лечения заболевания, обусловленного фактором коагулирования. Такое количество предпочтительно ниже токсичного для хозяина количества или количества, которое делает хозяина значительно более чувствительным к кровотечению. В целом предложено применять такое начальное фармацевтически эффективное количество ингибитора при его парентеральном введении, чтобы доза составляла примерно 0,01-100 мг/кг, предпочтительно примерно 0,1-20 мг/кг веса тела пациента в день, причем обычный начальный диапазон доз применяемого соединения составляет от 0,3 до 15 мг/кг/день. Соединение по изобретению вводят любым приемлемым путем, включая пероральный, местный, трансдермальный, парентеральный, подкожный, внутрибрюшинный, внутрилегочный и интриназальный и, если требуется местное иммуносупрессорое лечение, путем введения внутрь повреждения (включая перфузию или другой контакт трансплантата с ингибитором перед трансплантацией). Парентеральные перфузии включают внутримышечное, внутривенное, внутиартериальное, внутрибрюшинное или подкожное введение. Изобретение станет более понятным после изучения приведенных ниже примеров. Однако должно быть очевидно, что они не направлены на ограничение объема изобретения. Все патентные и литературные источники полностью включены в настоящее описание в качестве ссылки. Примеры Соединения по изобретению в целом могут быть получены с помощью приведенной ниже реакционной схемы. Соединения, отличные от конкретного приведенного продукта, получают согласно описанному выше процессу, используя соответствующие исходные продукты. Например, дополнительные соединения могут быть получены с использованием других исходных стиролов, которые могут быть легко синтезированы из поступающих в продажу исходных продуктов и с помощью стандартных реакций, которые хорошо известны в данной области. Пример 1 4-Бензилокси-3-метоксистирол (10 г, 42 ммоль) растворяли в дихлорметане (400 мл). Добавляли твердый бикарбонат калия (11 г, 110 ммоль) и реакционную смесь охлаждали до 0°С. Добавляли мета-хлорпербензойную кислоту (12 г, примерно 42 ммоль) и реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 ч. С помощью тонкослойной хроматографии проводили мониторинг реакции. Вносили дополнительное количество мета-хлорпербензойной кислоты (4 г) и реакционную смесь перемешивали в течение еще 4 ч до полного поглощения исходного продукта. Реакционную смесь сливали в делительную воронку и промывали сначала водой, затем бикарбонатом натрия и наконец NaOH. Органический слой отделяли и сушили над безводным сульфатом натрия. Раствор фильтровали и растворитель выпаривали в вакууме, получая примерно 11 г неочищенного продукта. Неочищенный продукт затем растворяли в ацетонитриле (60 мл) и добавляли перхлорат лития (8,5 г, 80 ммоль). Суспензию перемешивали в течение 5 мин, за это время реакционная смесь становилась гомогенной. Добавляли 4-аминобензонитрил (9,5 г, 80 ммоль) и реакционную смесь нагревали до 60°С в течение 12 ч. Тонкослойная хроматография показала присутствие нового продукта с более низким значением Rf. Растворитель удаляли в вакууме и остаток растворяли в этилацетате, промывали водой и сушили над безводным сульфатом натрия. Затем неочищенный продукт подвергали экспресс-хроматографии (гексан:этилацетат, 1:1), получая 6 г 4-[1-(4-бензилокси-3-метоксифенил)-2-гидроксиэтиламино] бензонитрила А. 1H-ЯМР (CDCl3): 7,3-7,45 (m, 7Н), 6,8 (m, 3Н), 6,5 (d, 2H), 5,18 (s, 2H), 4,42 (m, 1H), 3,95 (dd, 1H), 3,85 (s, 3Н), 3,8 (dd, 1H). 4-[1-(4-Бензилокси-3-метоксифенил)-2-гидроксиэтиламино] бензонитрил А-(470 мг, 1,25 ммоль), фталимид (1,47 г, 10 ммоль) и трифенилфосфин (787 мг, 3 ммоль) добавляли к 40 мл тетрагидрофурана. Смесь перемешивали в течение 10 мин и затем охлаждали до 0°С. Затем медленно добавляли диизопропилазодикарбоксилат (DIAD, 0,6 мл, 3 ммоль). Реакционной смеси давали перемешаться в течение 1 ч. ТСХ показала наличие нового продукта. Растворитель удаляли в вакууме и остаток растворяли в 50 мл этилацетата. Раствор промывали трижды 2н. гидроксидом натрия и дважды водой. Органический слой отделяли, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли в вакууме и остаток подвергали экспресс-хроматографии (гексан:этилацетат, 1:1), получая 478 мг 4-[1-(4-бензилокси-3-метоксифенил)-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино] бензонитрила Б (76%-ный выход). 1H-ЯМР (СDСl3): 7,85 (m, 2H), 7,75 (m, 2H), 7,23-7,45 (m, 9H), 6,9 (m, 3Н), 6,42 (d, 2H), 5,45 (d, 1H), 5,15 (s, 2H), 4,62 (m, 1H), 4,0 (m, 2H), 3,83 (s, 3H). 4-[1-(4-Бензилокси-3-метоксифенил)-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино]бензонитрил Б затем растворяли в этаноле (60 мл) и добавляли гидрат гидразина (2 г). Раствор нагревали до 60-70°С в течение 1,5 ч. ТСХ показала, что реакция завершена. Суспензию фильтровали для удаления побочного продукта и этанол удаляли в вакууме. Остаток подвергали экспресс-хроматографии на силикагеле (этилацетат:2н. NН3 в метаноле, 9:1), получая 372 мг 4-[2-амино-1-(4-бензилокси-3-метоксифенил)этиламино]бензонитрила Г (100%-ный выход). 1H-ЯМР (СDСl3): 7,3-7,45 (m, 7H), 6,32 (m, 3Н), 6,5 (d, 2H), 5,52 (d, 1H), 4,3 (q, 1H), 3,83 (s, 3Н), 3,08 (m, 2H), 1,95 (s, 2H). 4-[2-Амино-1-(4-бензилокси-3-метоксифенил)этиламино] бензонитрил Г (300 мг, 0,8 ммоль) растворяли в этаноле (3 мл) и добавляли гидрохлорид гидроксиламина (350 мг, 5 ммоль) и триэтиламин (1 мл, 5,7 ммоль). Реакционную смесь нагревали до 65-70°С в течение 2 ч. Остаток растворяли в этилацетате и воде. Органический слой отделяли, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли в вакууме и заменяли 4 мл метанола и 0,5 мл уксусной кислоты. Добавляли никель Ренея (примерно 30 мкл суспензии в гидроксиде натрия, фирма Aldrich) и реакционную смесь помещали в атмосферу водорода. Реакционную смесь интенсивно перемешивали в течение 3 ч, катализатор отфильтровывали и растворитель удаляли в вакууме. Неочищенный продукт очищали экспресс-хроматографией на силикагеле (этилацетат:ацетон:метанол:аммиак, 2:1:1:0,05), получая 160 мг 4-[2-амино-1-(4-бензилокси-3-метоксифенил)этиламино]бензамидина Д. МС (М+Н)=391. 4-[2-Амино-1-(4-бензилокси-3-метоксифенил)этиламино] бензамидин Д (20 мг, 0,03 ммоль) растворяли в ацетонитриле (2 мл), содержащем триэтиламин (17 мкл, 0,12 ммоль) и воду (0,3 мл). К этой смеси добавляли требуемый сульфонилхлорид формулы ClSO2R (0,03 ммоль) и реакционную смесь перемешивали в течение 4 ч. Растворитель удаляли в вакууме и соединения очищали препаративной ЖХВД с обращенной фазой (градиент ацетонитрила/воды с добавлением 0,1% трифторуксусной кислоты), получая после лиофилизации конечный продукт. Примеры 2а-2гг Работая аналогично, получали другие соединения по изобретению, включая: а) 4-[бензолсульфониламино-1-(4-бензилокси-3-метоксифенил) этиламино]бензамидин: МС (М+Н)=531, б) N-[4-[2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этилсульфамоил]фенил}ацетамид: МС (М+Н)=588, в) 4-[1-(4-бензилокси-3-метоксифенил)-2-(4-нитробензолсульфониламино)этиламино]бензамидин: МС (М+Н)=576, г) 4-[1-(4-бензилокси-3-метоксифенил)-2-(4-фторбензолсульфониламино)этиламино]бензамидин: МС (М+Н)=549, д) 4-[1-(4-бензилокси-3-метоксифенил)-2-(4-фторбензолсульфониламино)этиламино]бензамидин: МС (М+Н)=565, е) 4-[1-(4-бензилокси-3-метоксифенил)-2-(3-нитробензолсульфониламино)этиламино]бензамидин: МС (М+Н)=576, ж) 4-[1-(4-бензилокси-3-метоксифенил)-2-(2,5-дихлорбензолсульфониламино)этиламино]бензамидин: МС (М+Н)=599, з) 4-[1-(4-бензилокси-3-метоксифенил)-2-(2-бромбензолсульфониламино)этиламино]бензамидин: МС (М+Н)=609, 611, и) 4-[1-(4-бензилокси-3-метоксифенил)-2-(4-бромбензолсульфониламино)этиламино]бензамидин: МС (М+Н)=609, к) 4-[1-(4-бензилокси-3-метоксифенил)-2-(4-изопропилбензолсульфониламино)этиламино]бензамидин: МС (М+Н)=573, л) 4-[1-(4-бензилокси-3-метоксифенил)-2-фенилметансульфониламиноэтиламино]бензамидин: МС (М+Н)=545, м) 4-[1-(4-бензилокси-3-метоксифенил)-2-(4-карбоксибензолсульфониламино)этиламино]бензамидин: МС (М+Н)=575, н) 4-[1-(4-бензилокси-3-метоксифенил)-2-(3-карбоксибензолсульфониламино)этиламино]бензамидин: МС (М+Н)=575, о) 4-[1-(4-бензилокси-3-метоксифенил)-2-(2,4-динитробензолсульфониламино)этиламино]бензамидин: МС (М+Н)=621, п) 4-[1-(4-бензилокси-3-метоксифенил)-2-(2,3,5,6-тетраметилбензол-сульфониламино)этиламино]бензамидин: МС (М+Н)=587, р) 4-[1-(4-бензилокси-3-метоксифенил)-2-(3,5-дихлор-2-гидроксибензолсульфониламино)этиламино]бензамидин: МС (М+Н)=615, с) 4-[1-(4-бензилокси-3-метоксифенил)-2-(3,4-диметоксибензолсульфониламино)этиламино]бензамидин: МС (М+Н)=591, т) 4-[1-(4-бензилокси-3-метоксифенил)-2-(тиофен-2-сульфониламино) этиламино] бензамидин: МС (М+Н)=537, у) N-{5-[2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этилсульфамоил]-4-метилтиазол-2-ил}ацетамид: МС (М+Н)=595, ф) 4-[1-(4-бензилокси-3-метоксифенил)-2-(нафталин-2-сульфониламино)этиламино]бензамидин: МС (М+Н)=581, х) 4-[1-(4-бензилокси-3-метоксифенил)-2-(нафталин-1-сульфониламино)этиламино]бензамидин: МС (М+Н)=581, ц) 4-[1-(4-бензилокси-3-метоксифенил)-2-(2-фенилэтенсульфониламино)этиламино]бензамидин: МС (М+Н)=557, ч) 4-[1-(4-ензилокси-3-метоксифенил)-2-(3-трифторметилбензболсульфониламино)этиламино] бензамидин: МС (М+Н)=599, ш) 4-[1-(4-бензилокси-3-метоксифенил)-2-(2,3,4,5,6-пентафторбензол-сульфониламино)этиламино]бензамидин: МС (М+Н)=521, щ) 4-[1-(4-бензилокси-3-метоксифенил)-2-метансульфониламиноэтиламино]бензамидин: МС (М+Н)=469, э) 4-[1-(4-бензилокси-3-метоксифенил)-2-этансульфониламиноэтиламино]бензамидин: МС (М+Н)=483, аа) 4-[1-(4-бензилокси-3-метоксифенил)-2-пропансульфониламиноэтиламино]бензамидин: МС (М+Н)=497, бб) 4-[1-(4-бензилокси-3-метоксифенил)-2-бутансульфониламиноэтиламино]бензамидин: МС (М+Н)=511, вв) этиловый эфир [2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этилсульфамоил]уксусной кислоты: МС (М+Н)=541, гг) [2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этилсульфамоил]уксусную кислоту. Пример 3 4-[1-(4-Бензилокси-3-метоксифенил)-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино]бензонитрил Б (1,8 г) растворяли в смеси, содержащей этанол (50 мл), уксусную кислоту (3 мл), метанол (5 мл) и этилацетат (5 мл). Этот раствор вносили в колбу Парра, содержащую 10%-ный Pd/C (500 мг), и гидрировали в течение 16 ч при давлении 35 фунтов/кв. дюйм. Катализатор удаляли фильтрацией через целит и растворитель удаляли в вакууме, получая 1 г продукта, т.е. 4-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-1-(4-гидрокси-3-метоксифенил)этиламино]бензонитрила (68%). 4-[2-(1,3-Диоксо-1,3-дигидроизоиндол-2-ил)-1-(4-гидрокси-3-метоксифенил)этиламино]бензонитрил (1 г, 2,43 ммоль) растворяли в гетрагидрофуране (30 мл) и добавляли трифенилфосфин (1,27 г, 4,84 ммоль), (S)-2-хлор-1-фенилэтанол (1,13 г, 7,26 ммоль). Реакционную смесь охлаждали до 0°С и добавляли диэтилазодикарбоксилат (0,842 г, 4,8 ммоль). Реакционной смеси давали нагреться до комнатной температуры и реакционную змесь перемешивали в течение 2,5 ч. Растворитель удаляли в вакууме и остаток растворяли в этилацетате, промывали несколько раз 0,5 н. гидроксидом натрия, однократно промывали соляным раствором и сушили над безводным сульфатом натрия. Неочищенный продукт очищали экспресс-хроматографией на силикагеле (30% этилацетата в гексане), получая 1,1 г 4-[1-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино]бензонитрила (82%). 4-[1-[4-(2-Хлор-1-фенилэтокси)-3-метоксифенил]-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино]бензонитрил (0,5 г) растворяли в этаноле (40 мл) и добавляли гидрат гидразина (0,14 г). Реакционную смесь выдерживали в течение 2 ч при 65°С. Растворитель удаляли и заменяли этилацетатом. Раствор дважды промывали водой и один раз соляным раствором. Раствор сушили над безводным сульфатом натрия и растворитель удаляли в вакууме, получая 318 мг требуемого амина, т.е. 4-{2-амино-1-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]этиламино}бензнитрила (83%). 4-[2-Амино-1-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]этиламино}бензонитрил (0,1 г, 0,237 ммоль) растворяли в дихлорметане (6 мл) и добавляли триэтиламин (34 мкл, 0,3 ммоль). Добавляли фенилсульфонилхлорид (46 мкл, 0,26 ммоль) и реакционную смесь перемешивали в течение 90 мин. Реакционную смесь разбавляли дихлорметаном и промывали один раз насыщенным раствором бикарбоната натрия и один раз водой. Раствор сушили над сульфатом натрия и растворитель удаляли в вакууме. Продукт очищали на силикагеле, получая 100 мг требуемого продукта, т.е. N-[2-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]-2-(4-цианфениламино)этил]бензолсульфонамида. N-[2-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]-2-(4-цианфениламино)этил]бензолсульфонамид (100 мг, 0,18 ммоль) растворяли в этаноле (3 мл) и добавляли гидрохлорид гидроксиламина (62 мг, 0,89 ммоль). К этой смеси добавляли триэтиламин (90 мг, 0,89 ммоль), а затем карбонат калия (62 мг). Реакционную смесь нагревали до 80°С в течение 48 ч. Реакционную смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в этилацетате и промывали дважды водой и один раз соляным раствором. Раствор сушили над сульфатом натрия и удаляли растворитель. Неочищенный промежуточный продукт растворяли в метаноле (4 мл) и добавляли несколько капель уксусной кислоты. Добавляли приблизительно 50-100 мг никеля Ренея, суспендированного в гидроксиде натрия (фирма Aldrich) и реакционную смесь помещали в атмосферу водорода. Суспензию интенсивно перемешивали в течение 8 ч, отфильтровывали катализатор и растворитель удаляли в вакууме. Неочищенный продукт очищали хроматографией с обращенной фазой, получая 4-[2-бензолсульфониламино-1-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]этиламино}бензамидин (32 мг). МС (М+Н)=579. Пример 4 4-{2-Пропансульфониламино-1-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]этиламино}бензамидин получали аналогично примеру 2 за исключением того, что в реакции с 4-{2-амино-1-[4-(2-хлор-1-фенилэтокси)-3-метоксифенил]этиламино}бензонитрилом бензолсульфонилхлорид заменяли на пропансульфонилхлорид. МС (М+Н)=545. Пример 5 4-Бензилокси-5-метокси-2-нитробензальдегид (12,2 г, 42 ммоль) и 4-аминобензонитрил (5 г, 42 ммоль) растворяли в метаноле (165 мл) и перемешивали в течение 2 ч, а затем нагревали до 60°С в течение 30 мин. Реакционной смеси давали охладиться до комнатной температуры и добавляли бензилизонитрил (5 г, 42 ммоль). Реакционную смесь охлаждали до 0°С и в течение 5 мин по каплям добавляли трифторэфират бора (16 мл, 126 ммоль). Реакционную смесь перемешивали в течение 20 мин при 0°С и затем давали нагреться до комнатной температуры, после чего перемешивали при температуре окружающей среды в течение 2 ч. Добавляли воду (4 мл) и смесь перемешивали при комнатной температуре в течение ночи. На следующее утро образовывался осадок желтого цвета и твердый продукт отфильтровывали. Твердый продукт промывали метанолом и сушили на воздухе, получая 8 г требуемого продукта. Растворитель удаляли из фильтрата в вакууме и заменяли этилацетатом. Раствор промывали водой и насыщенным бикарбонатом натрия, сушили над безводным сульфатом магния и удаляли растворитель. Неочищенный продукт подвергали экспресс-хроматографии (гексан:этилацетат, 1:1), получая дополнительно 7 г требуемого продукта, т.е. метилового эфира (4-бензилокси-5-метокси-2-нитрофенил)-(4-цианфениламино) уксусной кислоты. 1H-ЯМР (CDCl3: 7,68 (s, 1H), 7,4 (m, 7H), 7,0 (s, 1H), 6,61 (d, 2H), 6,2 (s. 1H), 5,2 (s, 2H), 3,87 (s, 3Н), 3,75 (s, 3H). Метиловый эфир (4-бензилокси-5-метокси-2-нитрофенил)-(4-цианфениламино) уксусной кислоты (4,5 г, 10 ммоль) растворяли в диметоксиэтане и добавляли борогидрид лития (0,210 г, 10 ммоль). Реакционную смесь нагревали до температуры дефлегмации в течение 3 ч и охлаждали до комнатной температуры. Реакцию прекращали, добавляя уксусную кислоту, содержащую воду, и разбавляли этилацетатом. После переноса в делительную воронку органический слой промывали несколько раз водой. Органический слой сушили над безводным сульфатом натрия, фильтровали и удаляли растворитель. Затем неочищенный продукт подвергали экспресс-хроматографии, получая 3,1 г 4-[1-(4-бензилокси-5-метокси-2-нитрофенил)-2-гидроксиэтиламино]бензонитрила (74%). 1H-ЯМР (СDСl3): 7,77 (s, 1H), 7,3-7,5 (m, 7H), 7,15 (s, 1H), 6,42 (d, 2H), 5,4 (bs, 1H), 5,18 (dd, АВ сист., 2H), 4,15 (dd, 1H), 3,83 (s, 3H), 3,79-3,86 (dd, 1H). 4-[1-(4-Бензилокси-5-метокси-2-нитрофенил)-2-гидроксиэтиламино] бензонитрил (3,1 г, 7,4 ммоль) растворяли в тетрагидрофуране (120 мл) и добавляли трифенилфосфин (5,9 г, 22 ммоль) и фталимид (5,4 г, 37 ммоль). Реакционную смесь охлаждали до 0°С и по каплям добавляли диизопропилазадикарбоксилат (DIAD, 4,6 г). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли в вакууме и заменяли этилацетатом. Раствор несколько раз промывали 1н. NaOH и сушили над безводным сульфатом натрия. После экспресс-хроматографии (гексан:этилацетат, 1:1) получали требуемый продукт, в котором еще присутствовало некоторое количество DIAD. Твердый продукт промывали несколько раз этанолом, получая 3,2 г требуемого фталимида, т.е. 4-[1-(4-бензилокси-5-метокси-2-нитрофенил)-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино]бензонитрила (3,2 г). 1H-ЯМР (CDCl3: 7,83 (m, 2H), 7,75 (m, 2H), 7,37 (m, 7H), 6,91 (s, 1H), 6,41 (d, 2H), 6,17 (d, 1H), 5,65 (m, 1H), 5,18 (s, 2H), 4,27 (m, 2H), 3,61 (s, 3Н). 4-[1-(4-Бензилокси-5-метокси-2-нитрофенил)-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этиламино]бензонитрил (2,7 г, 5 ммоль) растворяли в этаноле (100 мл) и добавляли гидрат гидразина (0,65 мл, 20 ммоль). Реакционную смесь нагревали до 60°С в течение 3 ч и затем выдерживали при комнатной температуре в течение 48 ч. Осадившиеся твердые частицы отфильтровывали и остаток подвергали экспресс-хроматографии, получая 4-[2-амино-1-(4-бензилокси-5-метокси-2-нитрофенил)этиламино]бензонитрил (1,5 г). 1H-ЯМР (СDСl3): 7,75 (s, 1H), 7,3-7,5 (m, 7H), 7,05 (s, 1H), 6,45 (d, 2H), 5,80 (bs, 1H), 5,32 (m, 1H), 5,19 (s, 2H), 3,81 (s, 3Н), 3,25 (dd, 1H), 3,0 (dd, 1H), 1,65 (bs, 2H). 4-[2-Амино-1-(4-бензилокси-5-метокси-2-нитрофенил)этиламино]бензонитрил (0,227 г, 0,66 ммоль) растворяли в дихлорметане (4 мл) и триэтиламине (0,14 мл, 1 ммоль). Реакционную смесь охлаждали до 0°С и добавляли 1-пропансульфонилхлорид (0,085 мл, 0,75 ммоль). Реакционную смесь перемешивали в течение 20 мин и продукт очищали экспресс-хроматографией (гексан:этилацетат, 1:1), получая 260 мг требуемого продукта, т.е. [2-(4-бензилокси-5-метокси-2-нитрофенил)-2-(4-цианфениламино)этил]амида пропан-1-сульфоновой кислоты. 1H-ЯМР (СDСl3): 7,77 (s, 1H), 7,3-7,5 (m, 7H), 7,12 (s, 1H), 6,41 (d, 2H), 6,0 (d, 1H), 5,3 (m, 1H), 5,17 (dd, A-B, 2H), 4,75 (t, 1H), 3,85 (s, 3H), 3,65 (m, 1H), 3,04 (m, 2H), 1,83 (m, 2H), 1,05 (t, 3Н). [2-(4-Бензилокси-5-метокси-2-нитрофенил)-2-(4-цианфениламино)этил]амид пропан-1-сульфоновой кислоты (0,250 г) растворяли в этаноле (10 мл) и добавляли к Pt/C (5%). Реакционную смесь помещали в атмосферу водорода и интенсивно перемешивали в течение 3 ч. Катализатор отфильтровывали и продукт хроматографировали (гексан:этилацетат, 1:2), получая 133 мг [2-(2-амино-4-бензилокси-5-метоксифенил)-2-(4-цианфениламино)этил] амида пропан-1-сульфоновой кислоты. 1H-ЯМР (CDCl3): 7,3-7,45 (m, 7Н). 6,71 (s, 1H), 6,52 (d, 2H), 6,3 (s, 1H), 5,08 (s, 2H), 5,0 (t, 1H). 4,42 (q, 1H), 3,75 (s, 3Н), 3,70 (bs, 2H), 3.45 (t, 2H), 2,97 (m. 2H), 1,80 (m, 2H), 1,03 (t, 3H). [2-(2-Амино-4-бензилокси-5-метоксифенил)-2-(4-цианфениламино)этил]амид пропан-1-сульфоновой кислоты (133 мг, 0,27 ммоль) растворяли в дихлорметане и добавляли триэтиламин (0,05 мл, 0,35 ммоль). Реакционную смесь охлаждали и по каплям добавляли метансульфонилхлорид (0,023 мл, 0,3 ммоль). Реакционную смесь перемешивали в течение 2 ч и продукт очищали экспресс-хроматографией (гексан:этилацетат, 1:1). Затем продукт растворяли в этаноле и добавляли гидрохлорид гидроксиламина (35 мг, 0,5 ммоль). Добавляли этоксид натрия (48 мг, 0,7 ммоль) и реакционную смесь нагревали в течение 48 ч. Удаляли этанол и добавляли воду (4 мл). Твердый продукт отфильтровывали и промывали водой. Затем неочищенный продукт растворяли в 4 мл метанола, содержащего 0,5 мл уксусной кислоты. Добавляли никель Ренея (приблизительно 50 мг в виде суспензии в гидроксиде натрия, фирма Aldrich) и реакционную смесь помещали в атмосферу водорода. Реакционную смесь интенсивно перемешивали в течение 3 ч и отфильтровывали катализатор. Неочищенный продукт подвергали препаративной хроматографии с обращенной фазой, получая конечный продукт, т.е. 4-[1-(4-бензилокси-2-метансульфониламино-5-метоксифенил)-2-(пропан-1-сульфониламино)этиламино]бензамидин (12 мг): МС (М+Н)=590. Примеры 6а-6ж С помощью процесса, аналогичного описанному в примере 5, получали следующие соединения: а) 4-[2-бензолсульфониламино-1-(2-бензолсульфониламино-4-бензилокси-5-метоксифенил)этиламино]бензамидин. Процесс соответствовал процессу, описанному выше, за исключением того, что вместо пропансульфонилхлорида и метансульфонилхлорида использовали фенилсульфонилхлорид. МС:(М+H)=686. б) 4-[2-Бензолсульфониламино-1-(2-бензолсульфониламино-4-бензилокси-5-этоксифенил)этиламино]бензамидин. Процесс соответствовал процессу, описанному выше, за исключением того, что в качестве исходных продуктов использовали 3-этокси, 4-бензилокси, 6-нитробензальдегид. МС:(М+Н)=700. в) 4-[1-(4-Бензилокси-5-этокси-2-метансульфониламинофенил)-2-(пропан-1-сульфониламино)этиламино]бензамидин. Процесс соответствовал процессу, описанному выше, за исключением того, что в качестве исходных продуктов использовали 3-этокси, 4-бензилокси, 6-нитробензальдегид. МС:(М+Н)=604. г) 4-[1-(4,5-Диэтокси-2-метансульфониламинофенил)-2-(пропан-1-сульфониламино)этиламино]бензамидин. Процесс соответствовал процессу, описанному выше, за исключением того, что в качестве исходных продуктов использовали 3,4-диэтокси, 6-нитробензальдегид. МС:(М+Н)=542. д) Этиловый эфир [5-бензилокси-2-[1-(4-карбамимидоилфениламино)-2-(пропан-1-сульфониламино) этил]-4-метоксифенилсульфамоил} уксусной кислоты. Процесс соответствовал процессу, описанному выше, за исключением того, что вместо метансульфонилхлорида использовали этиловый эфир хлорсульфонилуксусной кислоты. МС: (М+Н)=676. е) {5-Бензилокси-2-[1-(4-карбамимидоилфениламино)-2-(пропан-1-сульфониламино)этил]-4-метоксифенилсульфамоил}уксусная кислота. Этиловый эфир [5-бензилокси-2-[1-(4-карбамимидоилфениламино)-2-(пропан-1-сульфониламино)этил]-4-метоксифенилсульфамоил} уксусной кислоты (10 мг) растворяли в воде (2 мл) и добавляли тетрагидрофуран (2 мл) и LiOH (3 мг). Смесь оставляли перемешиваться в течение ночи. Продукт очищали с помощью препаративной ЖХВД с обращенной фазой. Получали 3 мг продукта. МС (М+Н)=648. ж) 4-[1-(3,4-Диметокси-2-метансульфониламинофенил)-2-(пропан-1-сульфониламино)этиламино]бензамидин. Это соединение получали согласно процессу, аналогичному процессу, описанному выше, за исключением того, что вместо 4-бензилокси-5-метокси-2-нитробензальдегида использовали 2-бром-3,4-диметоксибензальдегид. МС:(М+Н)=499. Примеры 7а-7ж Соединения 7а-7ж в целом получают следующим образом. Соединение Е (20 мг, 0,03 ммоль) растворяют в ацетонитриле (2 мл), содержащем триэтиламин (17 мкл, 0,12 ммоль) и воду (0,3 мл). К этой смеси добавляют соответствующие ацилхлорид, алкилхлорформиат или изоцианат (0,03 ммоль) и реакционную смесь перемешивают в течение 4 ч. Растворитель удаляют в вакууме и соединения очищают с помощью препаративной ЖХВД с обращенной фазой (смесь ацетонитрил/вода, содержащая 0,1%-ную трифторуксусную кислоту), получая после лиофилизации конечный продукт. 7а) N-[2-(4-Бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]-2,2,2-трифторацетамид, МС (М+Н)=487. 7б) N-[2-(4-Бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]ацетамид, МС (М+Н)=433. 7в) N-[2-(4-Бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]бутирамид, МС (М+Н)=461. 7г) N-[2-(4-Бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]-2-хлорацетамид, МС (М+Н)=467. 7д) Метиловый эфир [2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]карбоновой кислоты, МС (М+Н)=449. 7е) Изобутиловый эфир [2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]карбоновой кислоты, МС (М+Н)=491. 7ж) 2,2,2-Трихлорэтиловый эфир [2-(4-бензилокси-3-метоксифенил)-2-(4-карбамимидоилфениламино)этил]карбоновой кислоты, МС (М+Н)=565. Пример 8 Метиловый эфир представленной выше кислоты (920 мг, 2,85 ммоль) суспендировали в смеси ТГФ/вода (3/1) (40 мл) и охлаждали до 0°С. Раствор обрабатывали 1 н. LlОH (7,1 мл, 7,1 ммоль) и перемешивали в течение ночи. Реакционную смесь подкисляли трифторуксусной кислотой до достижения значения рН 4,0. Растворитель удаляли в вакууме и неочищенный продукт очищали экспресс-хроматографией (этилацетат, содержащий 0,5%-ную уксусную кислоту), получая 1 г карбоновой кислоты. Карбонилдиимидазол (131 мг, 0,8 ммоль) растворяли в безводном ТГФ, (1,6 мл) и по каплям добавляли полученную выше карбоновую кислоту (251 мг, 0,8 ммоль) в виде раствора в ТГФ (1,6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, выдерживали при температуре дефлегмации в течение 30 мин и затем снова охлаждали до комнатной температуры. Добавляли н-пропилсульфонамид (100 мг) и перемешивали в течение 10 мин. Добавляли ДБУ (123 мг) в виде раствора в ТГФ (1,6 мл). Реакционную смесь подкисляли и экстрагировали этилацетатом. Растворитель удаляли и неочищенный продукт очищали экспресс-хроматографией (SiO2, этилацетат), получая 195 мг представленного выше ацилсульфонамида. Полученный выше нитрил (90 мг, 0,2 ммоль) растворяли в этаноле (2,5 мл). Добавляли диизопропилэтиламин (202 мг, 1,56 ммоль), а затем гидрохлорид гидроксиламина (83 мг, 1,2 ммоль). Реакционную смесь нагревали до 70°С в течение 21 ч. Реакционную смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в смеси 30% ацетонитрила/вода (4 мл) и очищали с помощью препаративной хроматографии с обращенной фазой (градиент воды/ацетонитрила с добавлением 0,1% ТФК), получая 14 мг представленного выше гидроксиамидина. Гидроксиамидин растворяли в этаноле (2 мл) и уксусной кислоте (8 капель). Добавляли никель Ренея (приблизительно 100 мг) и реакционную смесь интенсивно перемешивали в атмосфере водорода в течение 2 ч 45 мин. Продукт фильтровали через целит и целит промывали сначала смесью 30% ацетонитрила/вода, содержащей 0,1% ТФК, а затем ацетонитрилом. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью препаративной хроматографии с обращенной фазой (градиент воды/ацетонитрила с добавлением 0,1% ТФК), получая требуемый продукт (6 мг). М+Н=435. Пример 9. Синтез энантиомерно чистых 6-алкилсульфонаминосульфонамидов К диметилформамиду (600 мл) добавляли 3-этокси-4-гидроксибензальдегид (40 г), а затем карбонат калия (40 г, 1,2 экв.). Добавляли этилйодид (28,87 мл, 1,5 экв.) и раствор нагревали до 60°С в течение 6 ч. Раствор охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении. Раствор разбавляли этилацетатом (500 мл), промывали водой, соляным раствором, сушили над сульфатом магния и упаривали, получая неочищенный продукт 3,4-этоксибензальдегид (49 г, 104%). 3,4-Этоксибензальдегид (45 г) растворяли в этаноле (300 мл) и раствор охлаждали до 0°С. В отдельную колбу, содержащую этанол (300 мл), добавляли гидроксид калия (19,5 г, 1,5 экв.), а затем нитрометан (26 г, 1,5 экв.) и раствор перемешивали при комнатной температуре в течение 10 мин и охлаждали до 0°С. Раствор добавляли к 3,4-этоксибензальдегиду и перемешивали в течение 20 мин и сливали при 0°С на концентрированную соляную кислоту (200 мл). Этанол удаляли при пониженном давлении, раствор разбавляли водой (300 мл) и реакционную смесь экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и удаляли растворитель, получая неочищенный продукт. Неочищенный продукт очищали путем перекристаллизации из этилацетата (47 г, 87%). МС (М+Н)=238. К тетрагидрофурану (350 мл) добавляли 1-нитро-2-(3,4-диэтоксифенил)этилен (16,64 г) и 4-аминобензнитрил (9,12 г, 1,1 экв.) и охлаждали до 0°С. Медленно добавляли диизопропиламид лития (47,8 мл, 1,02 экв.) до получения устойчивой пурпурной окраски. Добавляли в виде одной порции цинк (50 г), а затем уксусную кислоту (35 мл). Раствор нагревали до комнатной температуры и перемешивали в течение 2 ч, после чего добавляли уксусную кислоту (35 мл) и цинк (10 г). Спустя еще 2 ч добавляли уксусную кислоту (25 мл) и реакционную смесь перемешивали в течение 1 ч. Добавляли концентрированную соляную кислоту (15 мл) и раствор перемешивали еще в течение 1 ч. Раствор фильтровали через подушку из целита и добавляли воду (250 мл). Раствор концентрировали при пониженном давлении до объема 300 мл и добавляли к лимонной кислоте (0,5 М, 500 мл) и смеси этилацетат/гексан (500 мл). Слой лимонной кислоты собирали и добавляли гидроксид аммония до тех пор, пока раствор не становился щелочным. Этот раствор экстрагировали этилацетатом (3×300 мл) и объединенные органические слои сушили над сульфатом магния и упаривали при пониженном давлении, получая неочищенный продукт. Неочищенный продукт разбавляли дихлорметаном (350 мл) и охлаждали до 0°С. Добавляли фосген (40,88 мл в виде 20%-ного раствора в толуоле, 1,1 экв.), а затем основание Хюнига (24,46 мл, 2 экв.). Раствор перемешивали в течение 10 мин и добавляли воду (200 мл). Дихлорметан собирали, сушили над сульфатом магния и очищали с помощью экспресс-хроматографии на силикагеле (смесь 80% этилацетата/20% гексана), получая продукт в виде твердого вещества белого цвета (9,76 г, 40%). МС (М+Н)=352. К тетрагидрофурану (200 мл) добавляли 4-[5-(3,4-диэтоксифенил)-2-оксоимидазолидин-1-илбензнитрил (2,10 г) и охлаждали до -78°С. По каплям добавляли н-бутиллитий (3,74 мл, 1 экв.) и раствор перемешивали в течение 10 мин. Добавляли в виде одной порции твердый (S)-(+)-2-(6-метокси-2-нафтил)пропионилхлорид (1,49 г, 1 экв.) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь нагревали до комнатной температуры и упаривали при пониженном давлении до объема 50 мл. Раствор разбавляли этилацетатом (300 мл) и промывали лимонной кислотой (0,5 М), водой, соляным раствором и сушили над сульфатом магния. Раствор упаривали при пониженном давлении и очищали с помощью экспресс-хроматографии на силикагеле (смесь 50% этилацетата/50% гексана), получая продукт 4-{5-(3,4-диэтоксифенил)-3-[2-(6-метоксинафталин-2-ил)пропионил]-2-оксоимидазолидин-1-ил}бензнитрил в виде одного диастереоизомера (1,20 г, 71%). Этот продукт добавляли к метанолу (200 мл), после чего добавляли гидроксид лития (1 мл в виде 10%-ного водного раствора) и перемешивали в течение 15 мин. Добавляли уксусную кислоту (10 капель), метанол удаляли при пониженном давлении и продукт очищали с помощью экспресс-хроматографии на силикагеле (смесь 80% этилацетата/20% гексана), получая продукт (0,71 г, 95%) в виде твердого вещества белого цвета. [ К тетрагидрофурану (20 мл) добавляли (R)-(-)-4-[5-(3,4-диэтоксифенил)-2-оксоимидазолидин-1-илбензнитрил (0,710 г) и охлаждали до -78°С. По каплям добавляли н-бутиллитий (1,27 мл в виде 1,6 М раствора в гексане, 1 экв.) и раствор перемешивали в течение 10 мин. Добавляли 1-пропилсульфонил-хлорид (0,275 мл, 1,2 экв.) и раствор перемешивали в течение 15 мин и нагревали до комнатной температуры. Добавляли уксусную кислоту (10 капель), растворитель удаляли при пониженном давлении и продукт очищали с помощью, экспресс-хроматографии на силикагеле (смесь 50% этилацетата/50% гексана), получая продукт (0,740 г, 80%). Этот продукт (0,300 г) разбавляли дихлорэтаном (10 мл) и охлаждали до 0°С. По каплям добавляли азотную кислоту (0,137 мл, 5 экв.) и раствор перемешивали в течение 1 ч. Раствор разбавляли дихлорэтаном (100 мл) и промывали водой, насыщенным бикарбонатом натрия, сушили над сульфатом магния и растворитель выпаривали при пониженном давлении. Этот продукт разбавляли метанолом (50 мл) и добавляли уксусную кислоту (1 мл) и платину (0,050 г, 5%-ная на угле). Раствор гидрировали в течение 1 ч, фильтровали через подушку из целита и растворитель выпаривали при пониженном давлении. Этот продукт растворяли в дихлорметане (5 мл) и добавляли основание Хюнига (0,177 мл, 1,5 экв.) и этиловый эфир хлорсульфонилуксусной кислоты (0,189 г, 1,5 экв.) и раствор перемешивали в течение 2 ч. Раствор очищали с помощью прямой экспресс-хроматографии на силикагеле (смесь 50% этилацетата/50% гексана), получая продукт (0,160 г) этиловый эфир (R)-[2-[3-(4-цианфенил)-2-оксо-1-(пропан-1-сульфонил)имидазолидин-4-ил]-4,5-диэтоксифенилсульфамоил} уксусной кислоты. МС (М+Н)=624. Этиловый эфир (R)-{2-[3-(4-цианфенил)-2-оксо-1-(пропан-1-сульфонил)имидазолидин-4-ил]-4,5-диэтоксифенилсульфамоил} уксусной кислоты (0,160 г) разбавляли этанолом (5 мл), а затем гидроксидом лития (1 мл в виде 10%-ного водного раствора, 10 экв.) и раствор перемешивали в течение 48 ч. Раствор очищали с помощью прямой экспресс-хроматографии на силикагеле (смесь 20% метанола/80% дихлорметана), получая продукт. Этот продукт разбавляли этанолом (1 мл) и добавляли гидроксиламин (0,035 г, 10 экв.) и основание Хюнига (0,088 мл, 10 экв.) и раствор нагревали до 60°С в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение 12 ч. Добавляли этанол (3 мл), уксусную кислоту (0,5 мл) и никель Ренея (0,025 г) и раствор гидрировали в течение 1 ч. Реакционную смесь фильтровали через подушку из целита и растворитель удаляли при пониженном давлении. Продукт очищали с помощью препаративной хроматографии с обращенной фазой, получая [2-[1-(4-карбамимидоилфениламино)-2-(пропан-1-сульфониламино)этил] -4,5-диэтоксифенилсульфамоил] уксусную кислоту (52 мг). [ Пример 10. 6-Алкоксизамещенные сульфонамиды Аминоацетонитрил (14,25 г, 92,5 ммоль) растворяли в 1,2-дихлорэтане (150 мл) и реакционную смесь помещали в атмосферу N2. Добавляли триэтиламин (32,76 г, 324 ммоль) и реакционную смесь охлаждали до 0°С. По каплям добавляли раствор пропан -1-сульфонилхлорида (13,19 г, 92,5 ммоль) в 1,2-дихлорэтане (20 мл), реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 ч. Анализ с помощью тонкослойной хроматографии выявил наличие нового продукта с более высоким Rf. Растворитель удаляли в вакууме и неочищенный продукт подвергали экспресс-хроматографии (метиленхлорид:этилацетат, 9:1), получая цианметиламид пропан-1-сульфоновой кислоты. 1H-ЯМР (СDСl3): 5,40 (s, 1Н), 4,11 (s, 2Н), 3,15 (m, 2H), 1,89 (q, 2H), 1,10 (t, 3Н). 3-Этокси-4-гидроксибензальдегид (фирма Aldrich, 100 г, 0,602 моль) растворяли в N,N-диметилформамиде (1 л). Реакционную смесь помещали в атмосферу N2. Добавляли твердый карбонат калия (103 г, 0,662 моль) и реакционную смесь перемешивали в течение 10 мин. Добавляли йодэтан (175 г, 1,26 моль) и реакционную смесь перемешивали в течение 16 ч при комнатной температуре. За ходом реакции наблюдали с помощью токослойной хроматографии, которая выявила полное поглощение фенола и образование нового продукта. Реакционную смесь фильтровали для удаления карбоната калия и растворитель удаляли в вакууме. Остаток растворяли в метиленхлориде и фильтровали. Добавляли силикагель (200 г) и дихлорметан удаляли в вакууме. Неочищенный продукт, адсорбированный на силикагеле, подвергали экспресс-хроматографии (гексан, 100%, 2 л, затем этилацетат:гексан, 1:3, 2 л, затем этилацетат:гексан, 1:1), получая 109,36 г 3,4-диэтоксибензальдегида. 1H-ЯМР (CDCl3): 9,83 (s, 1Н), 7,41 (m, 2Н), 6,96 (d, 1H), 4,17 (m, 4H), 1,50 (t, 3H), 1,47 (t, 3H). 3,4-Диэтоксибензальдегид (31,45 г, 0,162 моль) растворяли в метиленхлориде (500 мл). Добавляли 3-хлорпербензойную кислоту и реакционную смесь нагревали до температуры дефлегмации в течение 4 ч. Анализ с помощью тонкослойной хроматографии выявил поглощение альдегида. Реакционную смесь охлаждали до комнатной температуры, разбавляли метиленхлоридом и реакцию прекращали, добавляя насыщенный водный раствор карбоната калия. Разделяли слои и водный раствор экстрагировали метиленхлоридом. Метиленхлоридные экстракты объединяли, промывали водой, соляным раствором и сушили над безводным сульфатом натрия, фильтровали и раствритель удаляли в вакууме, получая неочищенное масло. Неочищенное масло растворяли в метаноле (300 мл) и добавляли 10%-ный водный раствор КОН (60 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Анализ с помощью тонкослойной хроматографии выявил поглощение образовавшегося промежуточного продукта. Метанол удаляли в вакууме и водный раствор подкисляли с помощью 1,2н. НСl. Раствор экстрагировали этилацетатом. Этилацетатные экстракты промывали водой и соляным раствором, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Остаток подвергали экспресс-хроматографии (гексан:этилацетат, 3:1), получая 22,06 г 3,4-диэтоксифенола. 1H-ЯМР (СDСl3): 6,56 (d, 1H). 6,25 (d, 1H), 6,10 (dd, 1H), 3,82 (m, 4H), 1,22 (t, 3H), 1,20 (t, 3H). Нитробензол (100 г) охлаждали до 0°С и насыщали газообразным НСl. К этому раствору добавляли 3,4-диэтоксифенол (11,64 г, 64 ммоль), цианметиламид пропан-1-сульфоновой кислоты (10,36 г, 64 ммоль) и хлорид цинка (17,45 г, 128 ммоль). Реакционную смесь перемешивали в течение 1 ч при 0°С и затем давали нагреться до комнатной температуры. Перемешивание продолжали в течение 2 ч. Анализ с помощью тонкослойной хроматографии выявил поглощение 3,4-диэтоксифенола. Осторожно добавляли воду (100 мл) и реакционную смесь нагревали в течение 1 ч до 100°С. Реакционную смесь охлаждали и экстрагировали метиленхлоридом. Экстракты промывали водой, соляным раствором, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Остаток подвергали экспресс-хроматографии (2,5-5%-ный этилацетат в метиленхлориде). Очищенный продукт растирали с простым эфиром, фильтровали и сушили на воздухе, получая 1,73 г [2-(4,5-диэтокси-2-гидроксифенил)-2-оксоэтил]амида пропан-1-сульфоновой кислоты. 1H-ЯМР (СDСl3): 11,86 (s, 1H), 6,93 (s, 1H), 6,46 (s, 1H), 5,26 (t, 1H)’, 4,55 (d, 2H), 4,14 (q, 2Н), 4,03 (q, 2H), 3,05 (m, 2H), 1,90 (m, 2H), 1,45 (m, 6Н), 1,07 (t, 3H). [2-(4,5-Диэтокси-2-гидроксифенил)-2-оксоэтил] амид пропан-1-сульфоновой кислоты (200 мг, 0,579 ммоль) растворяли в тетрагидрофуране (5 мл), помещали в атмосферу N2 и охлаждали до 0°С. По каплям добавляли комплекс боран: ТГФ (1,74 мл, 1,0 М в ТГФ, 1,74 ммоль). Реакционную смесь перемешивали в течение 15 мин и давали нагреться в течение 2 ч до комнатной температуры. Анализ с помощью тонкослойной хроматографии выявил поглощение исходного продукта и образование нового продукта с более низким Rf. Реакцию прекращали, добавляя 1,2н. водную НСl (2,5 мл), смесь разбавляли водой и экстрагировали этилацетатом. Экстракты промывали водой и соляным раствором, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме, получая 190 мг неочищенного масла. Неочищенное масло растворяли в ацетонитриле (5 мл). Добавляли 4-аминобензнитрил (194 мг, 1,64 ммоль) и перхлорат лития (233 мг, 2,19 ммоль). Реакционную смесь нагревали в течение 3,5 ч до 90°С и затем перемешивали в течение 18 ч при комнатной температуре. Анализ с помощью тонкослойной хроматографии выявил наличие нового продукта с более высоким значением Rf по сравнению с продуктом, полученным в результате восстановления. Реакцию прекращали, добавляя воду, и смесь экстрагировали этилацетатом. Этилацетатные экстракты промывали водой и соляным раствором, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Остаток подвергали экспресс-хроматографии (этилацетат:метиленхлорид, 1:9), получая 134 мг [2-(4-цианфениламино)-2-(4,5-диэтокси-2-гидроксифенил)этил]амида пропан-1-сульфоновой кислоты. 1H-ЯМР (СDСl3): 7,34 (d, 2Н), 6,71 (s, 1H), 6,61 (d, 2H), 6,41 (s, 1H), 5,58 (d, 1H), 4,79 (m, 1H), 4,57 (m, 1H), 3,99 (m, 4H), 3,48 (t, 2H), 3,00 (m, 2H), 1,80 (q, 2H), 1,41 (t, 3Н), 1,34 (t, 3Н), 1,03 (t, 3Н). [2-(4-Цианфениламино)-2-(4,5-диэтокси-2-гидроксифенил)этил]амид пропан-1-сульфоновой кислоты (100 мг, 0,233 ммоль) растворяли в диметилформамиде (1,5 мл) и обрабатывали твердым бикарбонатом калия (22 мг, 0,233 ммоль), а затем этилбромацетатом (0,37 мл, 0,233 ммоль). Реакционную смесь перемешивали в атмосфере N2 в течение 67 ч. Анализ с помощью тонкослойной хроматографии выявил наличие нового продукта с более высоким значением Rf. Реакцию прекращали, добавляя воду, и смесь экстрагировали этилацетатом. Этилацетатные экстракты промывали водой и соляным раствором, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Остаток подвергали экспресс-хроматографии (этилацетат:метиленхлорид, 1:9), получая 78 мг этилового эфира {2-[4-цианфениламино)-2-(пропан-1-сульфониламино)этил]-4,5-диэтоксифенок-си} уксусной кислоты. 1H-ЯМР (CDCl3): 7,35 (d, 2Н), 6,81 (s, 1H), 6,57 (d, 2H), 6,43 (s, 1H), 4,81 (t, 1H), 4,68 (ABq, 2H), 4,56 (t, 1H), 4,26 (q, 2H), 4,05 (q, 2H), 3,95 (m, 2H), 3,54 (t, 2H), 2,98 (m, 2H), 1,80 (m, 2H), 1,42 (t, 3H), 1,32 (t, 6H), 1,02 (t, 3Н). МС (М+Н)=534. Этиловый эфир [2-[4-цианфениламино)-2-(пропан-1-сульфониламино)этил]-4,5-диэтоксифенокси} уксусной кислоты (76 мг, 0,142 ммоль) растворяли в тетрагидрофуране (3 мл). Добавляли воду (1 мл) и реакционную смесь охлаждали до 0°С. К реакционной смеси добавляли водный LiOH (1,0 М, 0,42 мл, 0,42 ммоль). После перемешивания в течение 5 мин реакционной смеси давали нагреться до комнатной температуры и ее перемешивали в течение 16 ч при комнатной температуре. Поглощение эфира выявляли с помощью аналитической жидкостной хроматографии высокого давления. Добавляли дополнительную порцию LiOH (1,0 М, 0,14 мл). Реакционную смесь перемешивали при комнатной температуре еще в течение 8 ч. Поглощение эфира было неполным, поэтому добавляли свежеприготовленный LiOH (1,0 М, 0,14 мл) и реакционную смесь перемешивали в течение 24 ч. Добавляли еще одну порцию LiOH (1,0 М, 0,28 мл) и через 67 ч анализ с помощью аналитической ЖХВР-ОФ выявил поглощение эфира (общее количество 1,0 М LiOH составляло 0,98 мл, 6,9 экв.). Реакционную смесь подкисляли с помощью уксусной кислоты и ТГФ выпаривали в потоке N2. Раствор делали прозрачным путем добавления ацетонитрила и затем очищали с помощью ЖХФД с обращенной фазой (градиент ацетонитрила/воды с добавлением 0,1% трифторуксусной кислоты), получая после лиофилизации 44 мг кислого трифторацетата {2-[4-цианфениламино)-2-(пропан-1-сульфониламино)этил]-4,5-диэтоксифенокси}уксусной кислоты. 1H-ЯМР (СD3ОD): 7,15 (d, 2Н), 6,70 (s, 1H), 6,48 (d, 2H), 6,45 (s, 1H), 4,76 (t, 1H), 4,60 (s, 2H), 3,85 (q, 2H), 3,73 (m, 2H), 3,43 (dd, 1H), 3,26 (dd, 1H частично закрытый пиком растворителя СН3ОН), 2,76 (m, 2H), 1,52 (m, 2H), 1,18 (t, 3Н), 1,07 (t, 3Н), 0,77 (t, 3Н). МС (М+Н)=506. Кислый трифторацетат {2-[4-цианфениламино)-2-(пропан-1-сульфониламино) этил]-4,5-диэтоксифенокси] уксусной кислоты (44 мг, 0,071 ммоль) растворяли в этаноле (1 мл) и обрабатывали диизопропилэтиламином (0,89 мл, 0,515 ммоль) и твердым гидрохлоридом гидроксиламина (25 мг, 0,355 ммоль). Реакционную смесь помещали в атмосферу N2 и нагревали в течение 3 ч до 60°С. Через 3 ч анализ с помощью жидкостной хроматографии высокого давления выявил, что реакция еще не завершилась. Реакционную смесь охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение 16 ч и за процессом превращения наблюдали с помощью ЖХВД. Реакционную смесь выдерживали в течение 8 ч при 60°С, перемешивали при комнатной температуре в течение 16 ч, выдерживали в течение 8 ч при 70°С и перемешивали в течение 16 ч при комнатной температуре. Анализ с помощью ЖХВД выявил поглощение исходного продукта. Реакционную смесь подкисляли с помощью уксусной кислоты, добавляли никель Ренея и реакционную смесь гидрировали в течение 3 ч в сосуде с водородом при давлении приблизительно 1 атм. Анализ с помощью аналитической ЖХВД, выявил поглощение промежуточного продукта, представляющего собой гидроксиамидин. Отфильтровывали катализатор никель Ренея и растворитель удаляли в вакууме. Остаток очищали с помощью препаративной ЖХВД с обращенной фазой (градиент ацетонитрила/воды с добавлением 0,1% трифторуксусной кислоты), получая после лиофилизации 5,6 мг {2-[1-(карбамимидоилфениламино)-2-(пропан-1-сульфониламино)этил]-4,5-диэтоксифенокси}уксусной кислоты в форме бис-трифторацетата. МС (М+Н):523. Пример 11. Синтез 6-алкилсульфонилалкиламинозамещенного ацилсульфонамида 4,5-Диэтокси-2-нитробензальдегид (55,5 г, 206 ммоль) и 4-аминобензнитрил (23 г, 195 ммоль) растворяли в метаноле (700 мл) и перемешивали в течение 2 ч при 60°С. Реакционной смеси давали охладиться до 0°С и добавляли тозилметилизонитрил (45 г, 230 ммоль). По каплям в течение 10 мин добавляли трифторэфират бора (78 мл, 620 ммоль). Реакционную смесь перемешивали в течение 30 мин при 0°С, давали охладиться до комнатной температуры и затем перемешивали в течение 1,5 ч при температуре окружающей среды. Добавляли воду (18 мл) и смесь перемешивали при комнатной температуре в течение ночи. На следующий день метанол удаляли в вакууме и остаток растворяли в этилацетате. Органический слой промывали водой и затем сушили над безводным сульфатом натрия. Сульфат натрия отфильтровывали и этилацетат удаляли в вакууме. Неочищенный продукт подвергали экспресс-хроматографии (гексан:этилацетат, 2:1, затем 1:1), получая 46 г требуемого продукта, т.е. метилового эфира (4-этокси-5-этокси-2-нитрофенил)-(4-цианфениламино) уксусной кислоты. Метиловый эфир (4-этокси-5-этокси-2-нитрофенил)-(4-цианфениламино)уксусной кислоты (11 г, 27,5 ммоль) растворяли в этилацетате (300 мл) и в атмосфере азота добавляли в колбу, содержащую 5%-ный Pt/C (3 г). Азот удаляли и заменяли водородом (из баллона) и реакционную смесь интенсивно перемешивали в течение 6 ч. Катализатор отфильтровывали и растворитель удаляли в вакууме. Остаток растворяли в дихлорметане (приблизительно 300 мл) и добавляли пиридин (5,6 мл, 70 ммоль). Реакционную смесь охлаждали до 0°С и по каплям добавляли метансульфонилхлорид (2,5 мл, 33 ммоль). Реакционную смесь перемешивали в течение ночи. Раствор промывали водой и растворитель удаляли в вакууме. Неочищенный продукт хроматографировали на силикагеле с помощью экспресс-хроматографии (гексан:этилацетат, 1:1), получая 5 г требуемого продукта, т.е. метиловый эфир (4-цианфениламино)-[4,5-диэтокси-2-(метансульфониламино)фенил]уксусной кислоты. Продукт, представляющий собой метиловый эфир (4-цианфениламино)-[4,5-диэтокси-2-(метансульфониламино)фенил]уксусной кислоты (5 г, 10,7 ммоль), растворяли в безводном ДМФ (100 мл) и добавляли карбонат цезия (7,25 г, 22 ммоль) и йодметан (1 мл, 16 ммоль). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре и растворитель удаляли в вакууме. Остаток растворяли в этилацетате, подкисляли с помощью 1н. соляной кислоты и органический слой однократно промывали водой. Продукт сушили над безводным сульфатом натрия и растворитель удаляли в вакууме. Остаток подвергали экспресс-хроматографии (гексан:этилацетат, 1:1), получая 3,6 г требуемого продукта, т.е. метилового эфира (4-цианфениламино)-[4,5-диэтокси-2-(метансульфонилметиламино)фенил]уксусной кислоты. Полученный выше метиловый эфир (4-цианфениламино)-[4,5-диэтокси-2-(метансульфонилметиламино)фенил]уксусной кислоты (2,6 г, 5 ммоль) растворяли в метаноле. Добавляли 1н. LiOH (25 мл) и реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Метанол удаляли в вакууме и реакционную смесь подкисляли 1н. соляной кислотой. Продукт экстрагировали этилацетатом и промывали водой. С помощью экспресс-хроматографии (этилацетат с добавлением 5% уксусной кислоты) получали 1,9 г требуемой кислоты, т.е. (4-цианфениламино)-[4,5-диэтокси-2-(метансульфо-нилметиламино) фенил] уксусной кислоты. Полученную выше (4-цианфениламино)-[4,5-диэтокси-2-(метансульфонилметиламино) фенил] уксусную кислоту (350 мг, 0,75 ммоль) объединяли с карбонилдиимидазолом (610 мг, 3,77 ммоль) в безводном ТГФ (6 мл). Реакционную смесь выдерживали при 60°С в течение 1 ч и затем охлаждали до комнатной температуры. К этому раствору добавляли фенилсульфонамид (650 мг, 4,14 ммоль) и ДБУ (5 ммоль) в виде раствора в 5 мл ТГФ. Реакционную смесь перемешивали в течение 3 ч и ТГФ удаляли в вакууме. Остаток растворяли в этилацетате и подкисляли 1н. соляной кислотой. Органический слой отделяли, промывали водой и сушили над безводным сульфатом натрия. Неочищенный продукт очищали экспресс-хроматографией (гексан:этилацетат; 1:1, а затем этилацетатом с добавлением 5% уксусной кислоты), получая 302 мг требуемого продукта, т.е. N-{(4-цианфениламино)-[4,5-диэтокси-2-(метансульфонилметиламино)фенил]ацетил}бензолсульфонамида. Полученный выше N-{(4-цианфениламино)-[4,5-диэтокси-2-(метансульфонилметиламино)фенил]ацетил}бензолсульфонамид (126 мг, 0,21 ммоль) растворяли в этаноле (1,8 мл) и нагревали до 60°С. Добавляли диизопропилэтиламин (ДИПЭА; 260 мкл, 1,5 ммоль), а затем гидрохлорид гидроксиламина (74 мг, 1,04 ммоль). Реакционную смесь перемешивали при 60°С в атмосфере азота в течение 6 ч. Затем реакционной смеси давали охладиться до комнатной температуры. Раствор разбавляли метанолом (5 мл) и уксусной кислотой (2 мл) и добавляли никель Ренея 2800 (примерно 50 мг) в виде суспензии. Затем реакционную смесь интенсивно перемешивали в атмосфере водорода в течение 1 ч. Катализатор отфильтровывали и растворитель удаляли. Неочищенные продукты очищали с помощью препаративной ЖХВД с обращенной фазой, используя градиент воды-ацетонитрила (0,1% ТФК), получая 40 мг требуемого 4-{2-бензолсульфониламино-1-[4,5-диэтокси-2-(метансульфонилметиламино)фенил]-2-оксоэтиламино}бензамидина в виде соли трифторуксусной кислоты. МС: (М+Н)=604. Пример 12 4-Изопропокси-5-этоксибензальдегид (10,6 г, 50 ммоль) и 4-аминобензонитрил (5,9 г, 50 ммоль) растворяли в метаноле (150 мл) и перемешивали при 60°С в течение 1,6 ч. Реакционной смеси давали охладиться до 0°С и добавляли тозилметилизонитрил (9,75 г, 50 ммоль). В течение 10 мин по каплям добавляли трифторэфират бора (19 мл, 150 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 мин, давали нагреться до комнатной температуры и затем перемешивали при температуре окружающей среды в течение 1,5 ч. Добавляли воду (4,5 мл) и смесь перемешивали при комнатной температуре в течение 2 дней. Метанол удаляли в. вакууме и остаток растворяли в этилацетате. Органический слой промывали водой и затем сушили над безводным сульфатом натрия. Сульфат натрия отфильтровывали и этилацетат удаляли в вакууме. Неочищенный продукт подвергали экспресс-хроматографии (гексан:этилацетат, 1:1), получая 12,5 г требуемого продукта, т.е. метилового эфира (4-изопропокси-5-этоксифенил)-(4-цианфениламино)уксусной кислоты. Указанный выше продукт, т.е. метиловый эфир (4-изопропокси-5-этоксифенил)-(4-цианфениламино) уксусной кислоты (6 г, 16,3 ммоль) обрабатывали 1н. LiOH (примерно 50 мл) в ТГФ (примерно 150 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч и подкисляли 1н. соляной кислотой. ТГФ удаляли в вакууме и продукт экстрагировали этилацетатом. Неочищенный продукт очищали с помощью хроматографии с обращенной фазой (этилацетат, 3% уксусной кислоты), получая 4,85 г требуемой кислоты, т.е. (4-изопропокси-5-этоксифенил)-(4-цианфениламино) уксусной кислоты. Полученную выше (4-изопропокси-5-этоксифенил)-(4-цианфенилами-но) уксусную кислоту (200 мг, 0,57 ммоль) объединяли с карбонилдиимидазолом (200 мг, 1,2 ммоль) в безводном ТГФ (4 мл). Реакционной смеси давали перемешаться при комнатной температуре в течение 1 ч. В этому раствору добавляли соответствующий алкил или арилсульфонамид (2,2 ммоль) и ДБУ (2,2 ммоль) в виде раствора в 3 мл ТГФ. Реакционную смесь перемешивали в течение ночи и ТГФ удаляли в вакууме. Остаток растворяли в этилацетате и, подкисляли уксусной кислотой. Органический слой отделяли, промывали водой и сушили над безводным сульфатом натрия. Неочищенный продукт очищали экспресс-хроматографией (гексан:этилацетат, 1:2), получая требуемый продукт, т.е. N-{(4-цианфениламино)-[4-изопропоксил-5-этоксифенил]ацетил](алкил или арил)сульфонамид. Полученный выше N-{(4-цианфениламино)-[4-изопропоксил-5-этоксифенил]ацетил} (алкил или арил)сульфонамид (примерно 0,24 ммоль) растворяли в этаноле (1-3 мл) и нагревали до 60°С. Добавляли диизопропилэтиламин (ДИПЭА; 260 мкл, 1,5 ммоль, 6 экв.), а затем гидрохлорид гидроксиламина (84 мг, 1,25 ммоль, 5 экв.). Реакционную смесь перемешивали при 60°С в атмосфере азота в течение примерно 6 ч. Затем реакционной смеси давали охладиться до комнатной температуры. Раствор разбавляли метанолом (5 мл) и уксусной кислотой (2 мл) и добавляли никель Ренея (примерно 50 мг) в виде суспензии. Затем реакционную смесь интенсивно перемешивали в атмосфере водорода в течение 1-6 ч. Катализатор отфильтровывали и растворитель удаляли. Неочищенные продукты очищали с помощью препаративной ЖХВД с обращенной фазой, используя градиент воды-ацетонитрила (0,1% ТФК) или с помощью экспресс-хроматографии (этилацетат:ацетон:вода:уксусная кислота, 6:2:1:1), получая требуемый 4-{2-(алкил или арил)сульфониламино-1-[4-изопропокси,5-этоксифенил]-2-оксоэтиламино]бензамидин в виде соли трифторуксусной кислоты или уксусной кислоты. Используя аналогичный процесс, получали следующие соединения, имеющие различные R1-группы: R1 обозначает этил: 4-{2-этилсульфониламино-1-[4-изопропокси, 5-этоксифенил]-2-оксоэтиламино} бензамидин: МС (М+Н)=463. R1 обозначает пропил: 4-{2-пропилсульфониламино-1-[4-изопропокси,5-этоксифенил]-2-оксоэтиламино}бензамидин: МС (М+Н)=477. R1 обозначает н-бутил: 4-{2-бутилсульфониламино-1-[4-изопропокси,5-этоксифенил]-2-оксоэтиламино}бензамидин: МС (М+Н)=491. R1 обозначает СН2СН2СО2Ме: метиловый эфир 3-[(4-карбамимидоилфениламино)-(3-этокси-4-изопропоксифенил)ацетилсульфамоил]пропоновой кислоты: МС (М+Н)=521. R1 обозначает фенил: 4-{2-бензолсульфониламино-1-[4-изопропокси,5-этоксифенил]-2-оксоэтиламино} бензамидин: МС (М+Н)=511. Пример 13. Ацилсульфонамид с замещением на аминобензамидиновом кольце 2-Гидрокси-4-нитробензонитрил (11,2 г, 68 ммоль) растворяли в ДМФ (200 мл). Добавляли карбонат калия (11 г, 80 ммоль) и бензилбромид (9 мл, 75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. ДМФ удаляли в вакууме и остаток растворяли в этилацетате и воде. Органический слой отделяли, промывали 1н. NaOH, затем водой и сушили над сульфатом натрия. Неочищенный продукт (5 г) растворяли в этилацетате (75 мл) и добавляли в колбу, содержащую 5%-ный Pt/C (500 мг). Реакционную смесь помещали в атмосферу водорода (баллон) и интенсивно перемешивали в течение нескольких часов до завершения реакции (по данным ТСХ). Катализатор отфильтровывали и растворитель удаляли. Продукт очищали экспресс-хроматографией, получая 4,12 г 4-амино-2-бензилоксибензонитрила. 4,5-Диэтоксибензальдегид (3,6 г, 17,8 ммоль) и 4-амино-2-бензилокси-бензонитрил (3,7 г, 17,8 ммоль) растворяли в метаноле (40 мл) и перемешивали в течение 2 ч. Добавляли тозилметилизонитрил (3,48 г, 17,8 ммоль). Реакционную смесь охлаждали до 0°С и по каплям добавляли трифторэфират бора (6,7 мл, 54 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 мин, давали нагреться до комнатной температуры и затем перемешивали при температуре окружающей среды в течение 3,5 ч. Добавляли воду (1,6 мл) и смесь перемешивали при комнатной температуре в течение 2 дней. Метанол удаляли в вакууме и остаток растворяли в этилацетате. Органический слой промывали водой и затем сушили над безводным сульфатом натрия. Сульфат натрия отфильтровывали и этилацетат удаляли в вакууме. Неочищенный продукт подвергали экспресс-хроматографии (гексан: этилацетат, 1:1), получая 4,2 г требуемого продукта, т.е. метилового эфира (3-бензилокси-(4-цианфениламино)-3,4-этоксифенил)уксусной кислоты. Полученный выше продукт обрабатывали LiOH (1,96 г) в воде (50 мл), метаноле (100 мл) и ТГФ (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и подкисляли уксусной кислотой. Растворитель удаляли в вакууме и продукт экстрагировали этилацетатом. Неочищенный продукт очищали с помощью хроматографии с обращенной фазой (этилацетат, 3% уксусной кислоты), получая 5 г требуемой кислоты, т.е. (3-бензилокси-4-цианфениламино)-3,4-этоксифенил)уксусной кислоты. (3-Бензилокси-4-цианфениламино)-3,4-этоксифенил)уксусную кислоту (750 мг, 1,68 ммоль) растворяли в безводном ТГФ (3 мл) и добавляли карбонилдиимидизол (КДИ; 545 мг, 3,36 ммоль). Реакционную смесь нагревали до 40°С в течение 1 ч и затем охлаждали до комнатной температуры. К смеси добавляли раствор бензолсульфонамида (1,05 г, 6,7 ммоль), ДБУ (1,02 г, 6,72 ммоль) в ТГФ (3 мл). Реакционную смесь перемешивали в течение ночи и затем добавляли 1н. соляную кислоту. ТГФ удаляли в вакууме и продукт очищали экспресс-хроматографией (этилацетат, 2% уксусной кислоты), получая требуемый продукт. Этот продукт (215 мг) растворяли в этаноле (5 мл), содержащем уксусную кислоту (1 каплю), и добавляли к 5%-ному Pd/C (100 мг). Реакционную смесь помещали в атмосферу водорода и интенсивно перемешивали до завершения реакции. Катализатор отфильтровывали и затем растворитель удаляли в вакууме. Неочищенный продукт очищали экспресс-хроматографией, получая 150 мг требуемого продукта, т.е. N-[(3-гидрокси-4-цианфениламино)-(3,4-диэтоксифенилацетил]бензолсульфонамида. Полученный выше N-[(3-гидрокси-4-цианфениламино)-(3,4-диэтоксифенилацетил]бензолсульфонауид (75 мг, 0,15 ммоль) растворяли в этаноле (2 мл) и нагревали до 60°С. Добавляли диизопропилэтиламин (ДИПЭА; 185 мкл, 1 ммоль), а затем гидрохлорид гидроксиламина (53 мг, 0,75 ммоль). Реакционную смесь перемешивали при 60°С в атмосфере азота в течение примерно 6 ч. Затем реакционной смеси давали охладиться до комнатной температуры. Раствор разбавляли уксусной кислотой (1 мл) и метанолом (1 мл). Добавляли никель Ренея 2800 (примерно 50 мг) в виде суспензии. Затем реакционную смесь интенсивно перемешивали в атмосфере водорода в течение 0,5 ч. Катализатор отфильтровывали и растворитель удаляли. Неочищенные продукты очищали с помощью препаративной ЖХВД с обращенной фазой, используя градиент воды-ацетонитрила (0,1% ТФК), получая требуемый амидин, т.е. 4-[2-бензолсульфониламино-1-(3,4-диэтоксифенил)-2-оксоэтиламино]-2-гидроксибензамидин. МС (М+Н)=513. С помощью аналогичного процесса из 2-хлор-4-нитробензонитрила и 2-бром-4-нитробензонитрила могут быть получены соответствующие галогенсодержащие соединения. Пример 14. Анализ антагонистической активности в отношении тканевого фактора/фактора VIIa Этот анализ может применяться для определения константы ингибирования (Кi) репрезентативного соединения по изобретению. Материалы:
Процедура анализа: 1. Приготовить раствор, содержащий 2 мкг/мл (90 нМ) тканевого фактора и 1,5 мкг/мл (30 нМ) фактора VIIa в буфере для анализа, включающем CaCl2. 2. Инкубировать в течение 15 мин при комнатной температуре. 3. Добавить 50 мкл образца в каждую лунку. 4. Добавить 50 мкл раствора тканевого фактора/фактора VIIa в каждую лунку. 5. Инкубировать в течение 15 мин при комнатной температуре, осторожно встряхивая. 6. Добавить 50 мкл субстрата в каждую лунку. 7. Встряхивать планшет в течение 20-25 с. 8. Определять абсорбцию при 405 нм каждые 10 с в течение всего 5 мин при комнатной температуре. 9. Рассчитать значение vmax по 10 точкам. Пример 15. Анализы Фактора Ха, тромбина и плазменного калликреина Эти анализы могут применяться для определения константы ингибирования (Кi) репрезентативного соединения по изобретению. Материалы:
Процедура анализа: 1. Добавить 50 мкл образца в каждую лунку. 2. Добавить 50 мкл соответствующим образом разбавленного фактора коагулирования в каждую лунку. 3. Инкубировать в течение 5 мин при комнатной температуре, осторожно встряхивая. 6. Добавить 50 мкл соответствующим образом разбавленного субстрата в каждую лунку. 7. Встряхивать планшет в течение 20-25 с. 8. Определять абсорбцию при 405 нм каждые 10 с в течение всего 5 мин при комнатной температуре. 9. Рассчитать значение Vmax по 10 точкам. Анализ – фермент, субстрат и конечные концентрации
Формула изобретения
1. Соединение, имеющее приведенную ниже структуру: где А и В независимо друг от друга обозначают СН или СR3, Х обозначает С=O или (CR4aR4b)m, где m равно 1 или 2; Y обозначает S(O)n-R1, где n равно 1 или 2, S(O)n-NR2R2, где n равно 1 или 2, S(O)n-OR2, где n равно 1 или 2; N1 и N2 обозначают атомы азота; Q и R1 независимо друг от друга обозначают (1) необязательно замещенный алкил, имеющий от 1 до 10 атомов углерода; (2) необязательно замещенный аралкил, содержащий арильный фрагмент, имеющий от 6 до 10 кольцевых атомов углерода, который связан с алкильным фрагментом, включающим от 1 до 10 атомов углерода; (3) необязательно замещенный аралкенил, содержащий арильный фрагмент, имеющий от 5 до 10 кольцевых атомов, который связан с алкенильным фрагментом, включающим от 1 до 10 атомов углерода; (4) необязательно замещенный арил, имеющий от 6 до 10 кольцевых атомов углерода; (5) необязательно замещенный гетероарил, имеющий от 5 до 10 кольцевых атомов, где кольцевые атомы выбирают из группы, включающей атомы углерода и гетероатомы, причем гетероатомы представляют собой серу; каждый R2 обозначает Н; R3 обозначает Н; R4a, R4b, R5 и R6 независимо друг от друга обозначают Н; R7 и R8 независимо друг от друга обозначают Н или С1-С6алкил; и его кислотно-аддитивные соли и соли присоединения оснований. 2. Соединение по п.1, в котором Q обозначает фенил, необязательно замещенный одним или более заместителями, выбранными из ряда, включающего галоген, нитро, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, NR7R8, OR7, SR7, С1-С6алкил-С(O)ОR7, ОС1-С6алкил-С(O)ОR7, C1-C6алкил-OR7, ОС1-С6алкил-ОR7, С1-С6алкил-NR7R8, ОС1-С6алкил-NR7R8, C1-C6алкил-C(O)NR7R8, ОС1-С6алкил-С(O)NR7R8, С1-С6алкил-С(O)R7, ОС1-С6алкил-С(O)R7, С1-С6галоалкил, O-аралкил, C(O)OR7, C(O)NR7R8, OC(O)NR7R8, NHC(O)R7, NHC(O)NR7R8, NR7S(O)2R1, NR7S(O)2R7, NR7S(O)2С1-С6алкил-С(O)ОR7, S(O)2R7 и S(O)2NR7R8. 3. Соединение по п.1, в котором Q имеет структуру где R9 и R10 независимо обозначают Н, С1-С6алкокси или О-С1-С10алкил-С6-С10арил; R11 обозначает Н, NR7S(O)2R7 или NR7S(O)2С1-С6алкил-С(O)ОR7; Z1 обозначает Н; Z2 обозначает Н или С1-С6алкокси. 4. Соединение по п.3, в котором Z1 и Z2 обозначают водород; Z1, Z2 и R11 обозначают водород; или Z1, R10 и R11 обозначают водород. 5. Соединение по п.1, в котором R9 и R10 независимо обозначают ОСН3, OEt, OiPr, OiBu, ОСН2Рh или OCH(CH2Cl)Ph. 6. Соединение по п.1, в котором Х обозначает карбонильную группу, С=O. 7. Соединение по любому из пп.1-6, где Y означает S(O)nR1, где n = 1 или 2. 8. Фармацевтическая композиция для ингибирования серинпротеазных ферментов, включающая соединение по любому из пп.1-7 и носитель или эксципиент. 9. Способ ингибирования активности ТФ/фактора VIIa, фактора Ха, тромбина или калликреина, предусматривающий приведение ТФ/фактора VIIa, фактора Ха, тромбина или калликреина в контакт с эффективным количеством композиции по п.8. 10. Способ лечения заболевания, опосредуемого ТФ/фактором VIIa, фактором Ха, тромбином или калликреином, предусматривающий введение млекопитающему, которое нуждается в этом, эффективного количества композиции по п.8. 11. Способ предупреждения тромбоза или лечения аномального тромбоза, предусматривающий введение млекопитающему, которое нуждается в этом, эффективного количества композиции по п.8.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
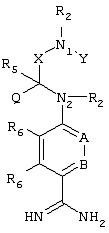
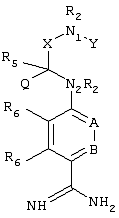
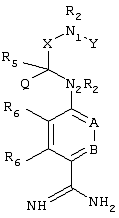
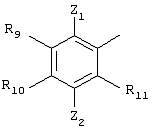
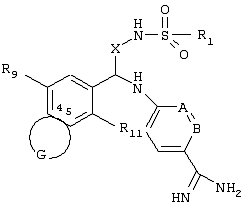
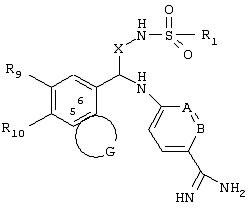
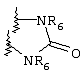
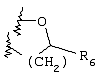
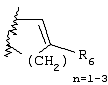
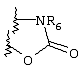
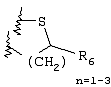
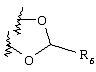
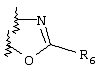
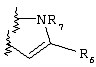
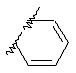
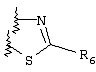
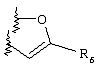
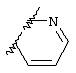
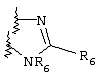
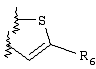
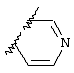
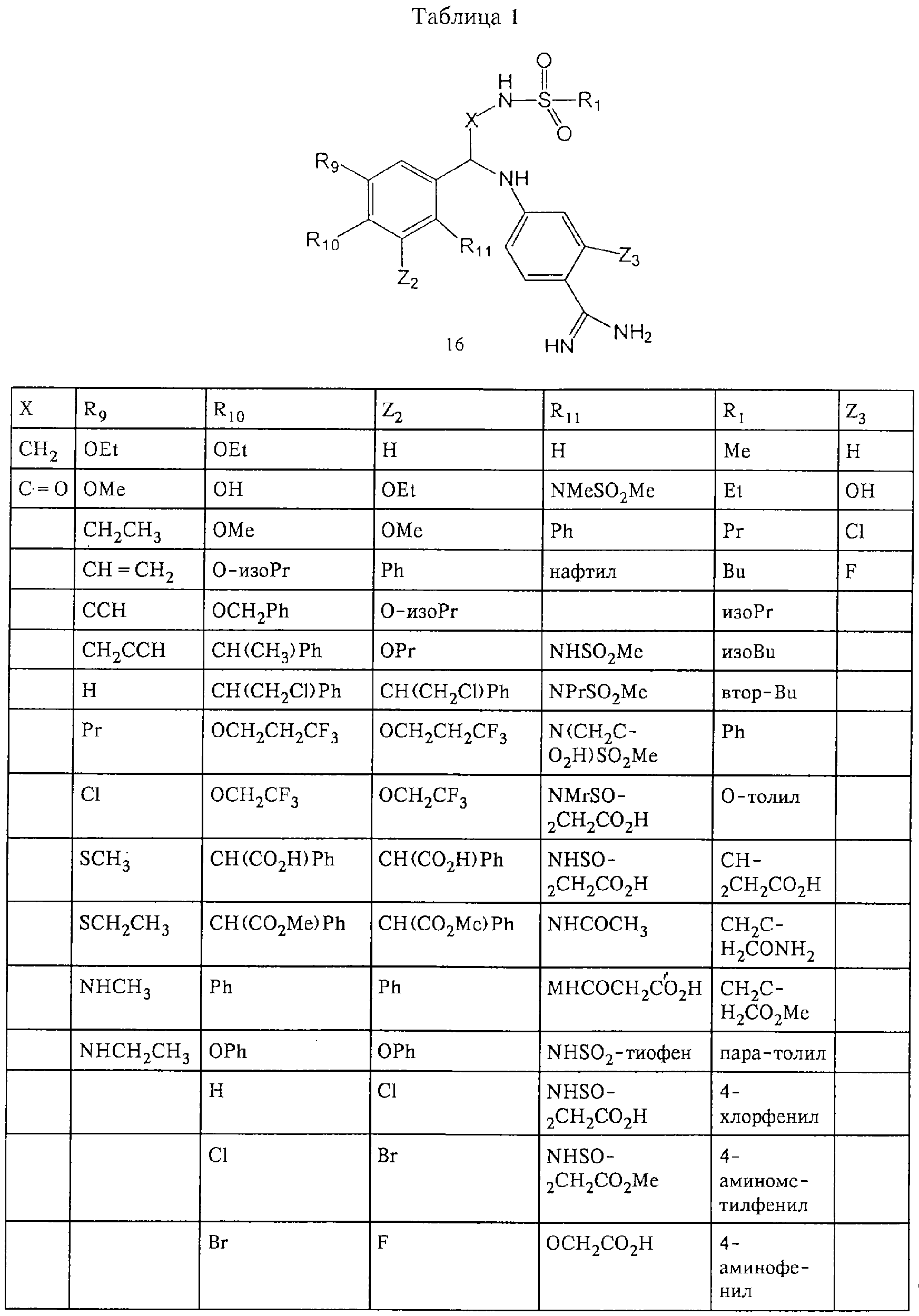
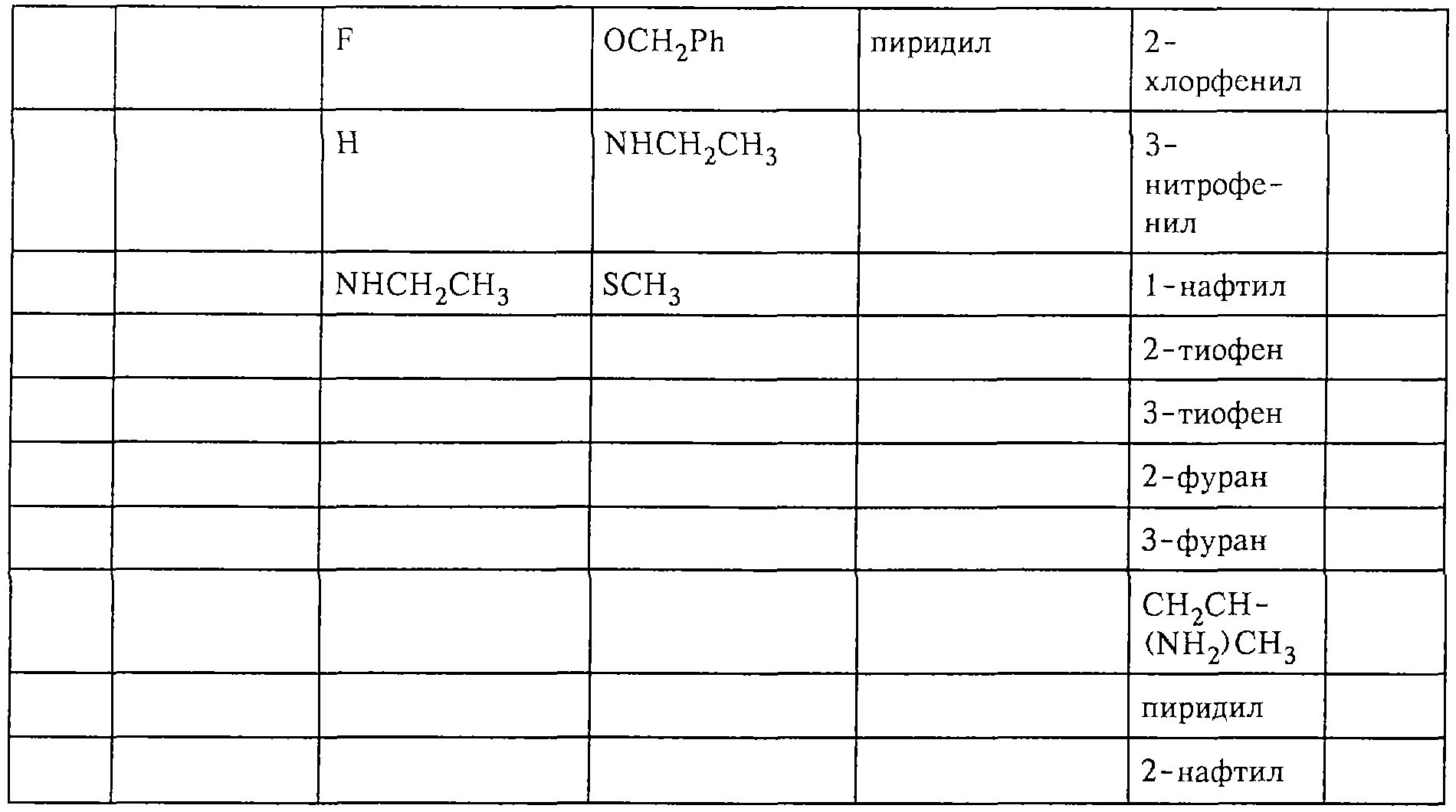
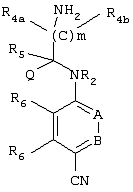
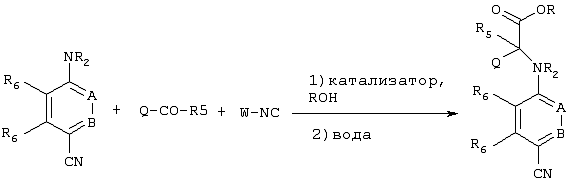
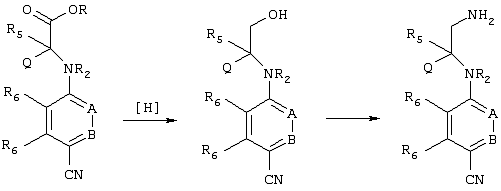
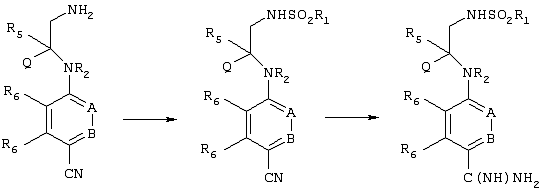
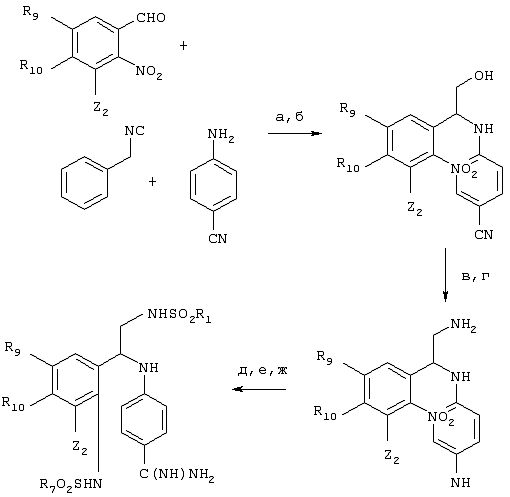
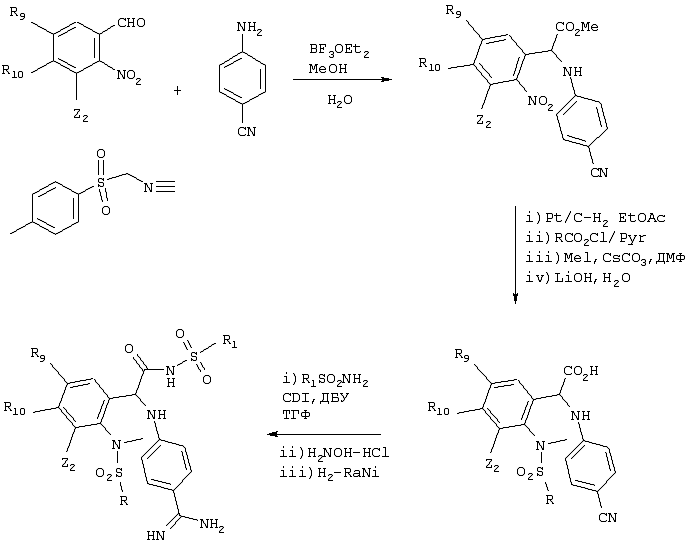
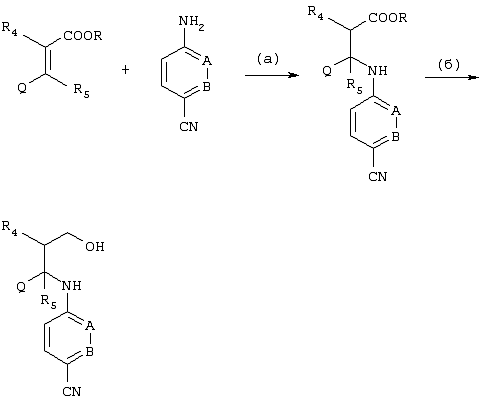
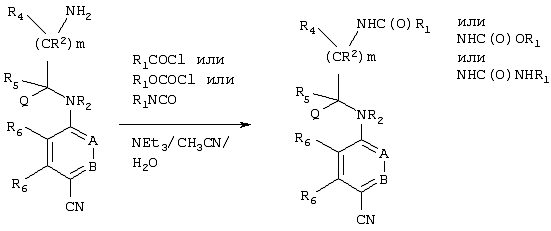
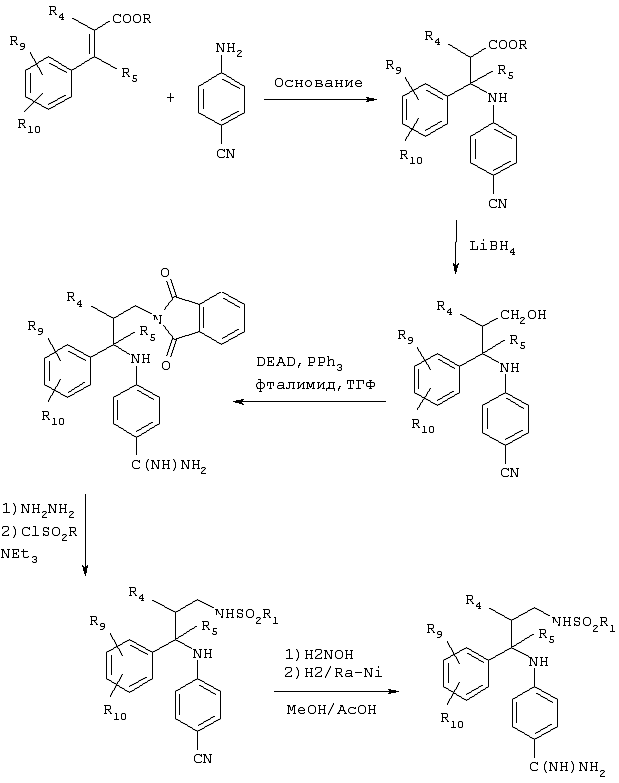
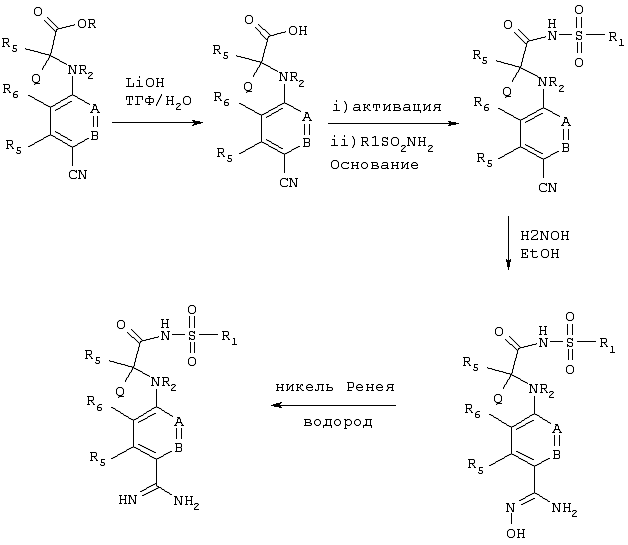
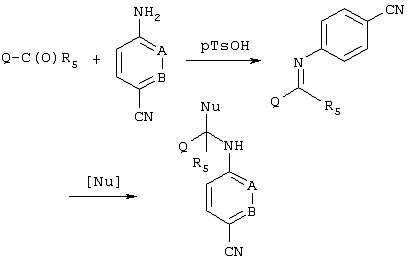
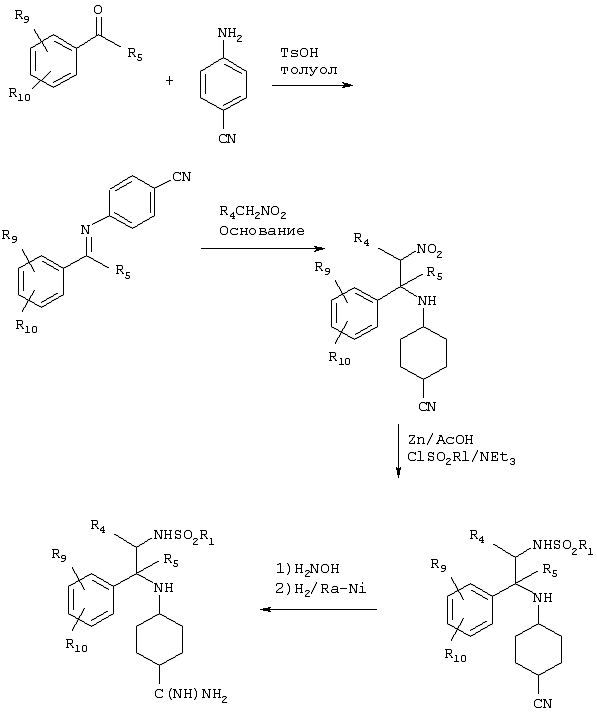
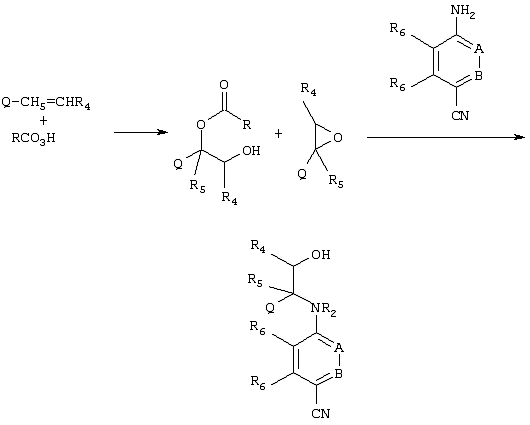
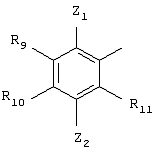
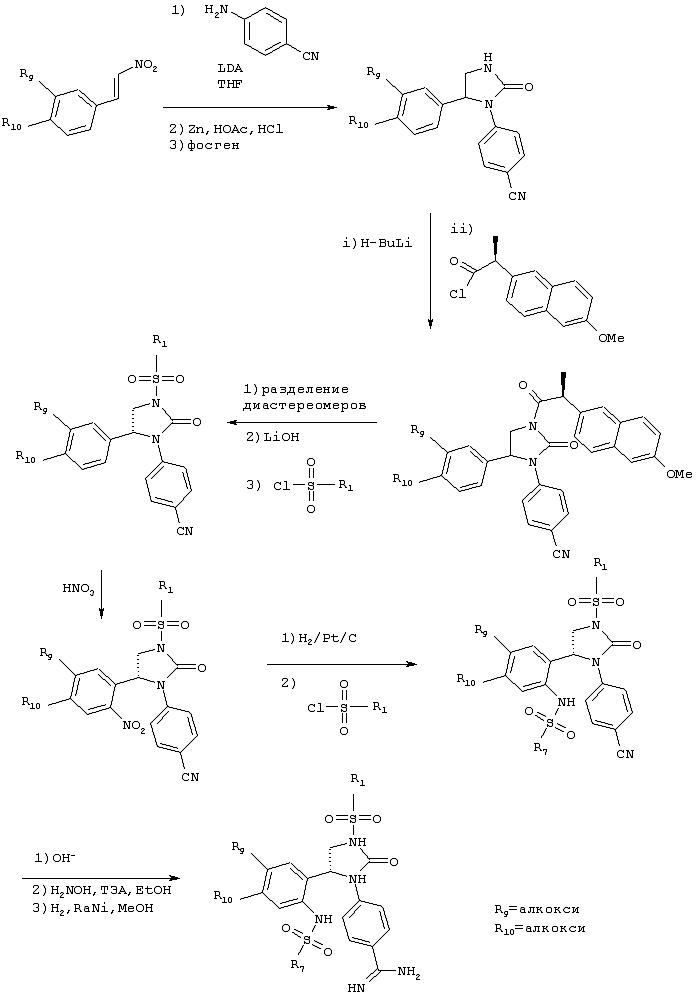
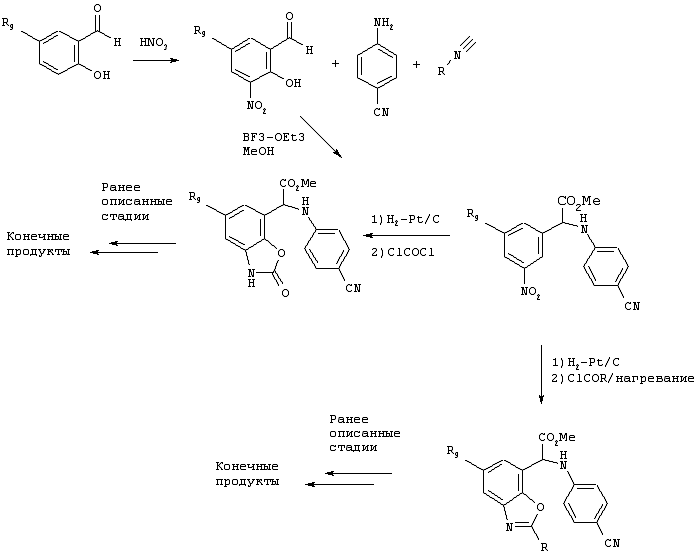
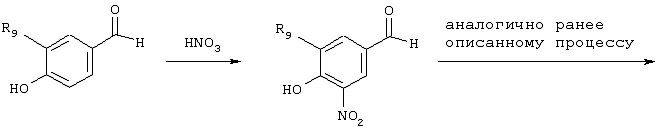
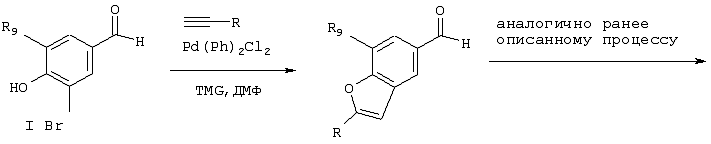
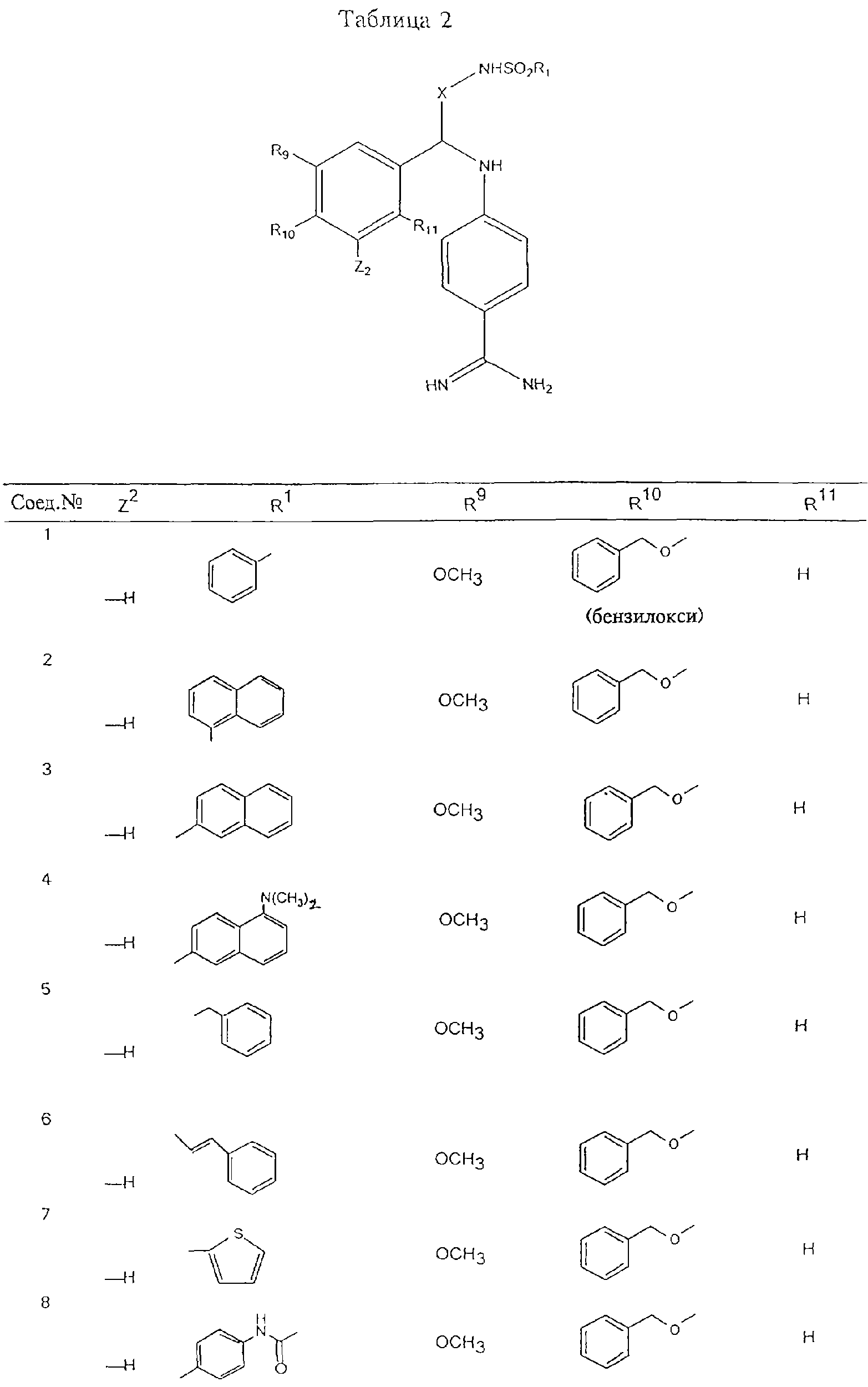
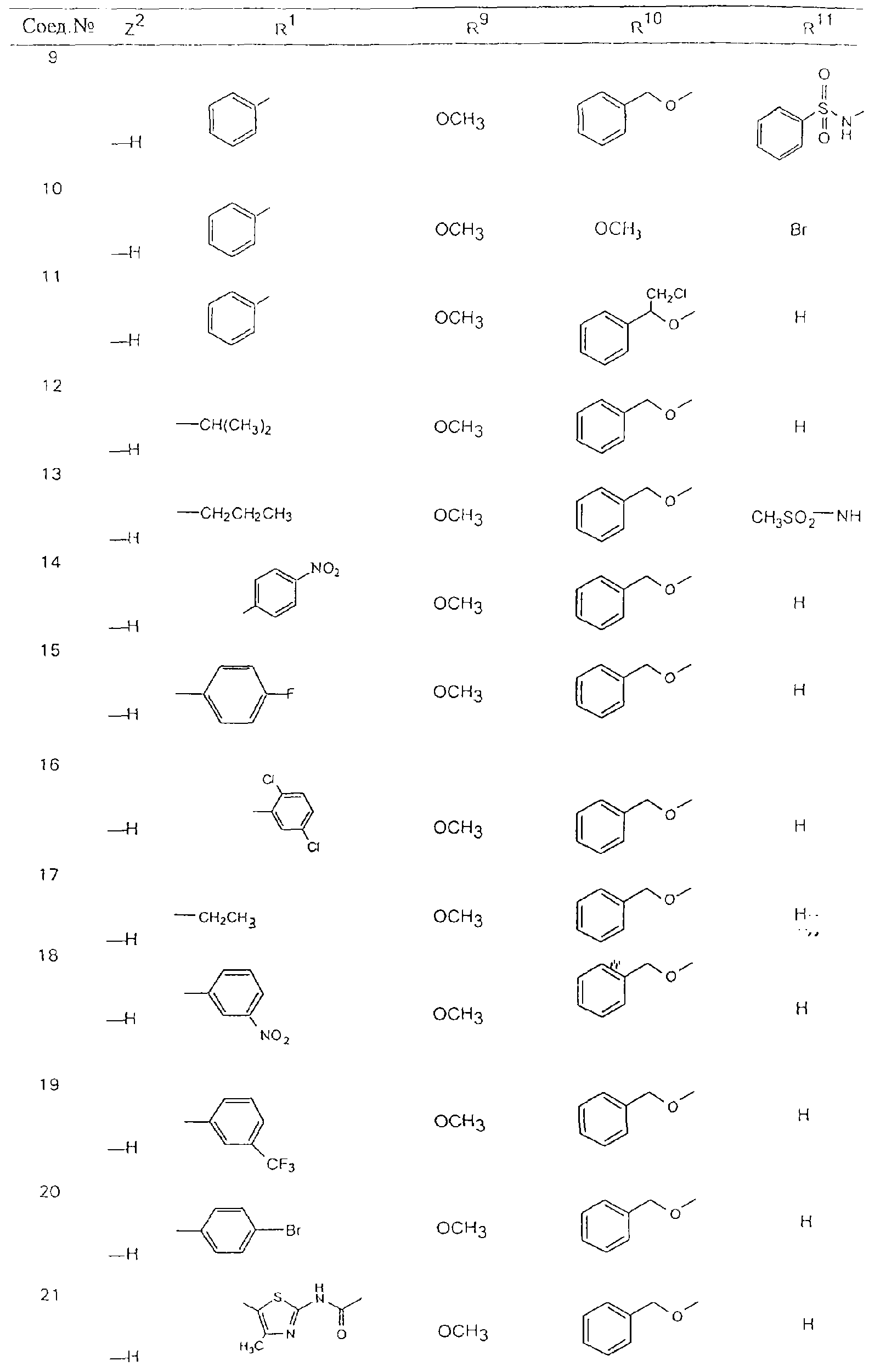
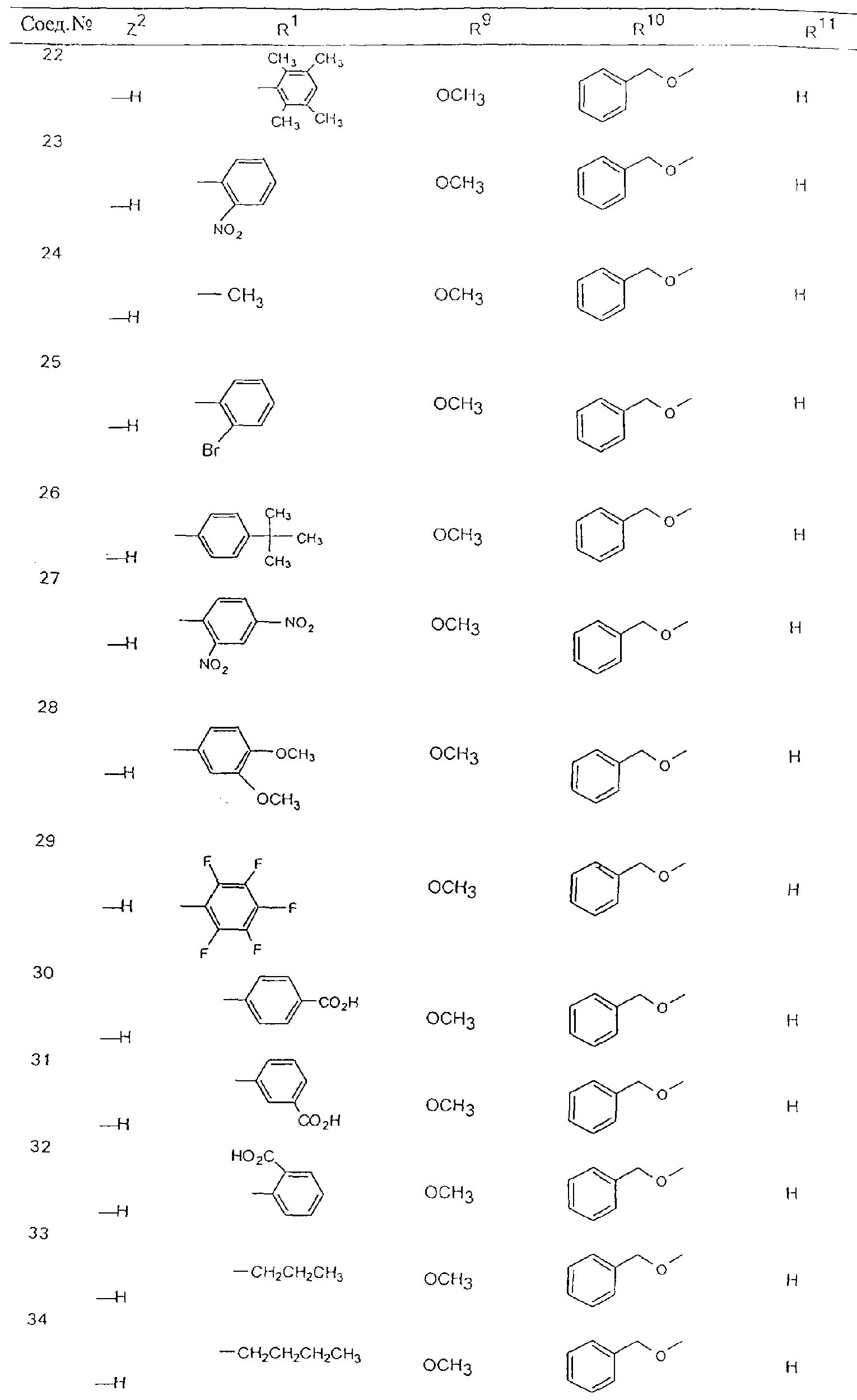
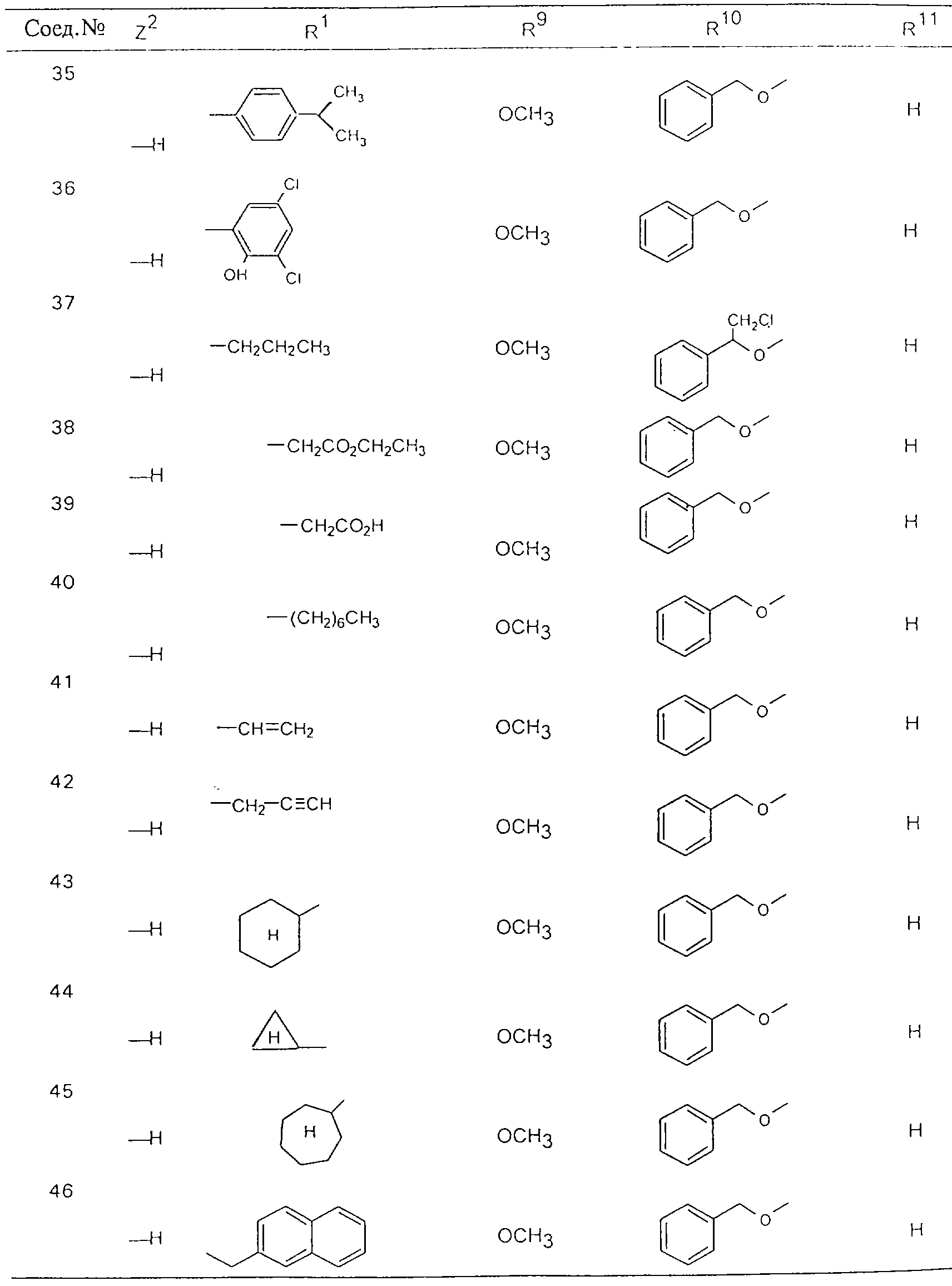
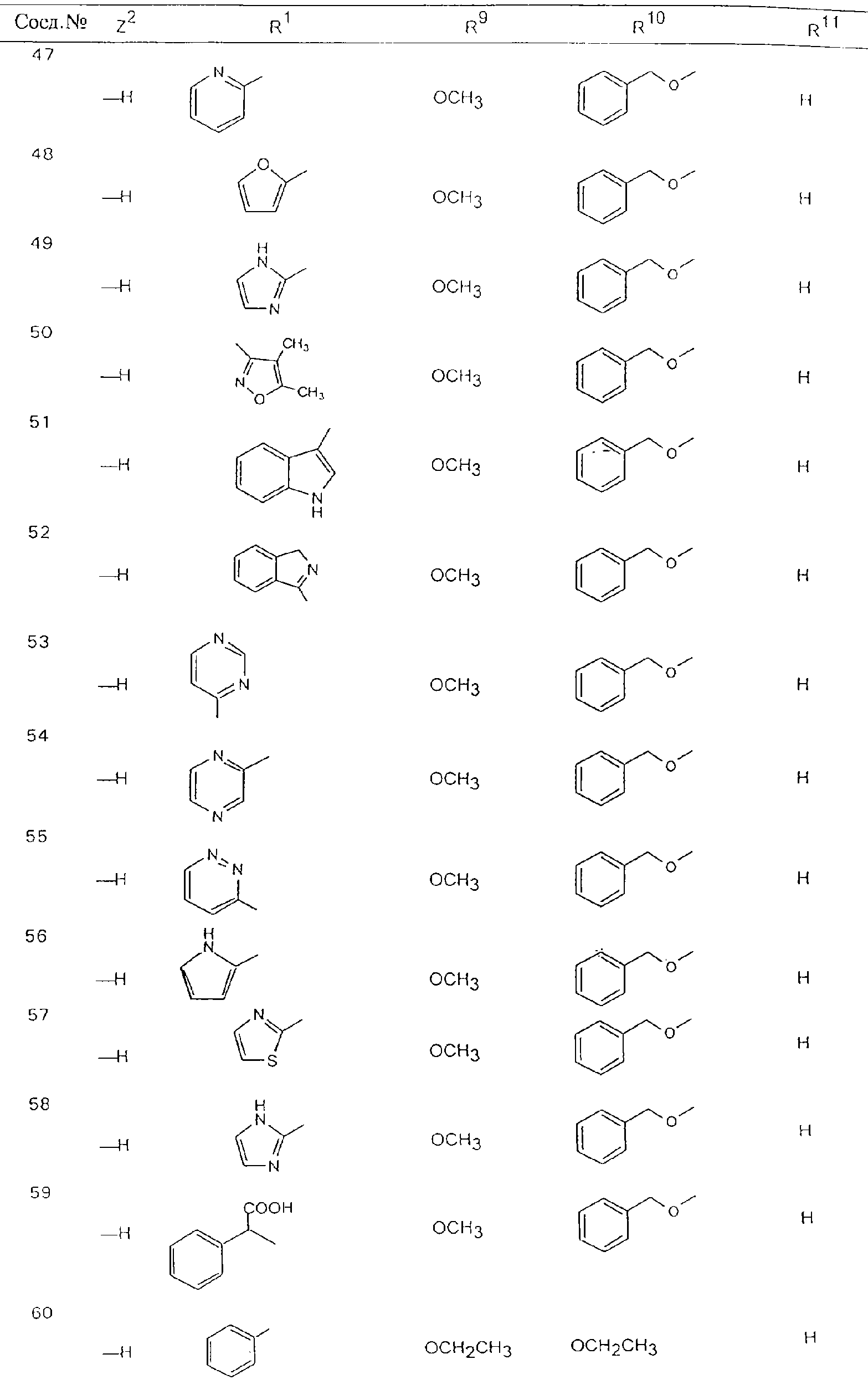
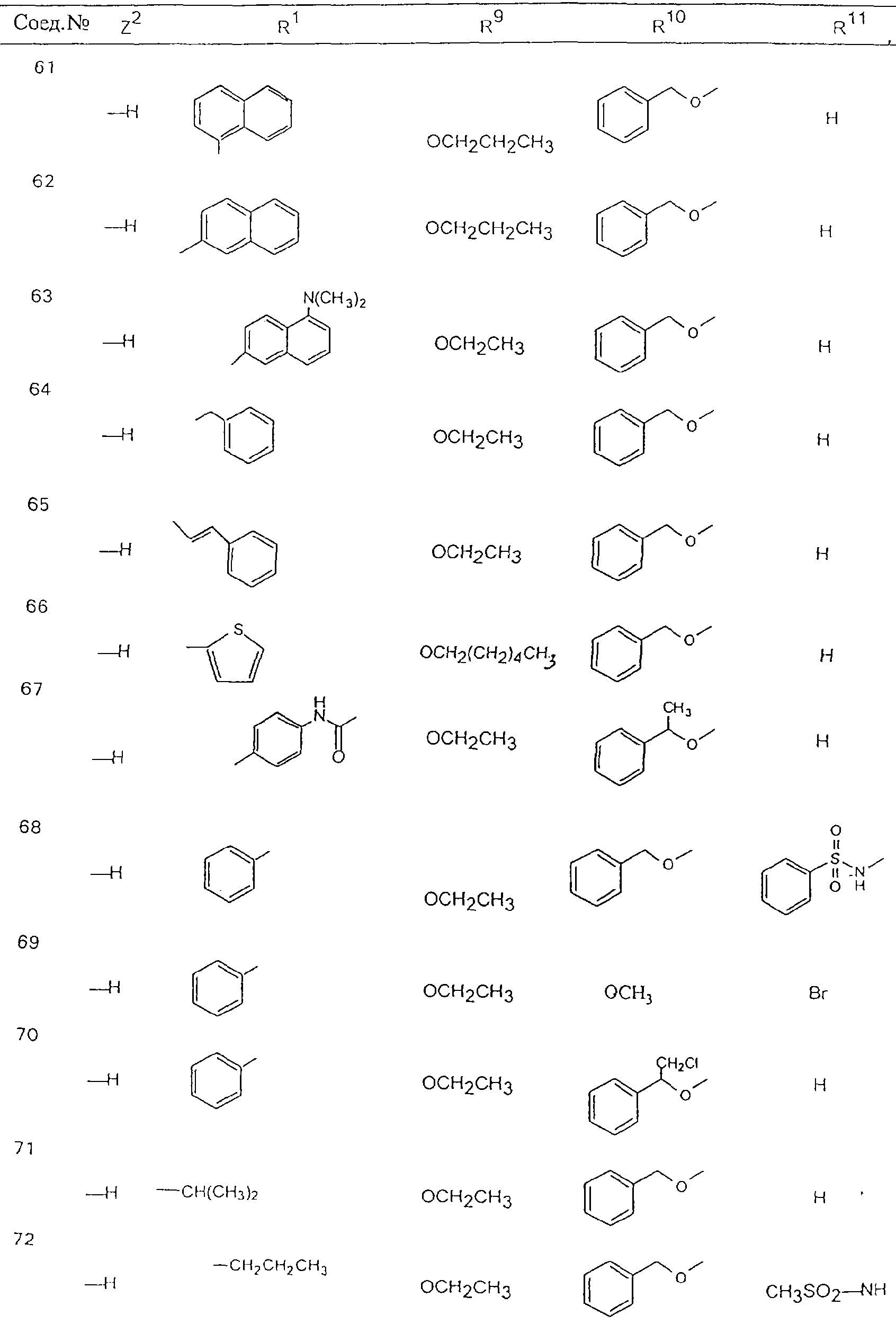
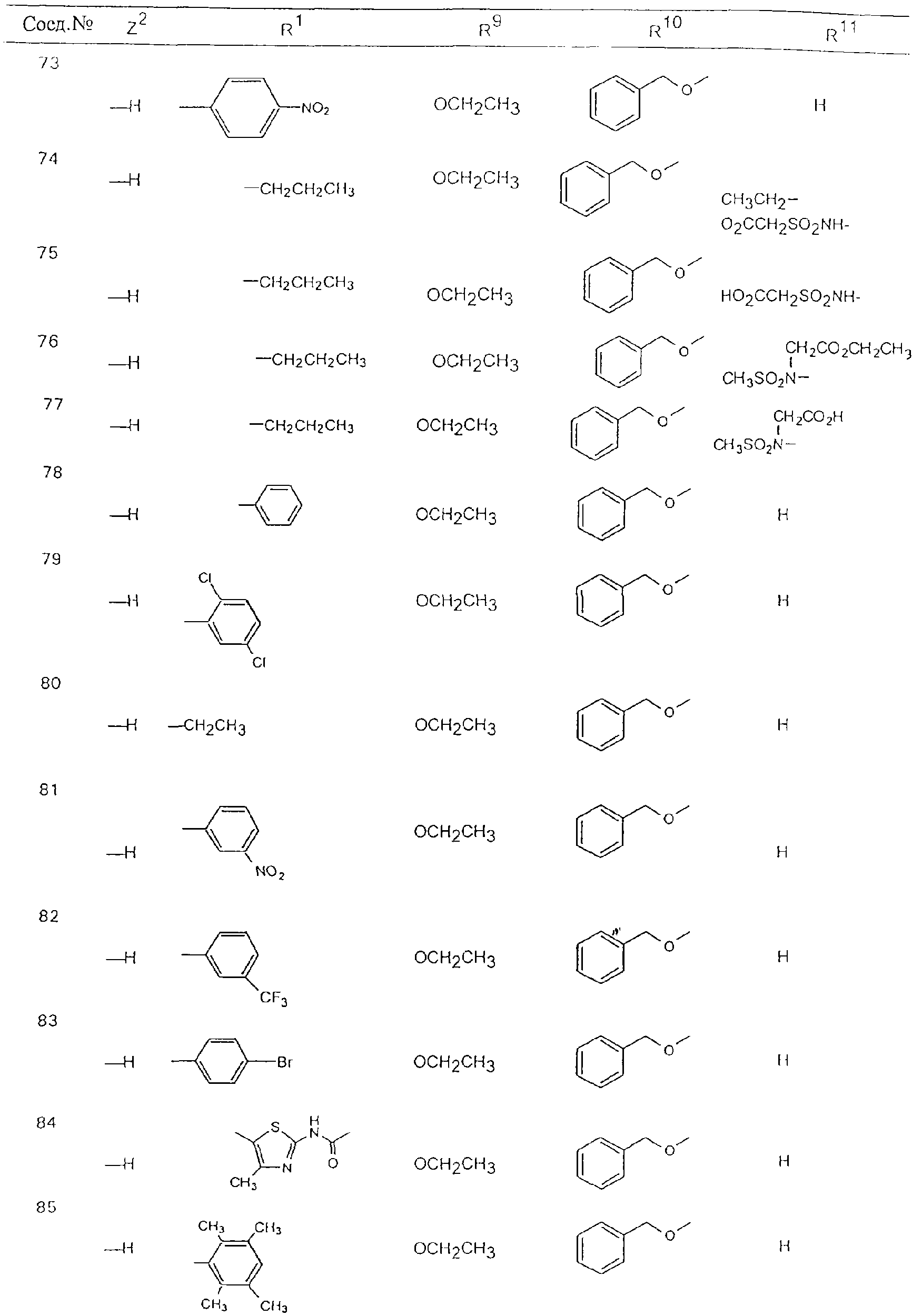
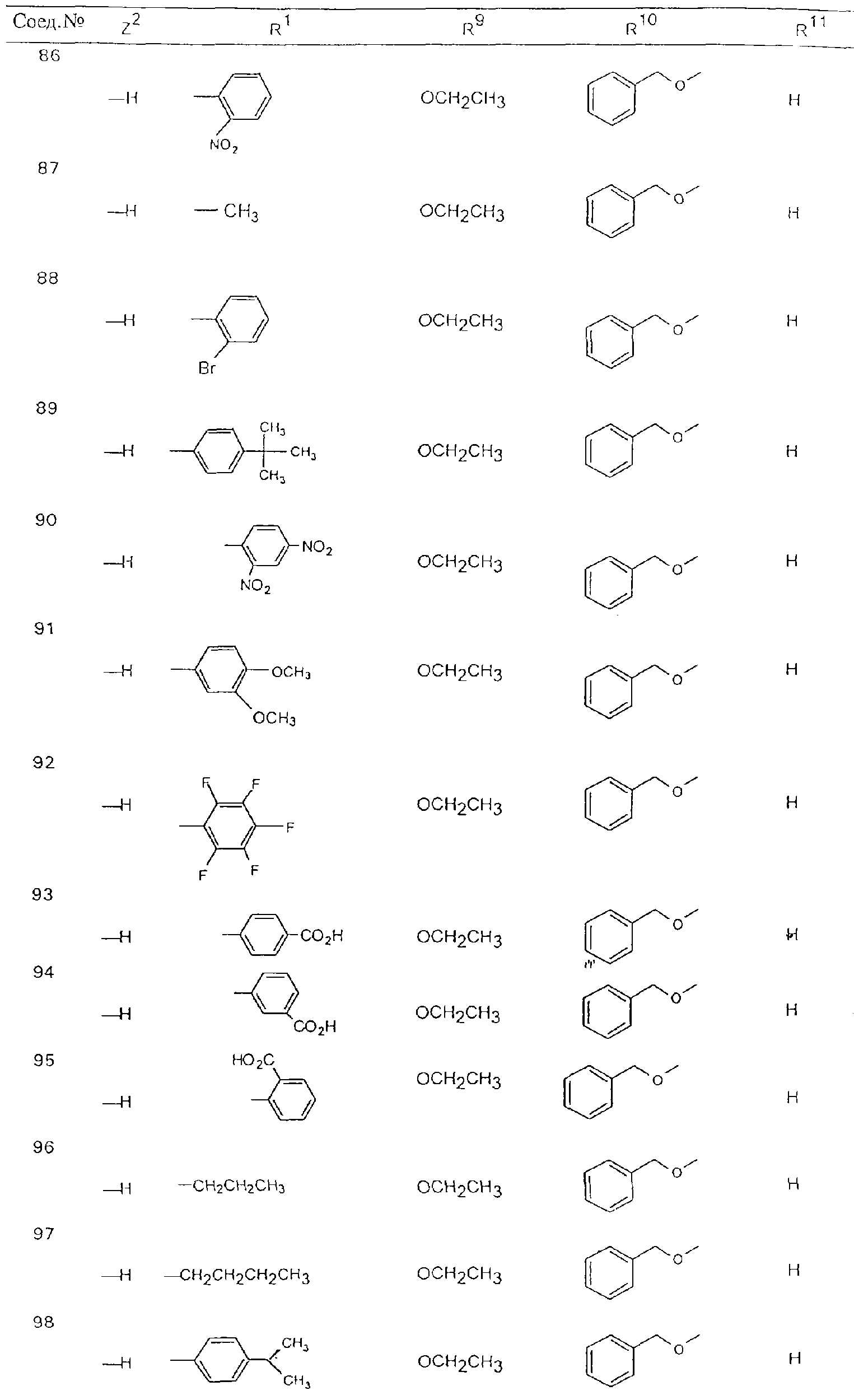
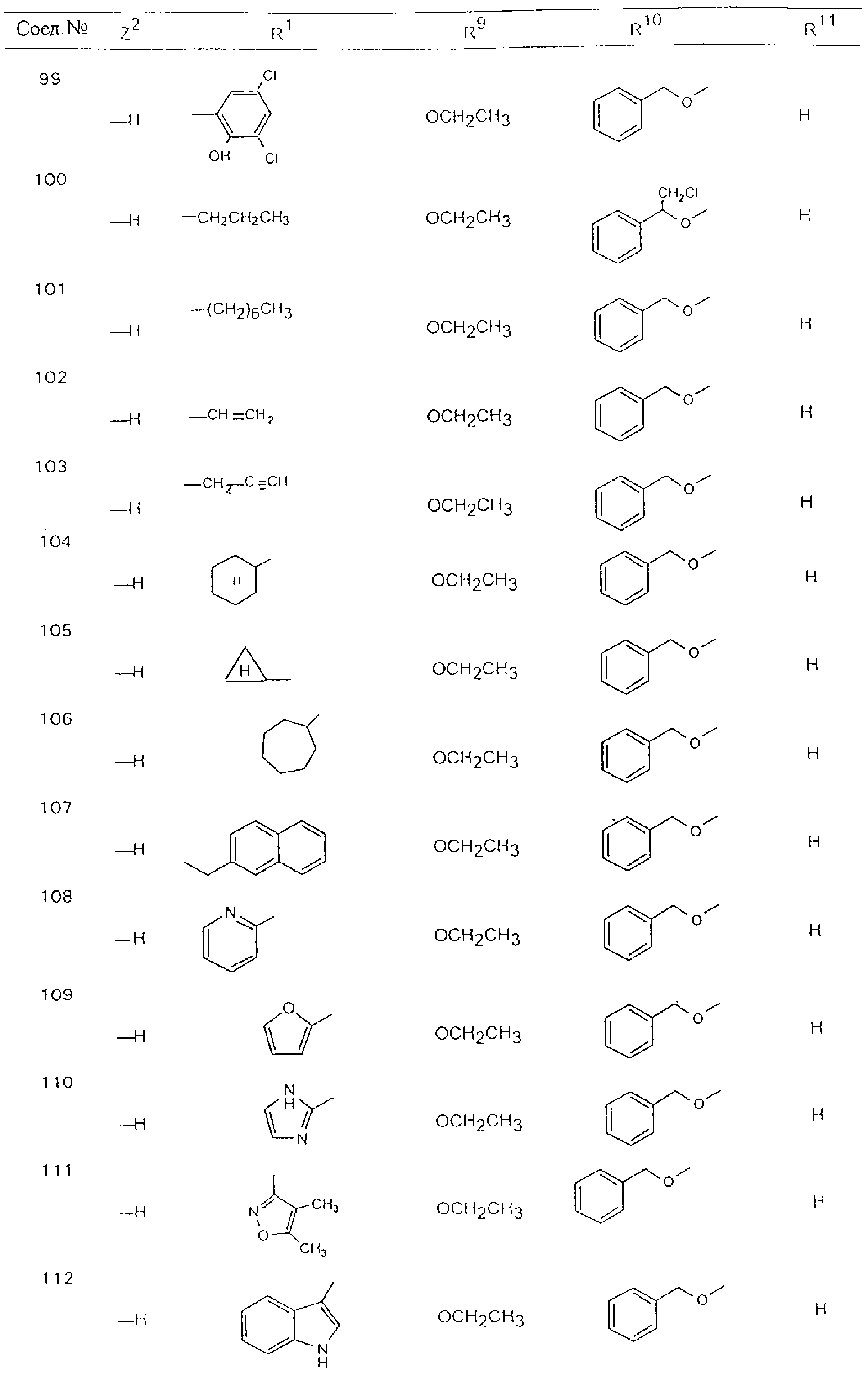
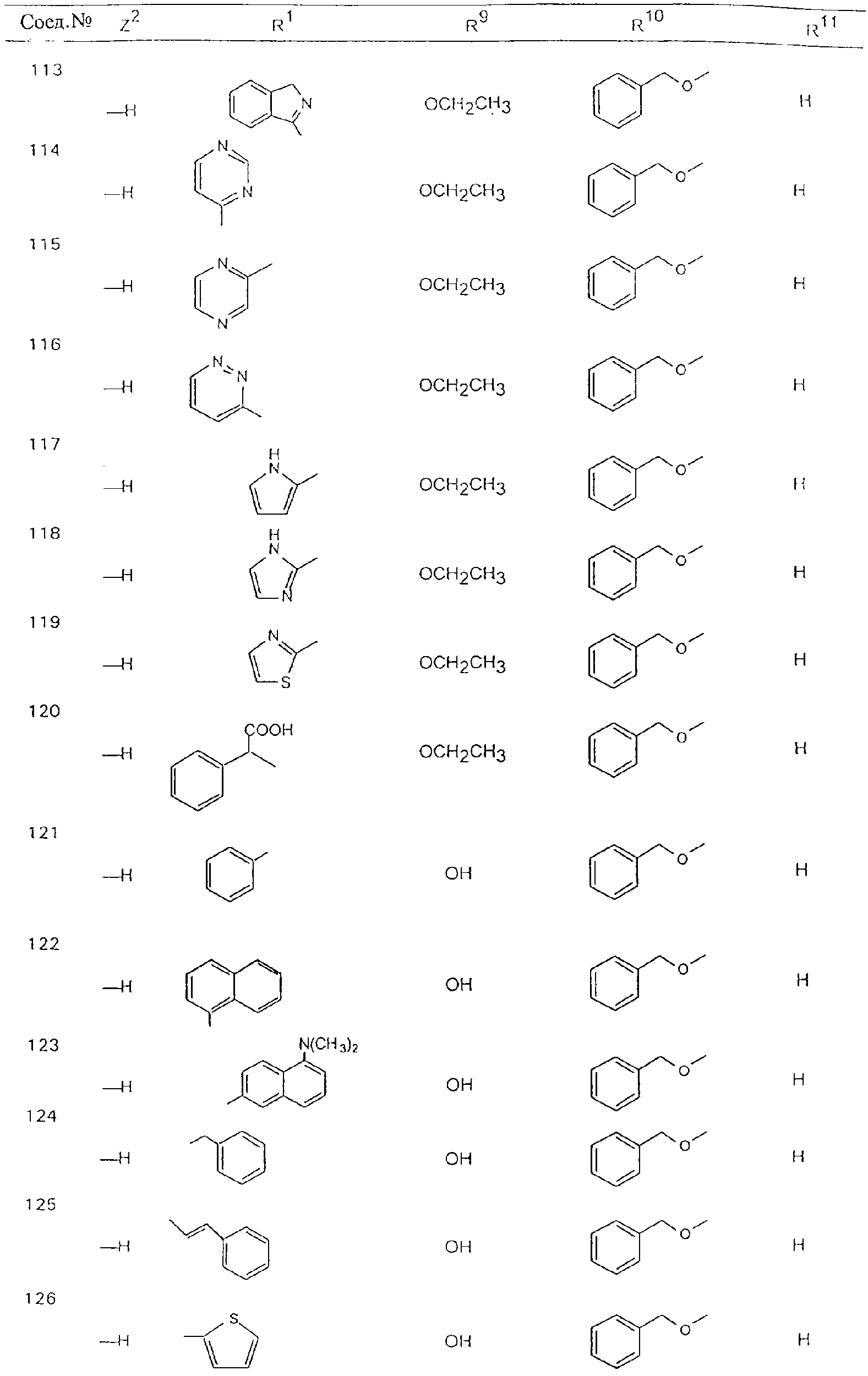
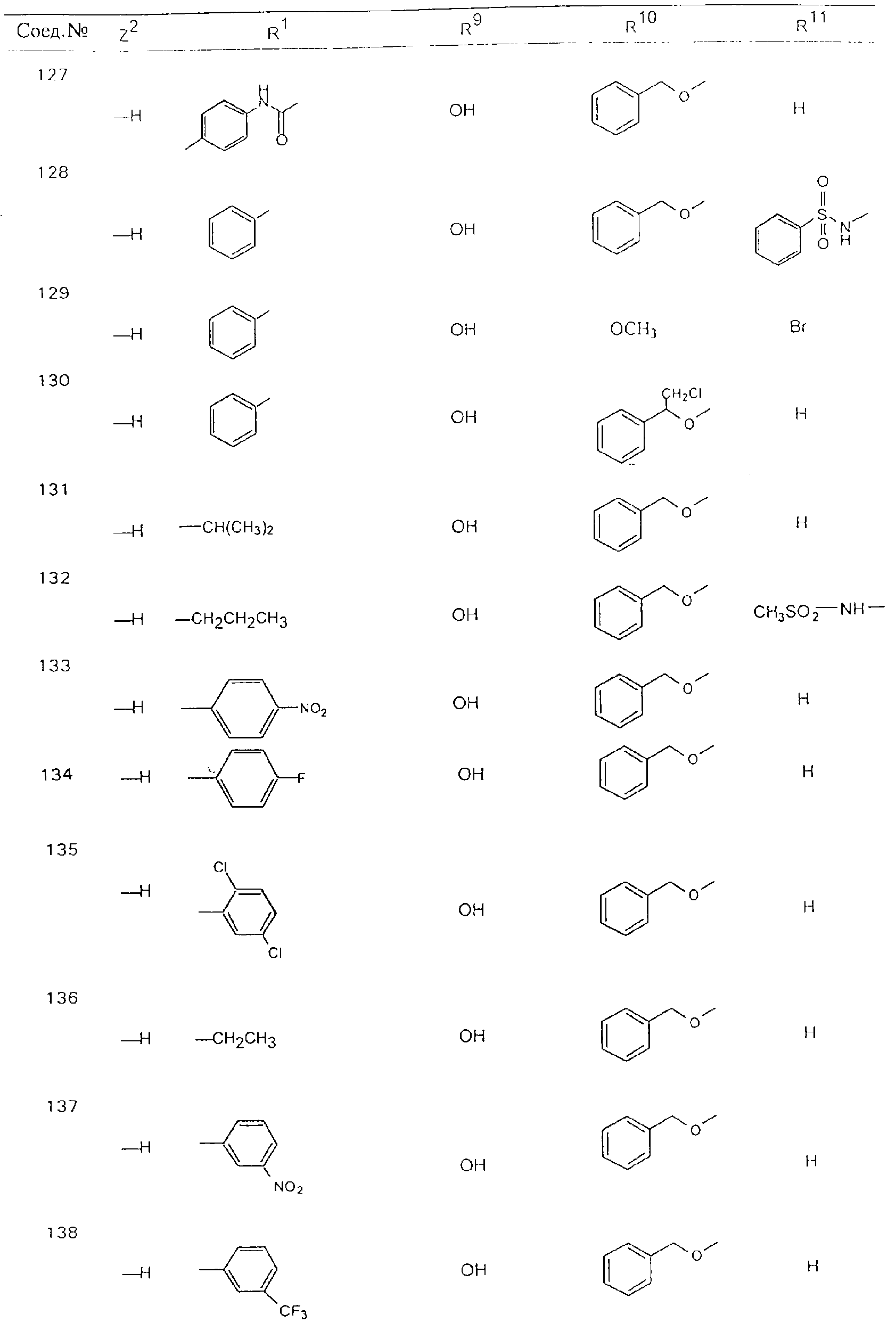
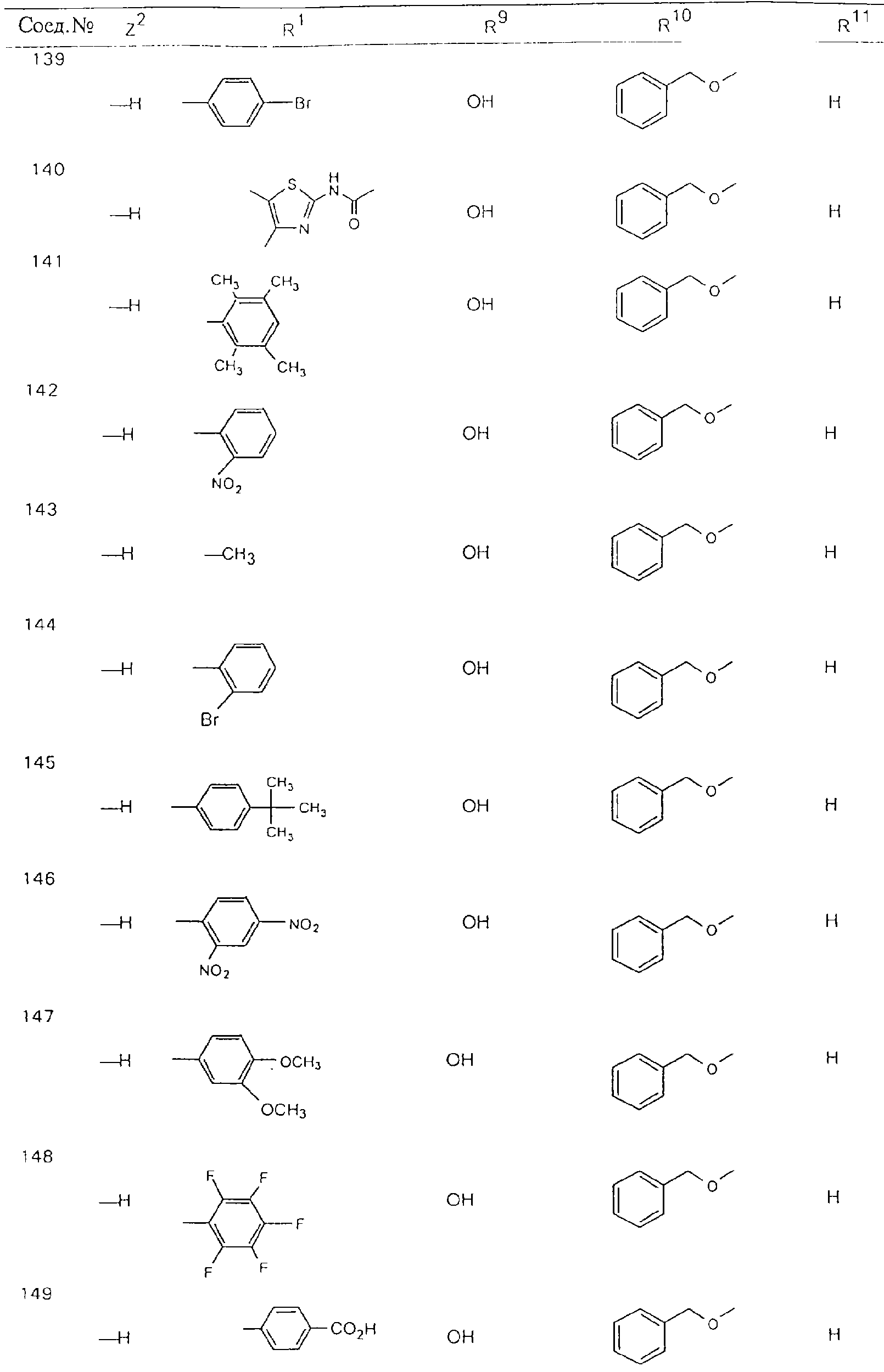
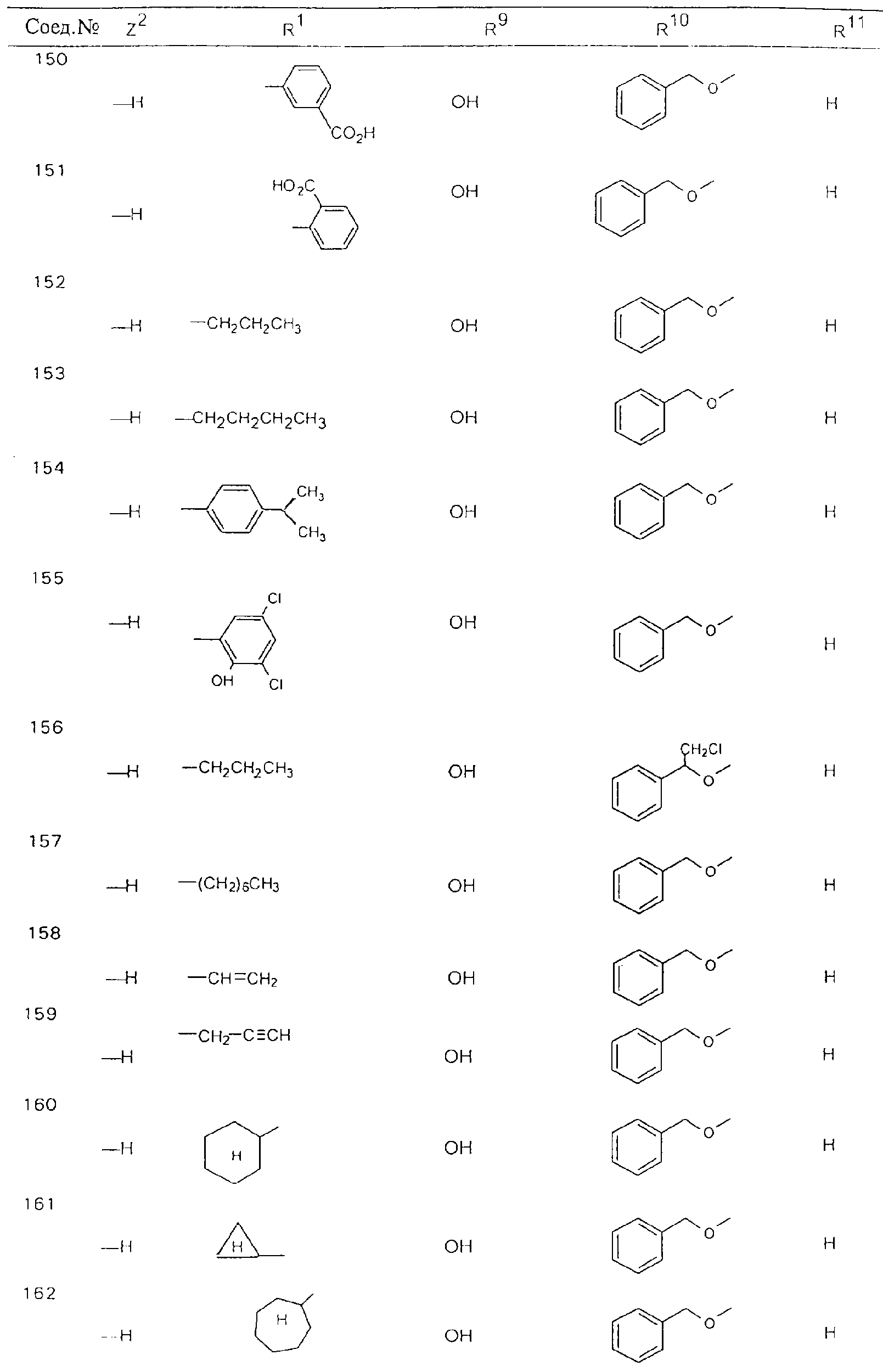
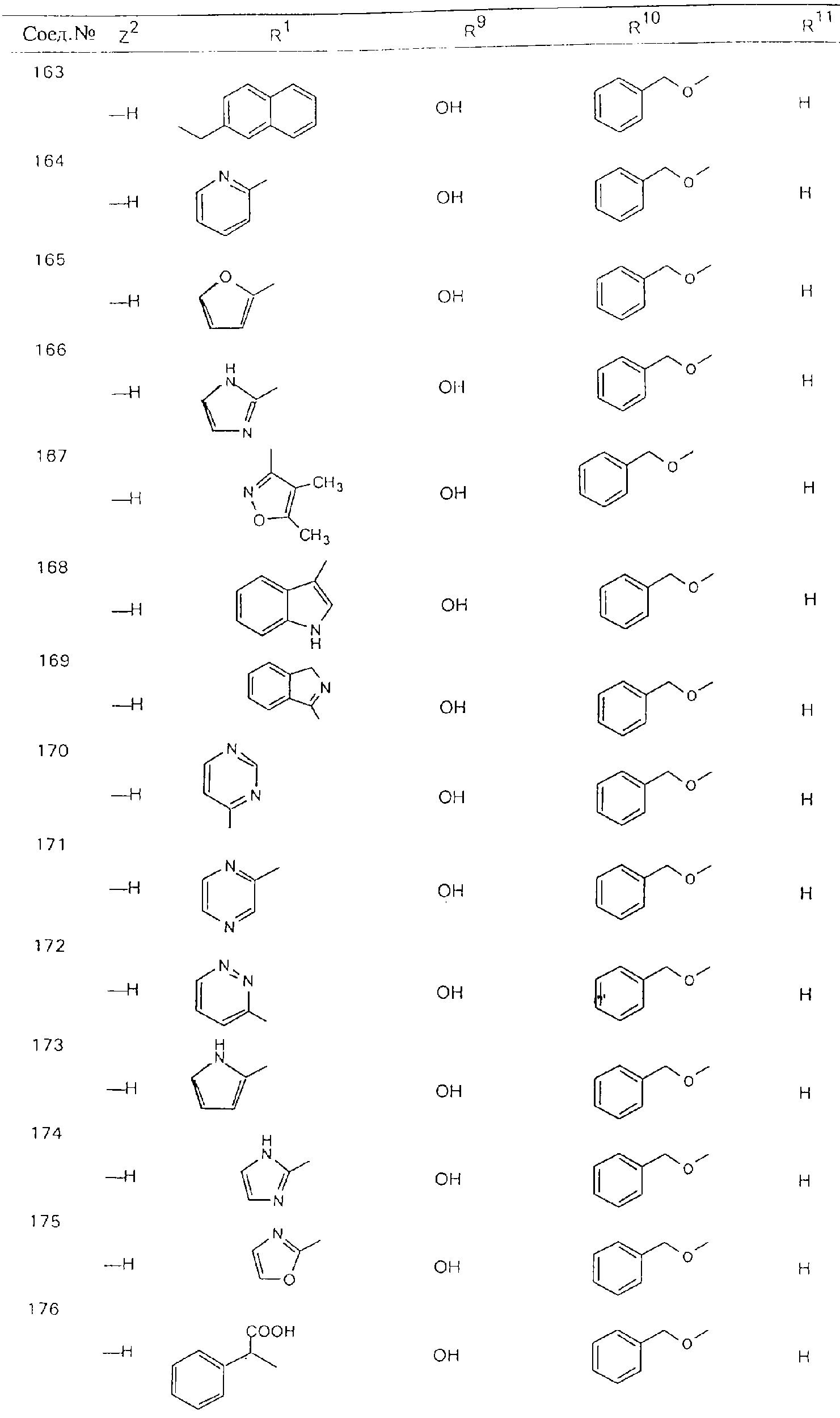
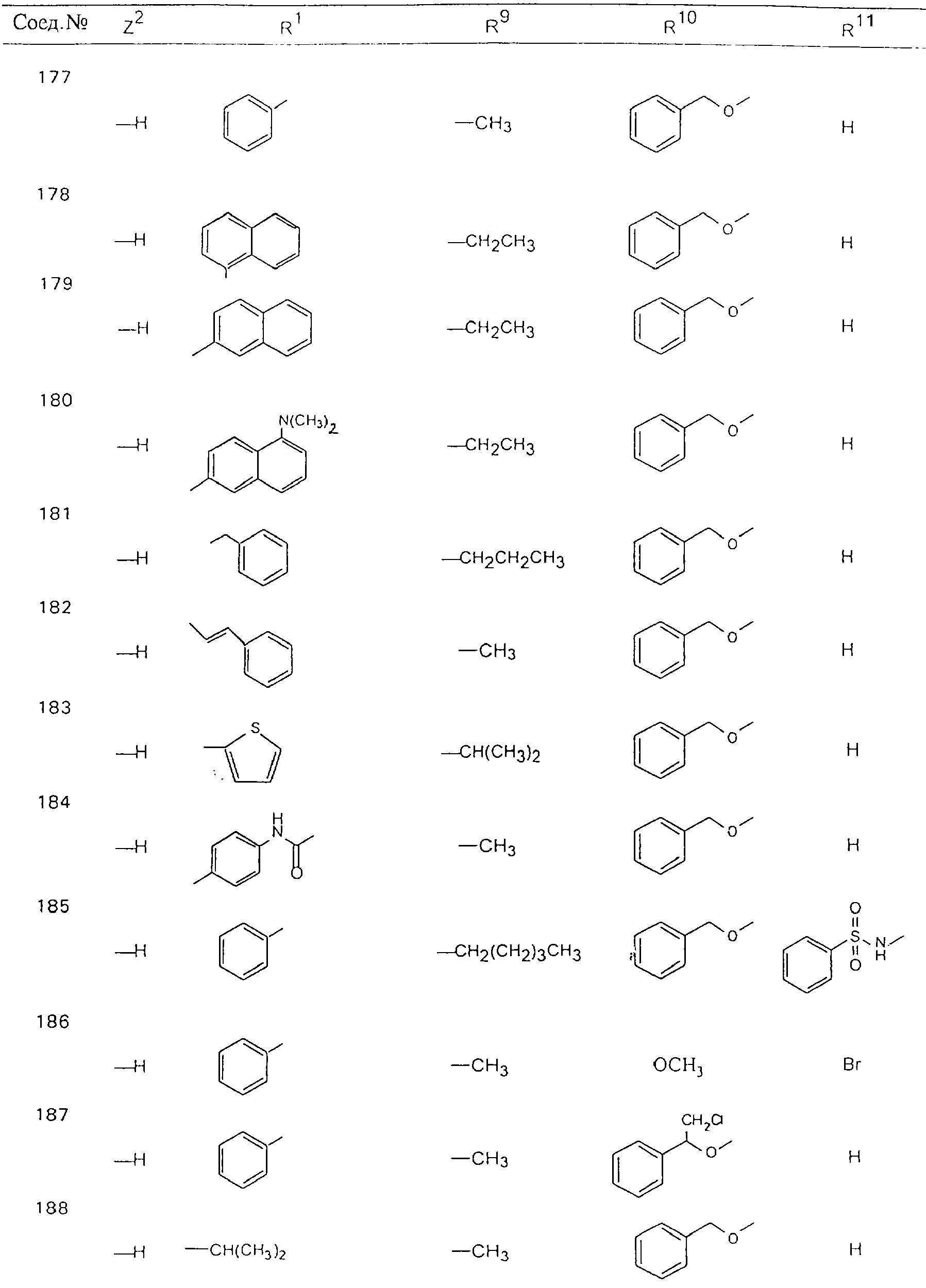
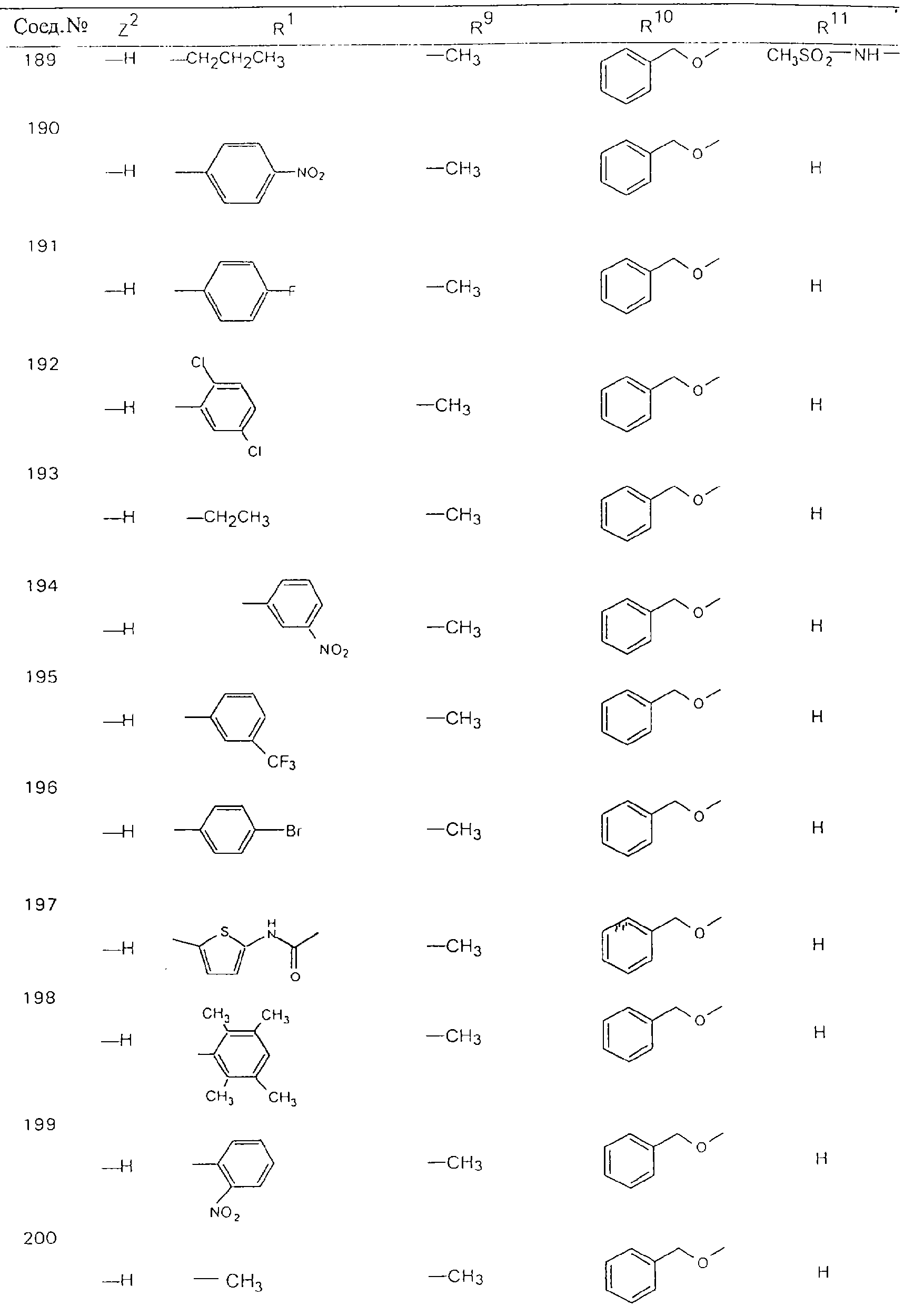
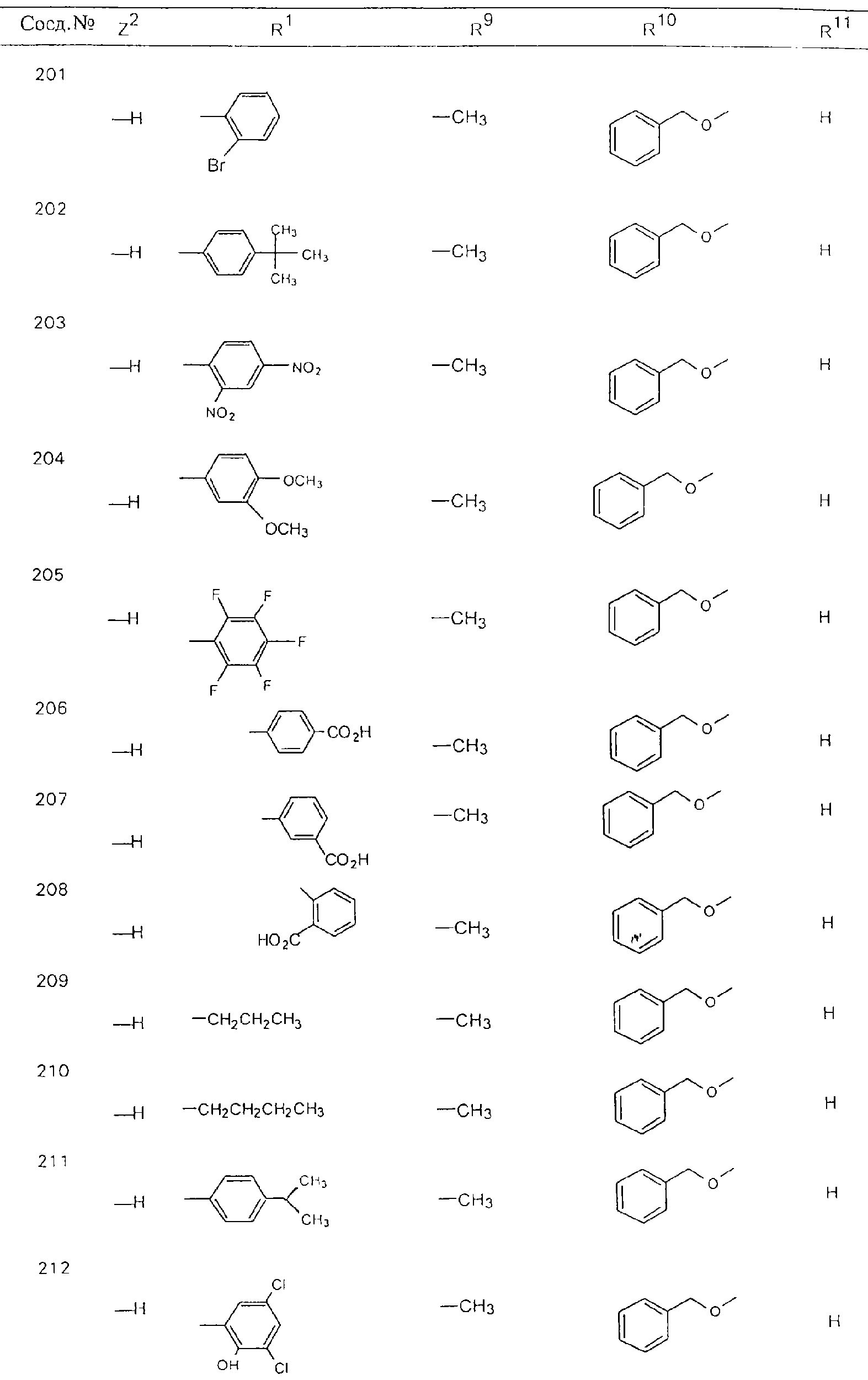
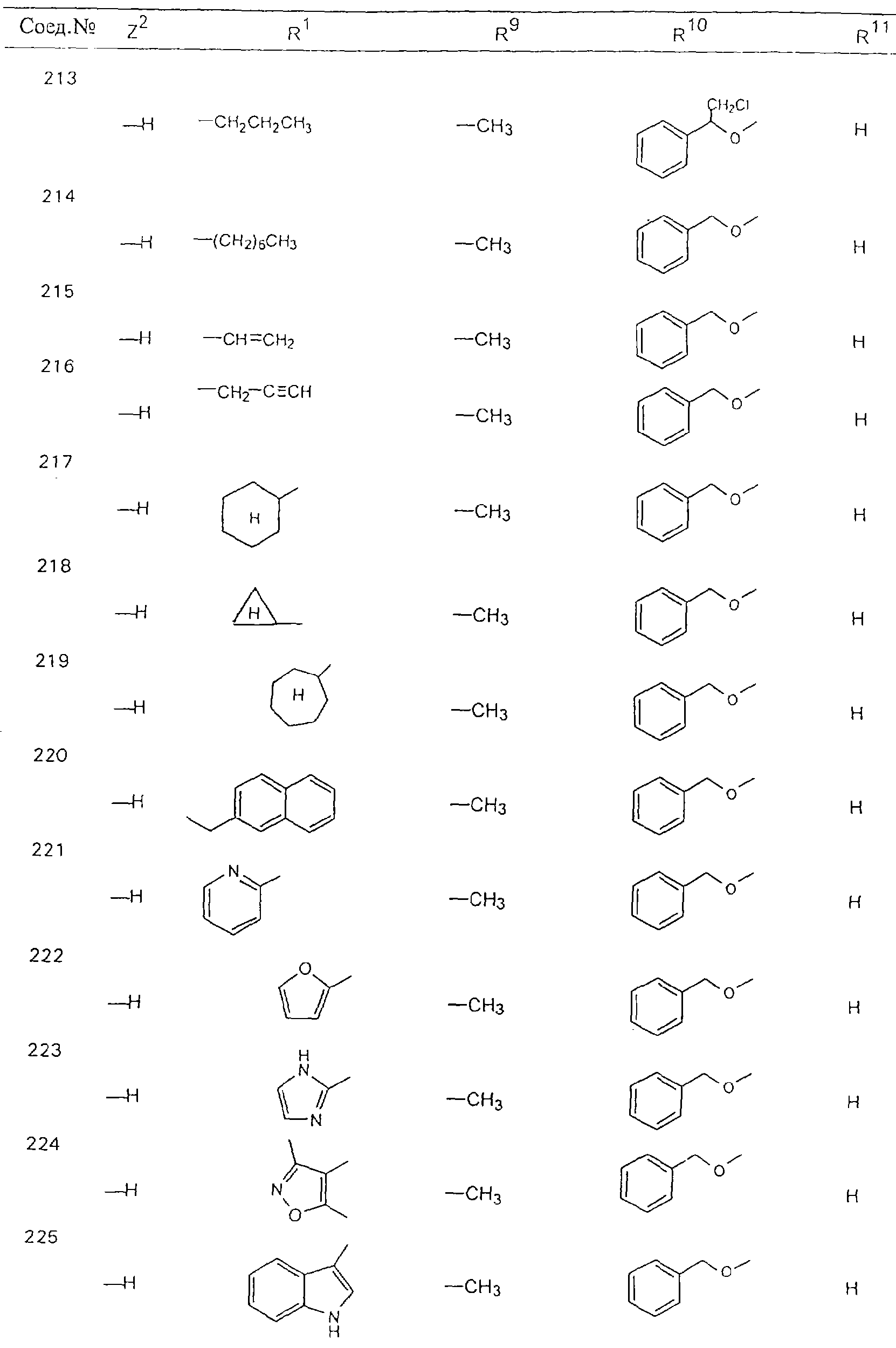
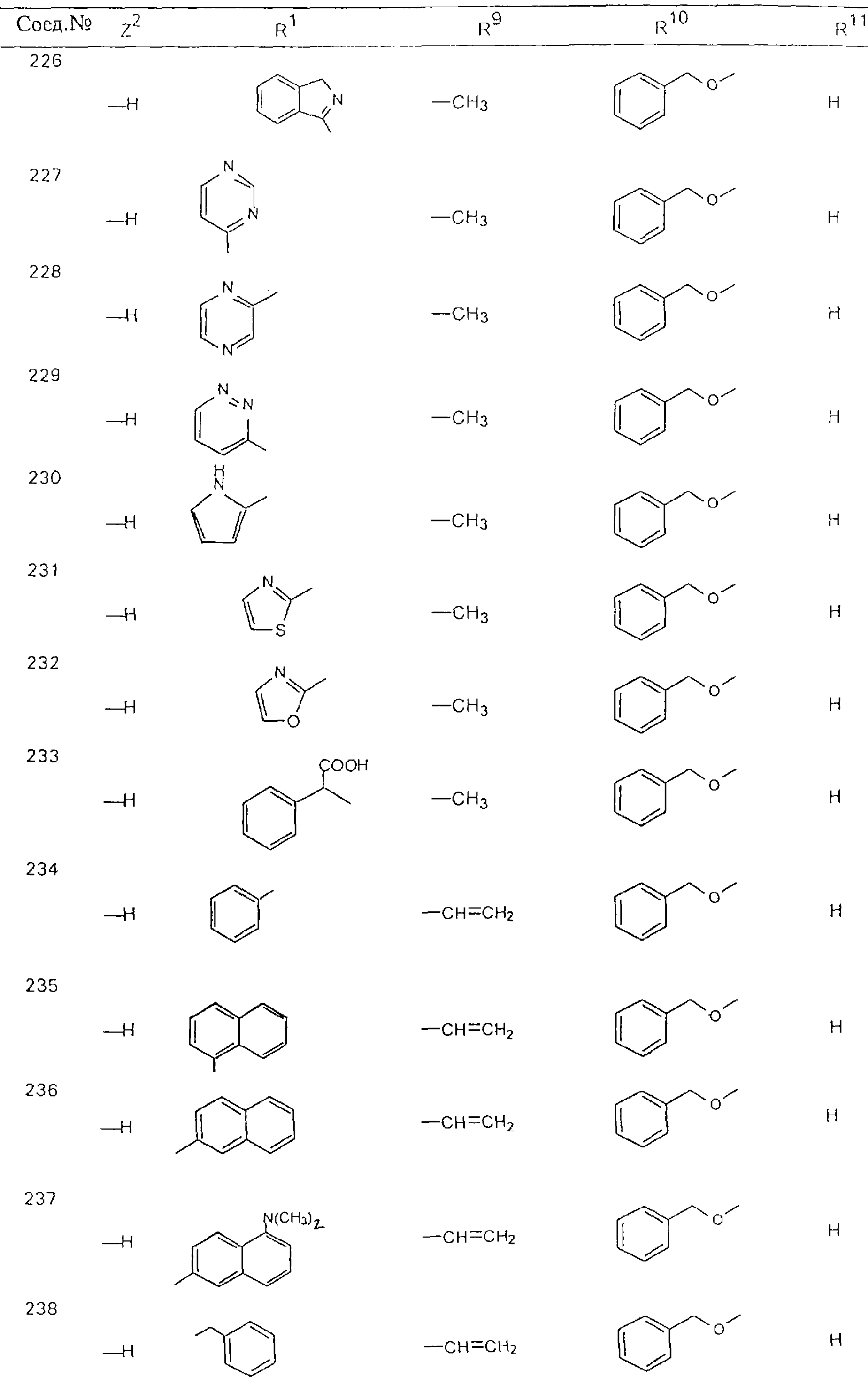
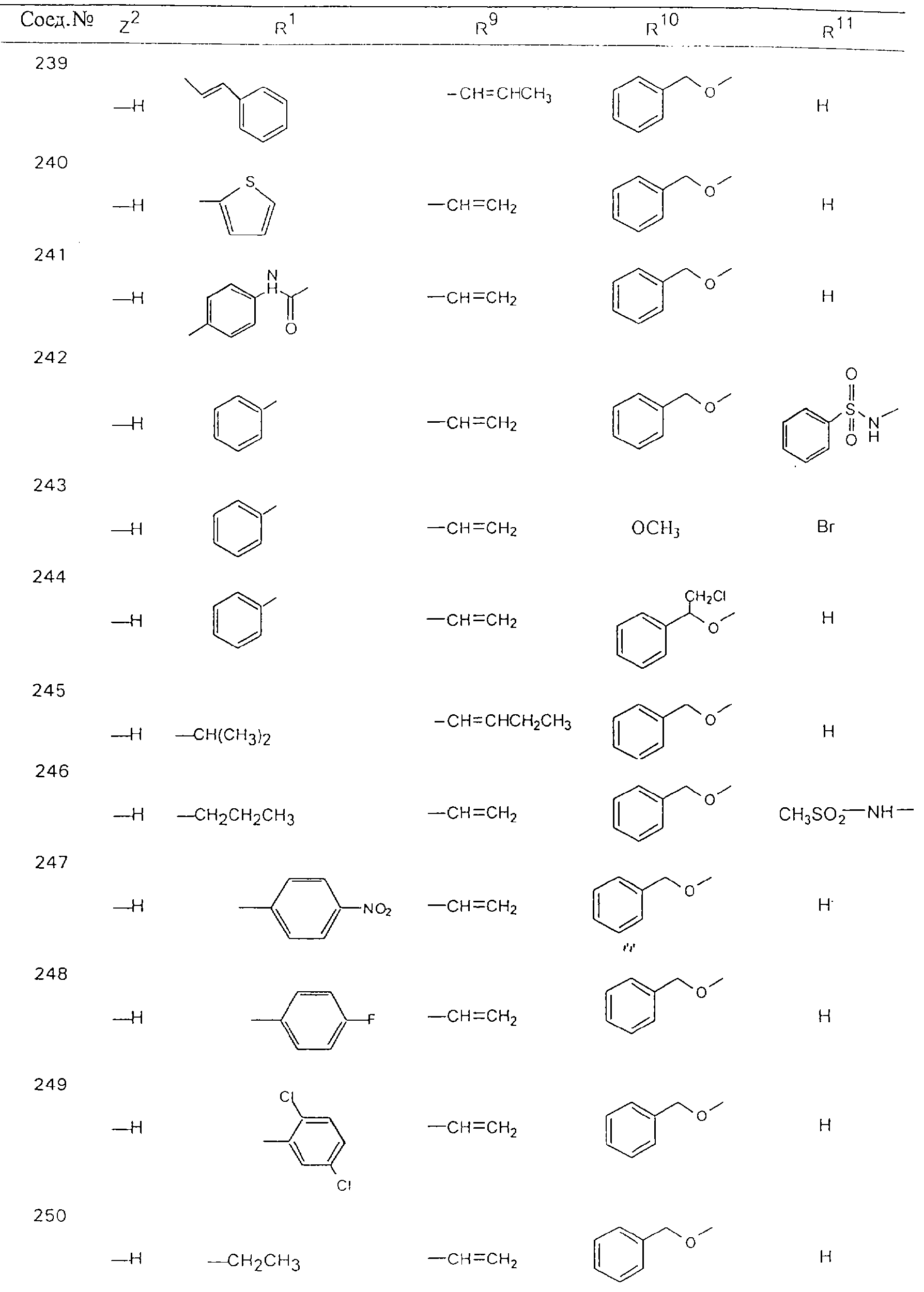
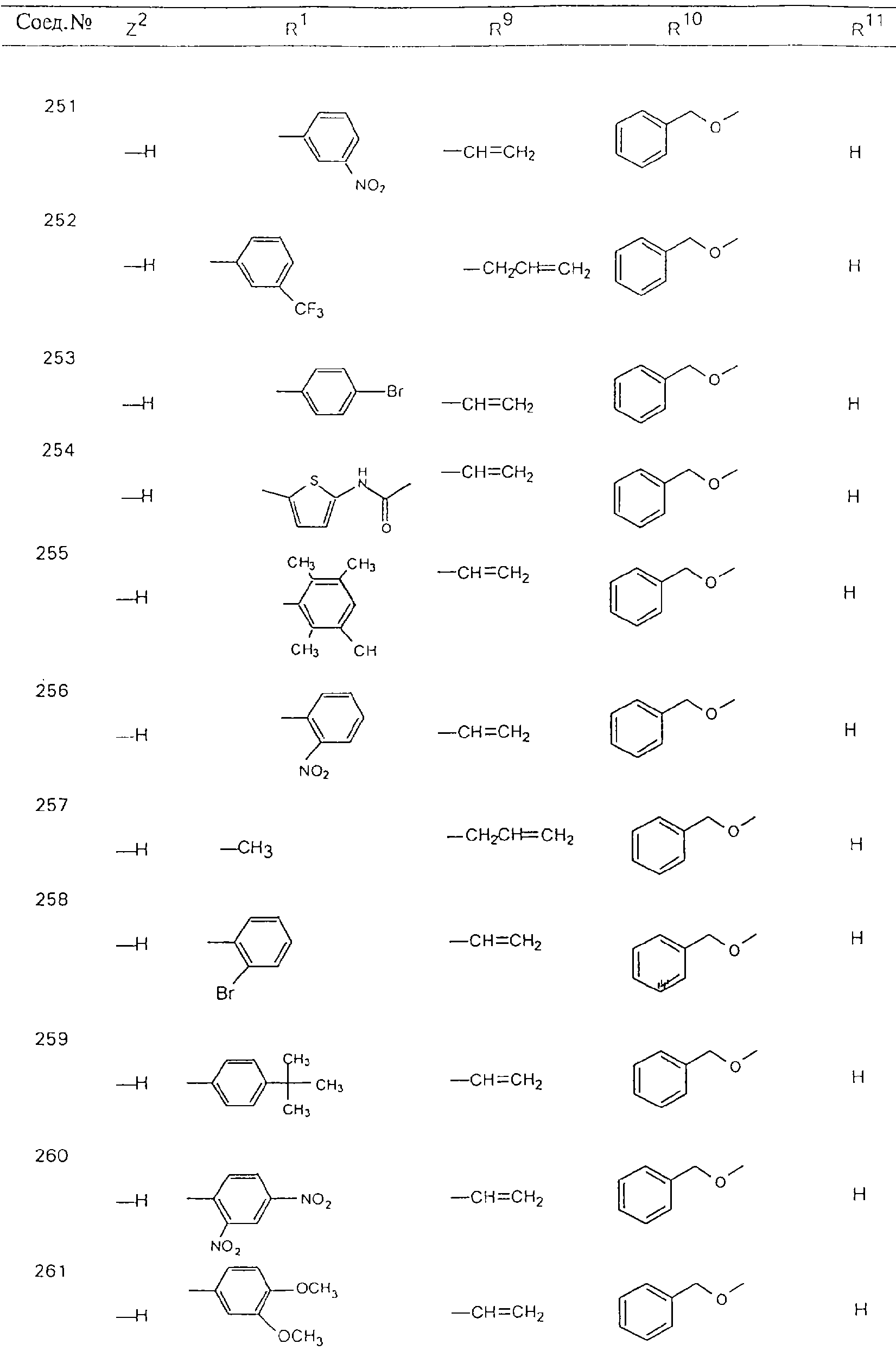
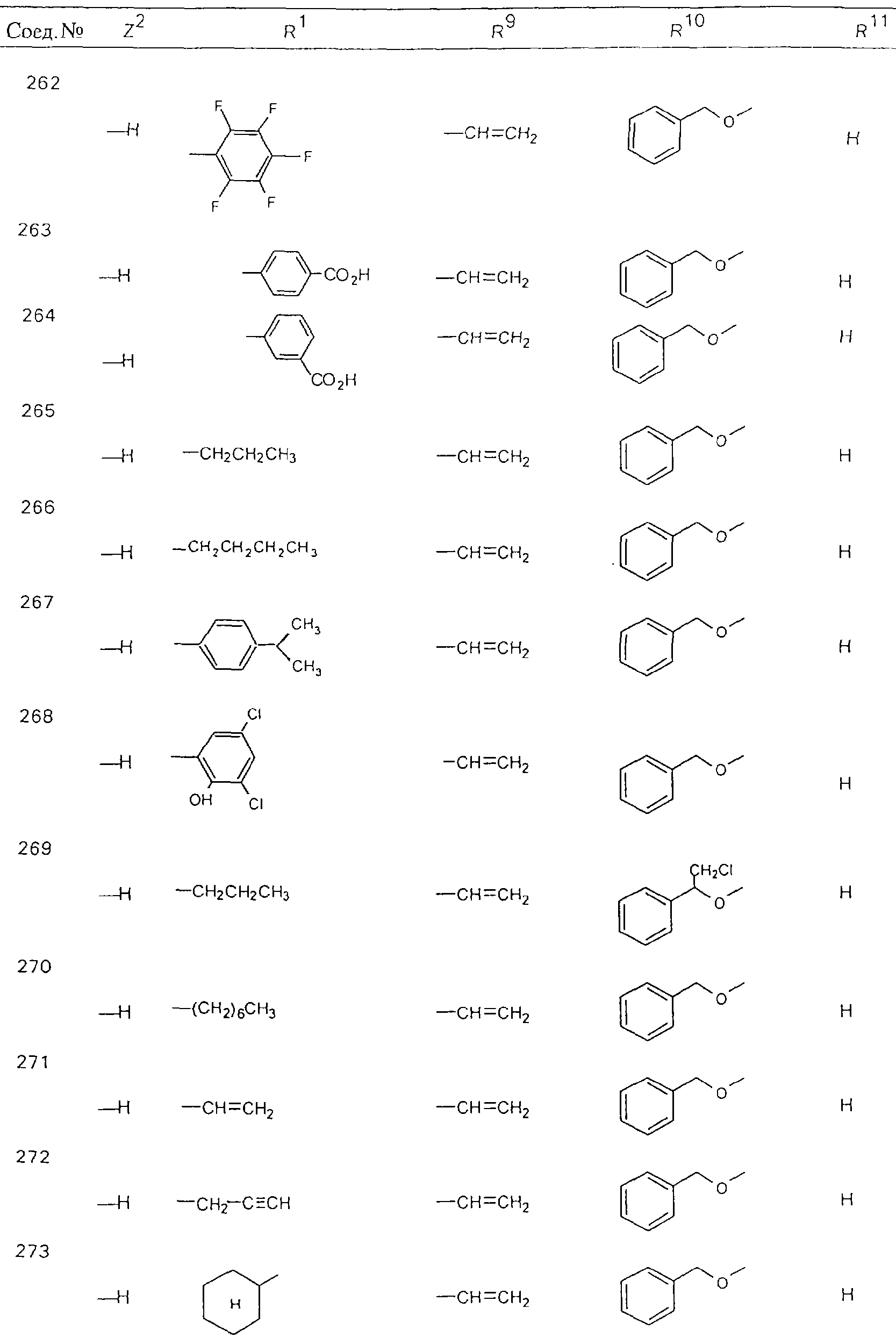
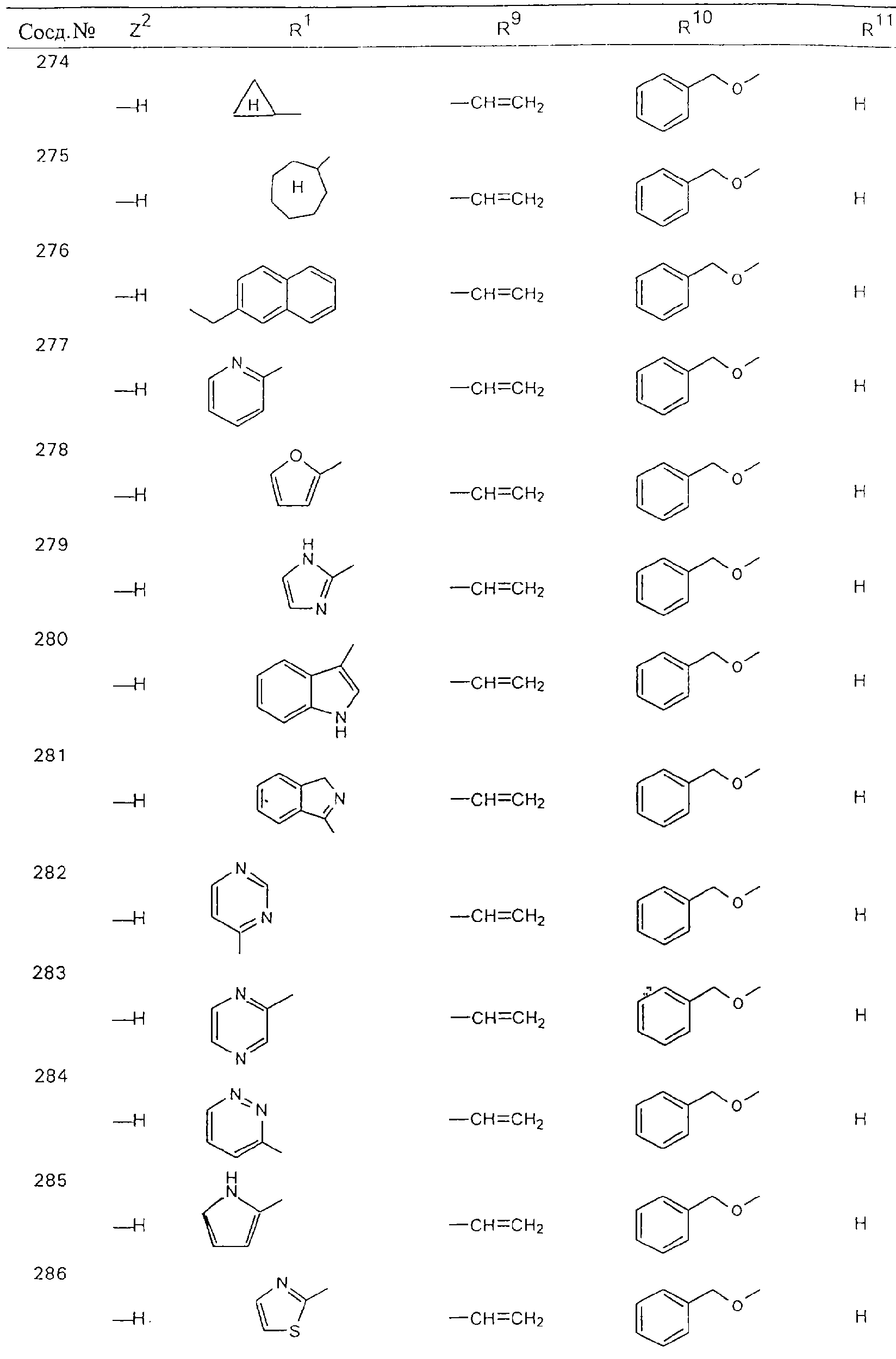
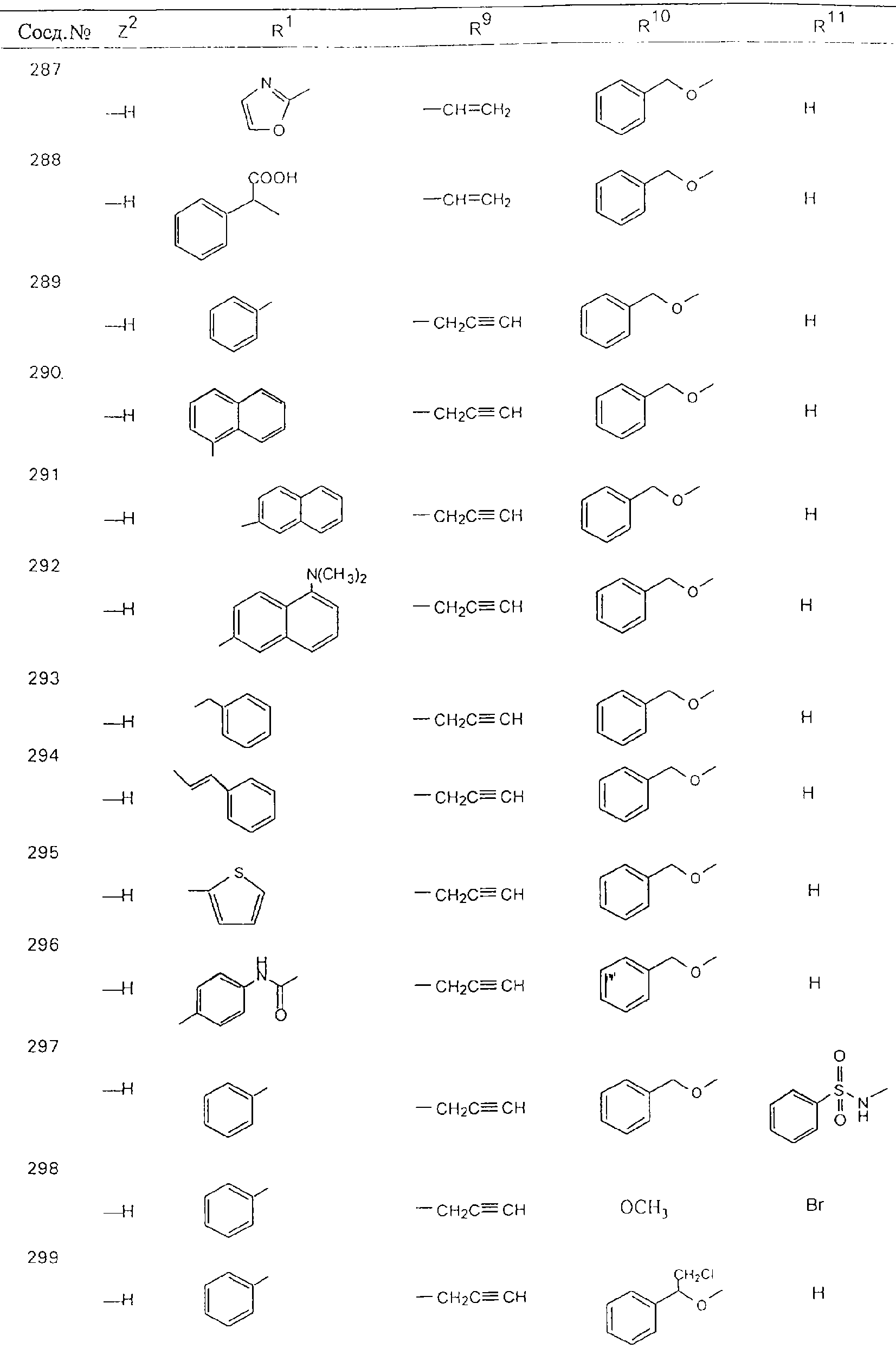
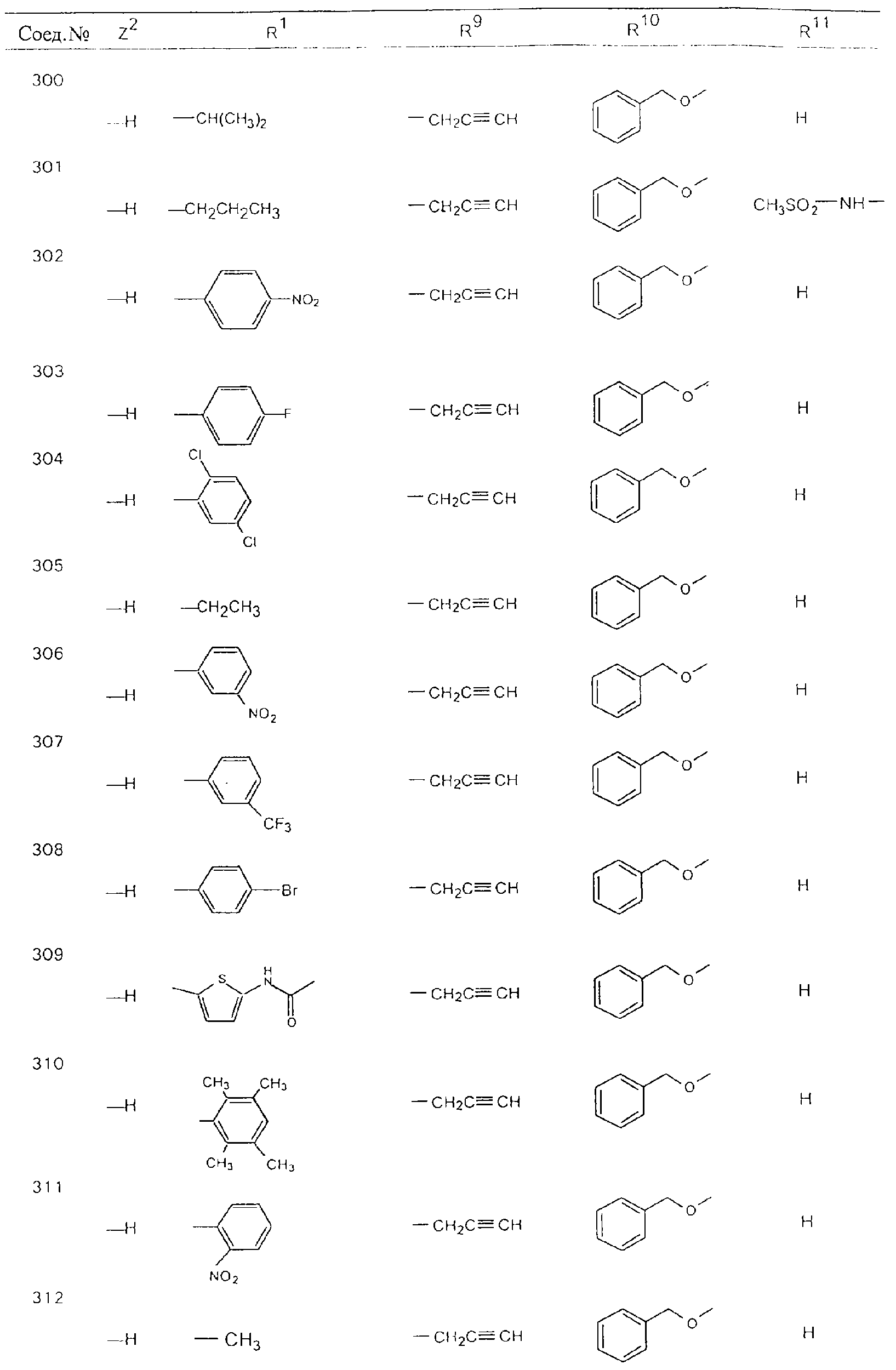
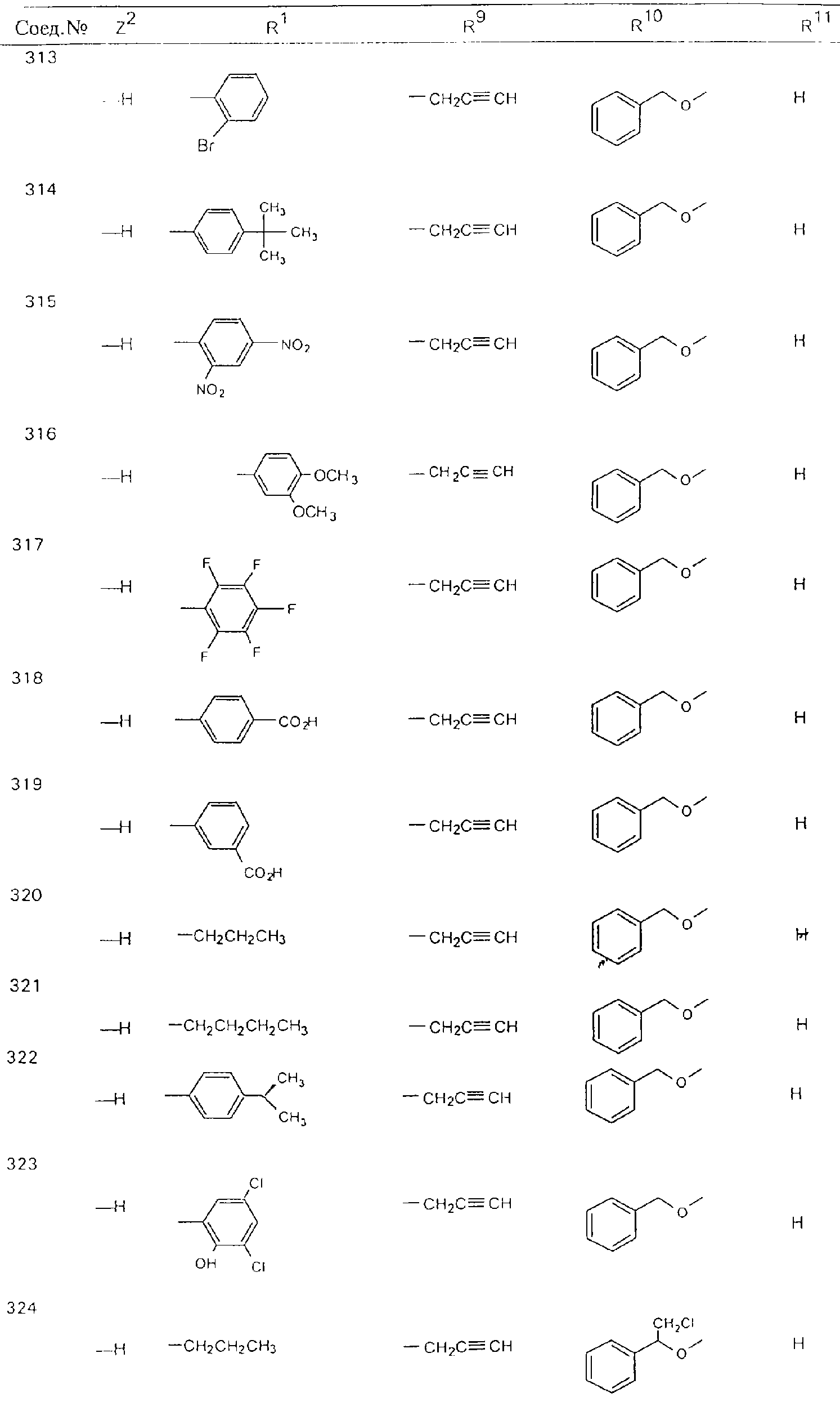
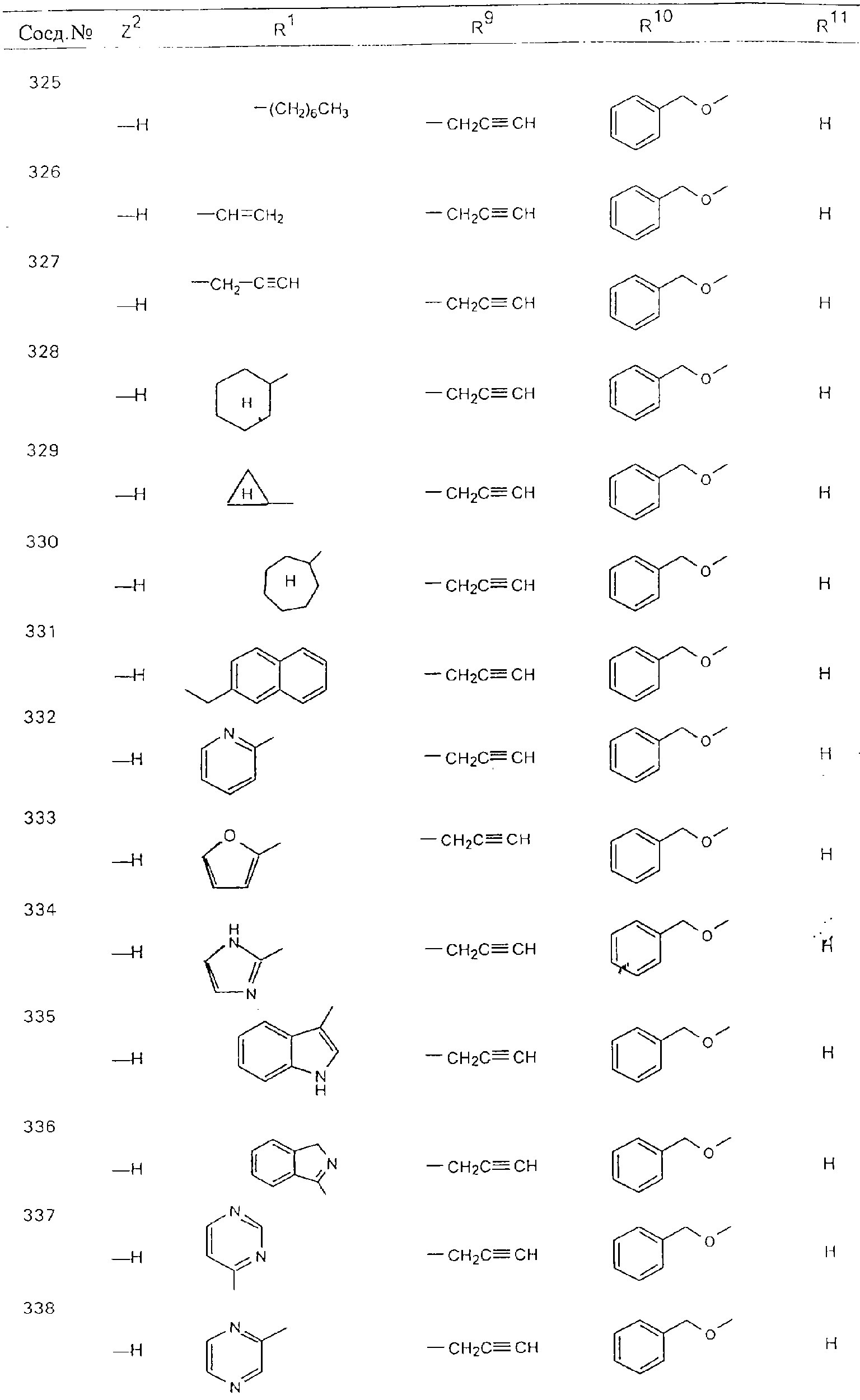
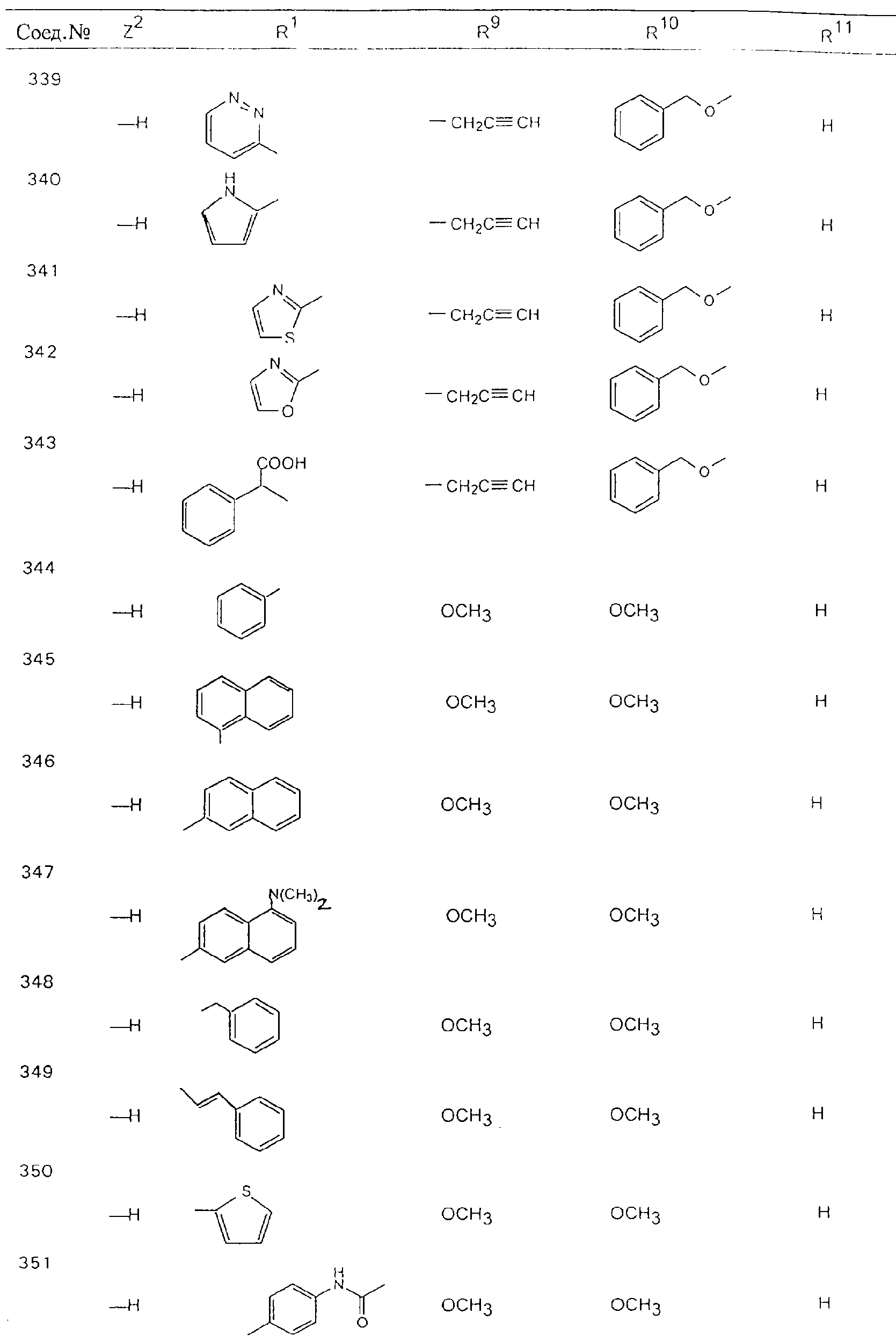
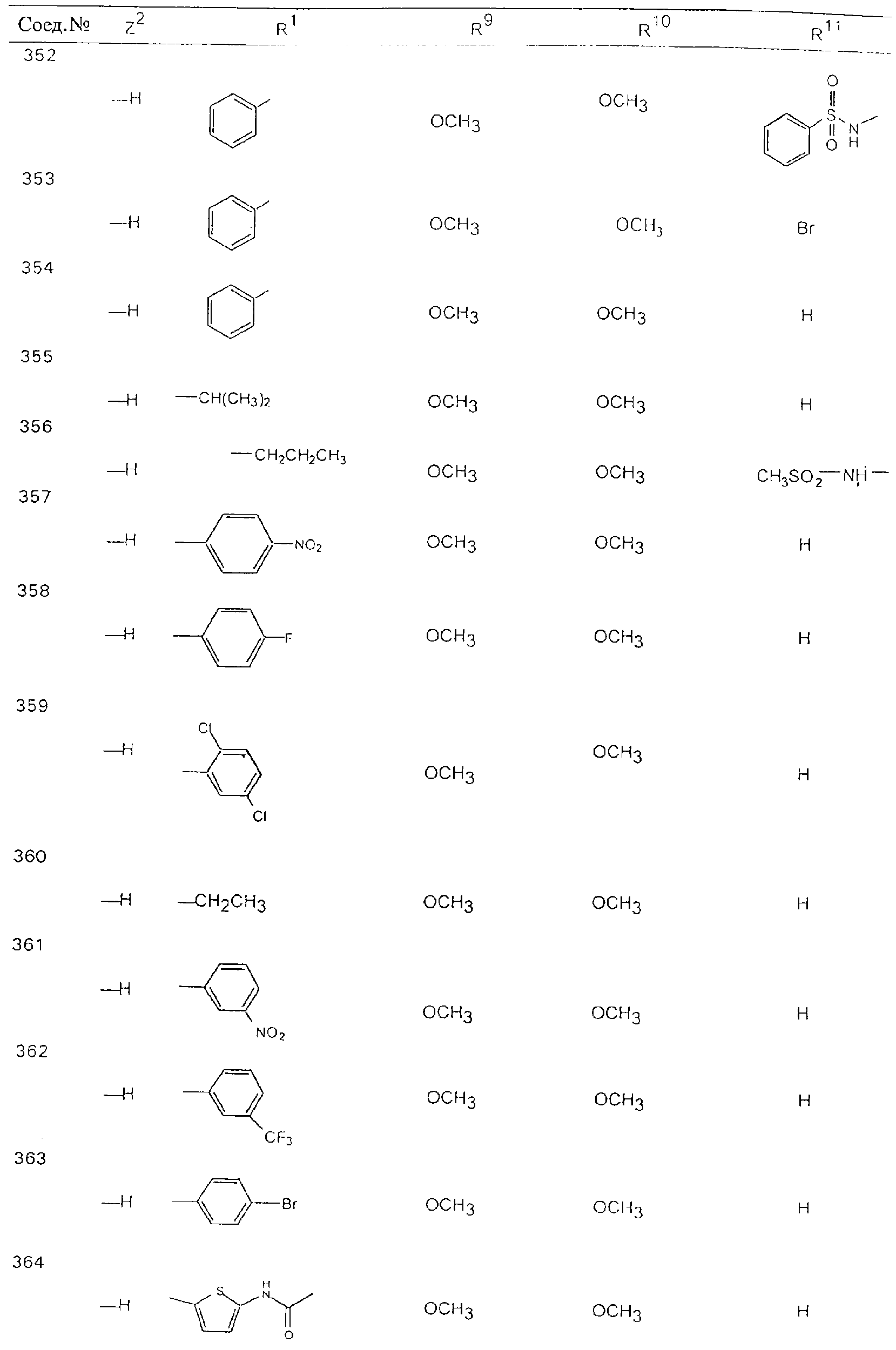
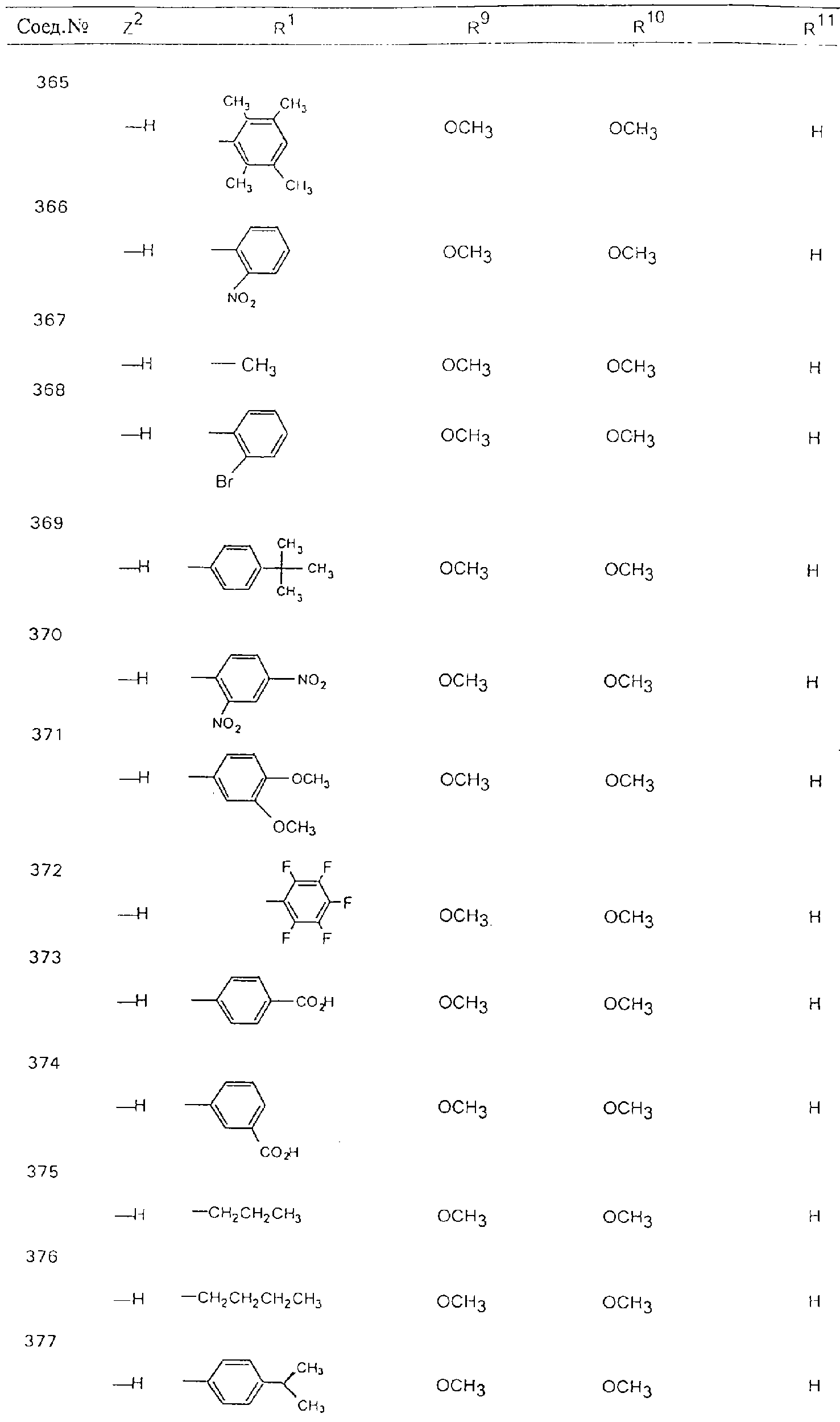
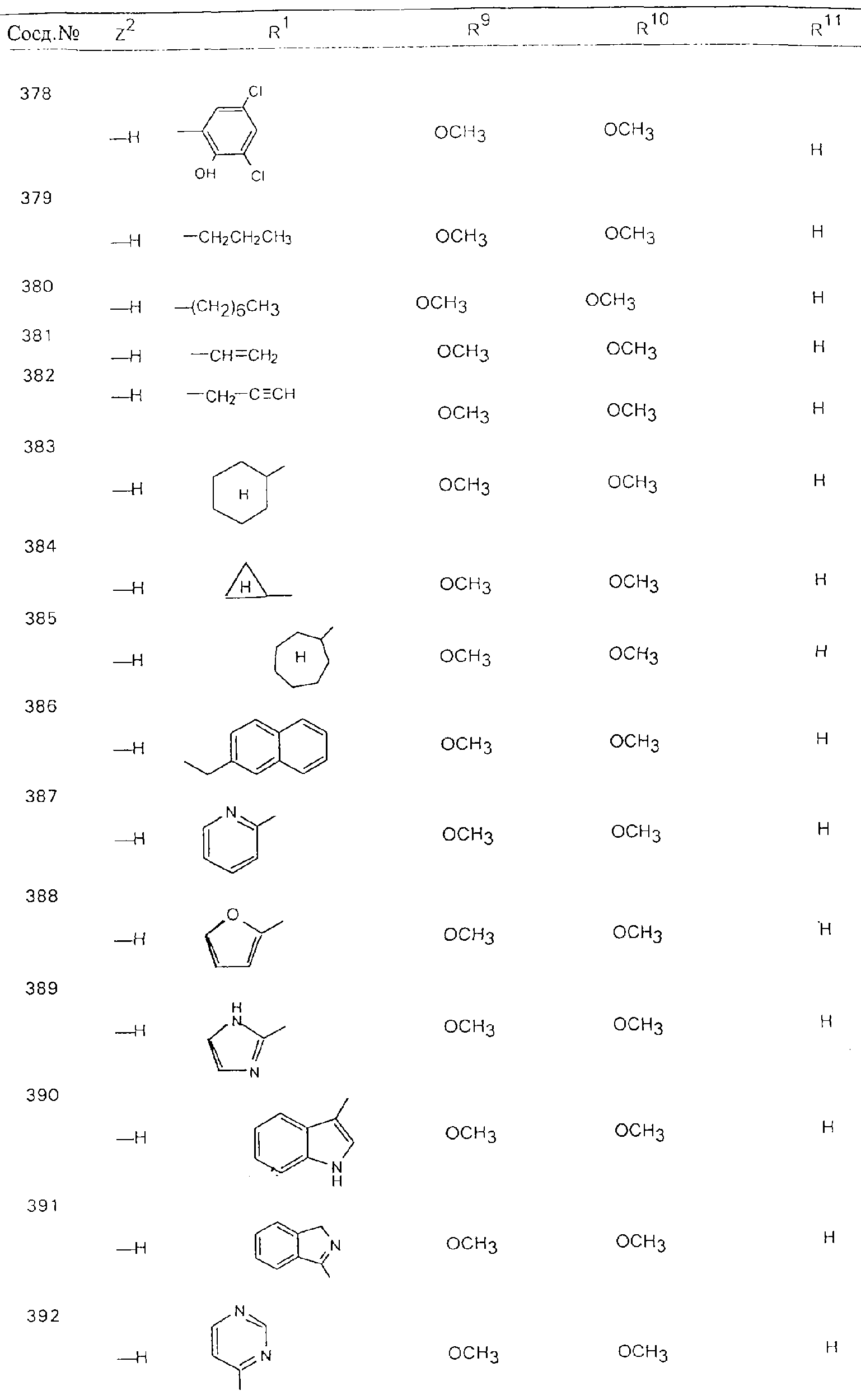
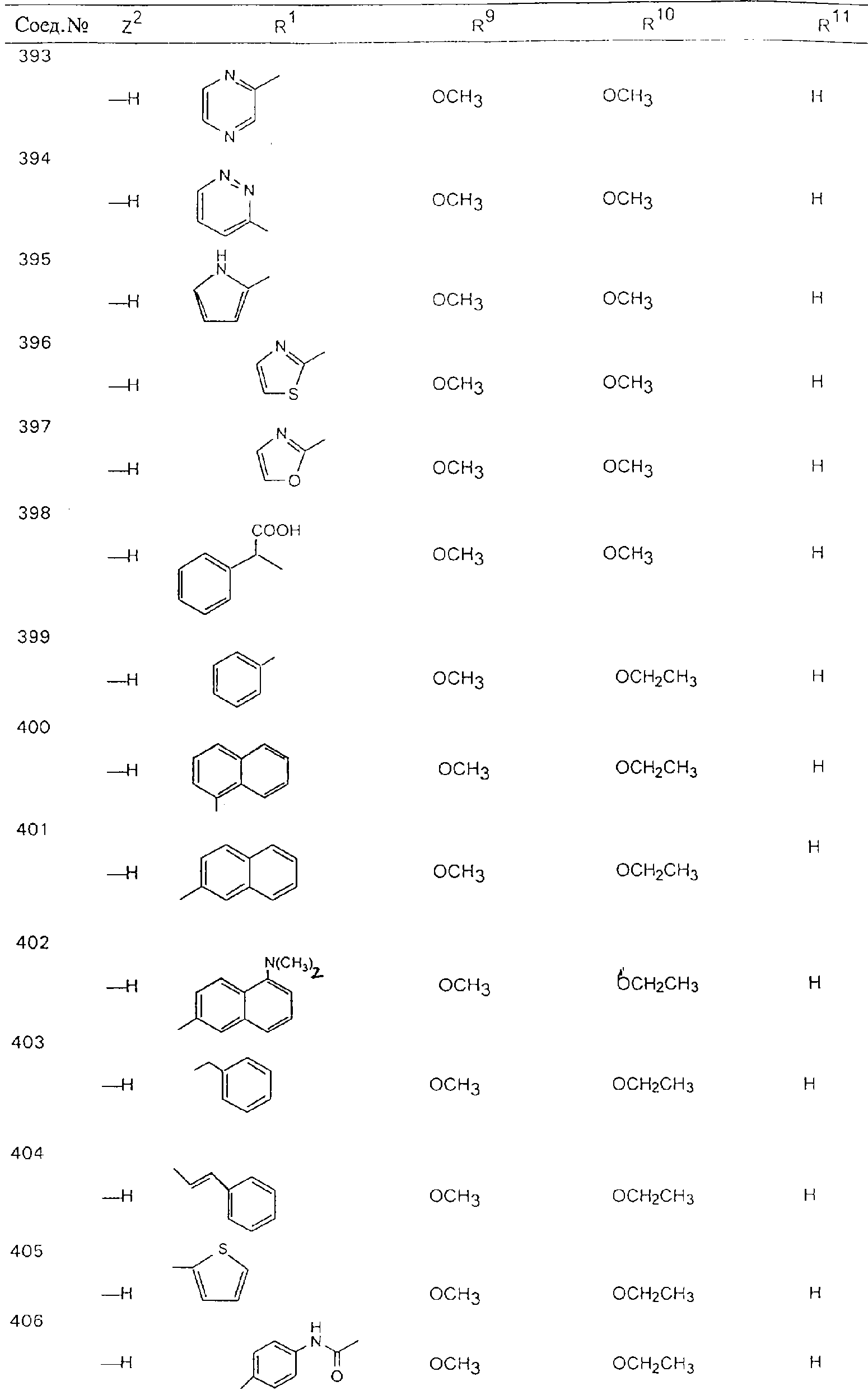
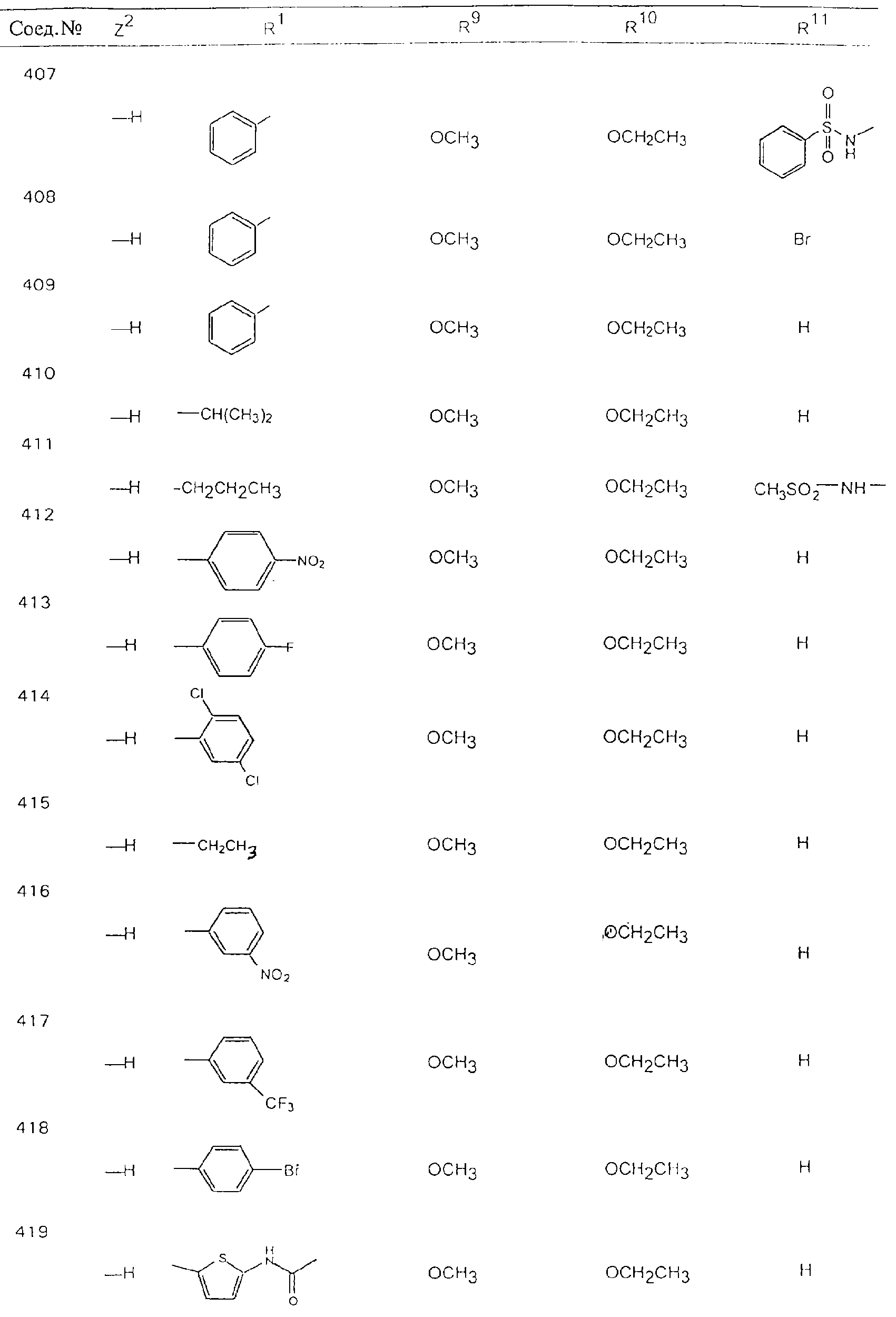
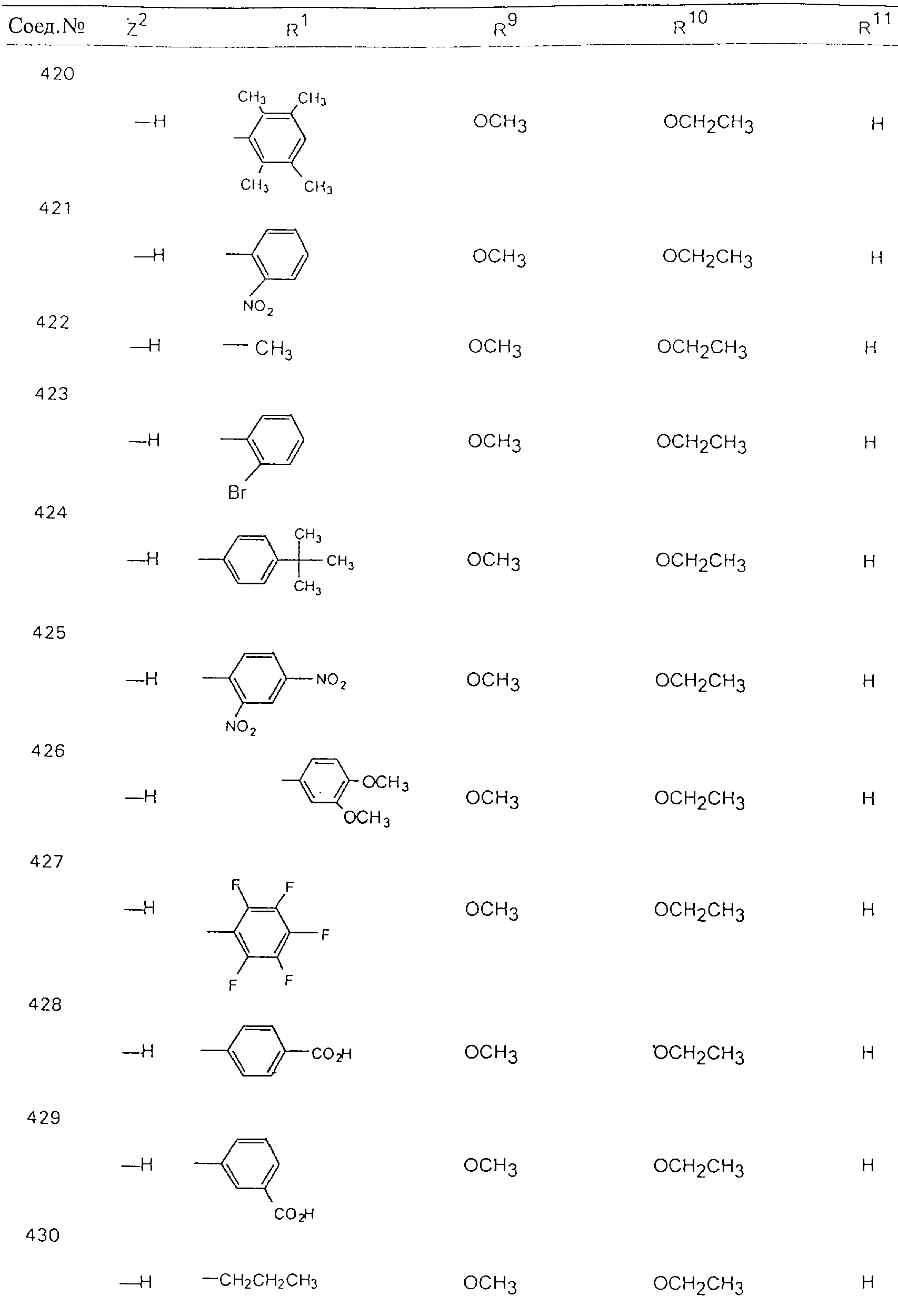
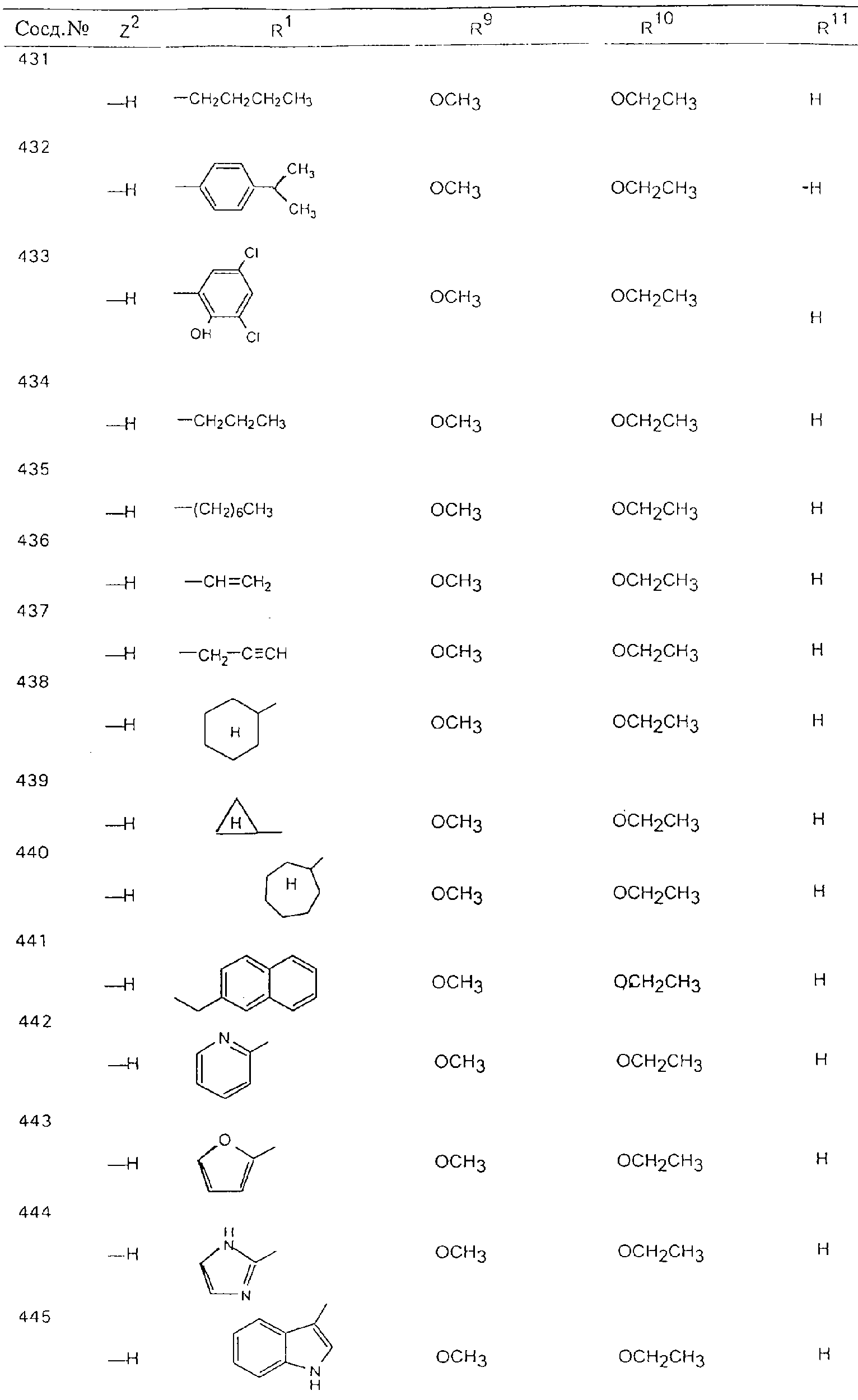
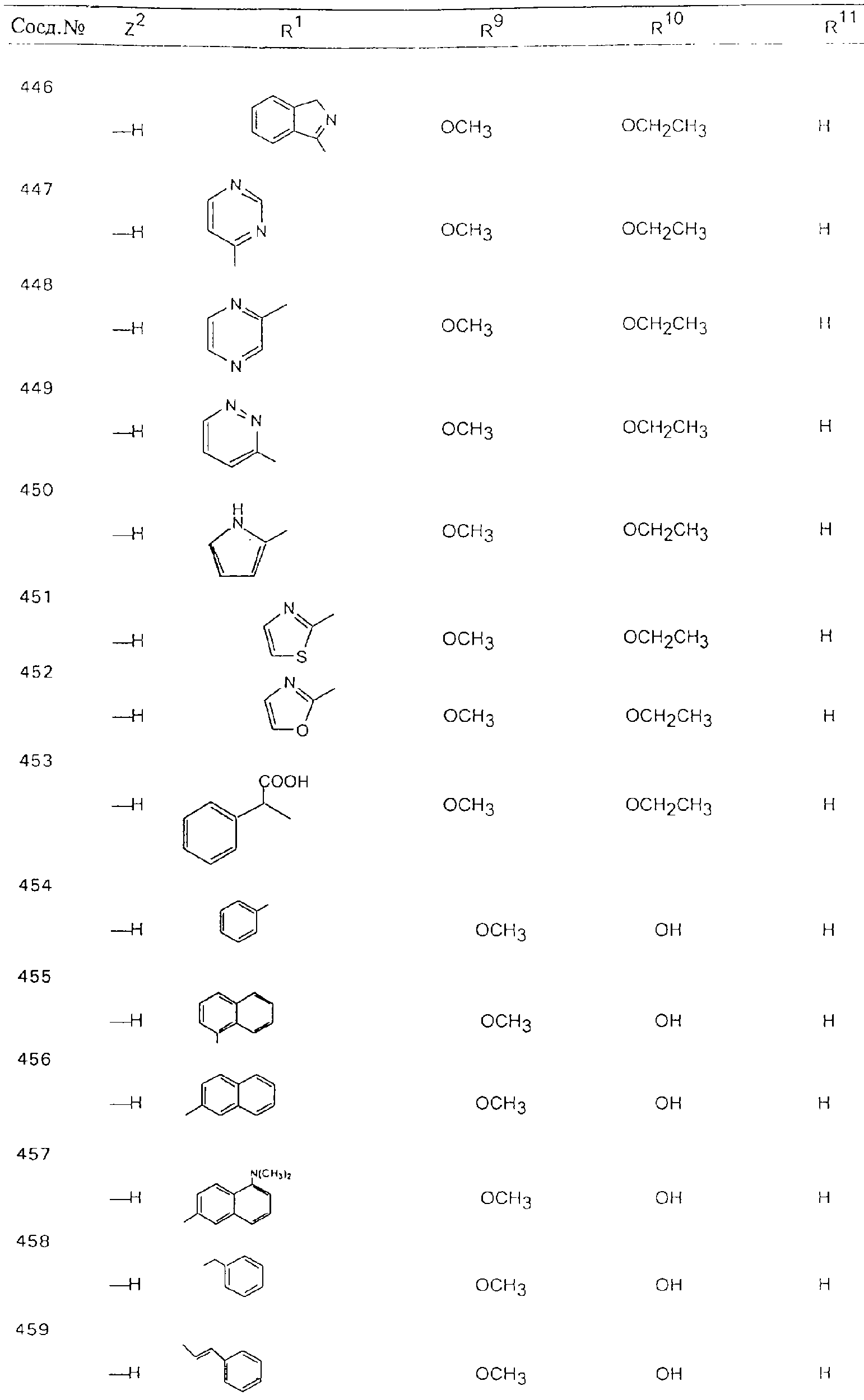
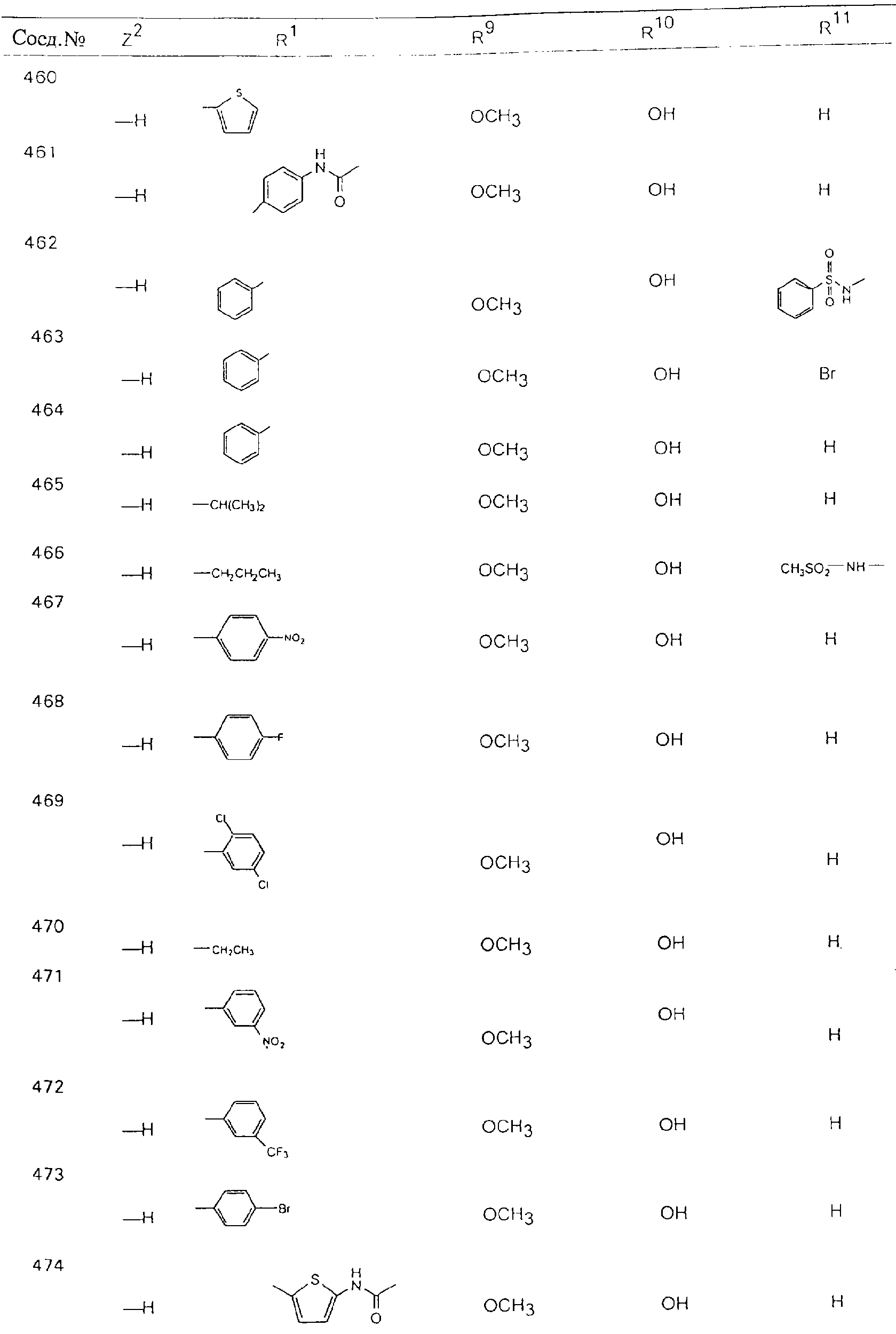
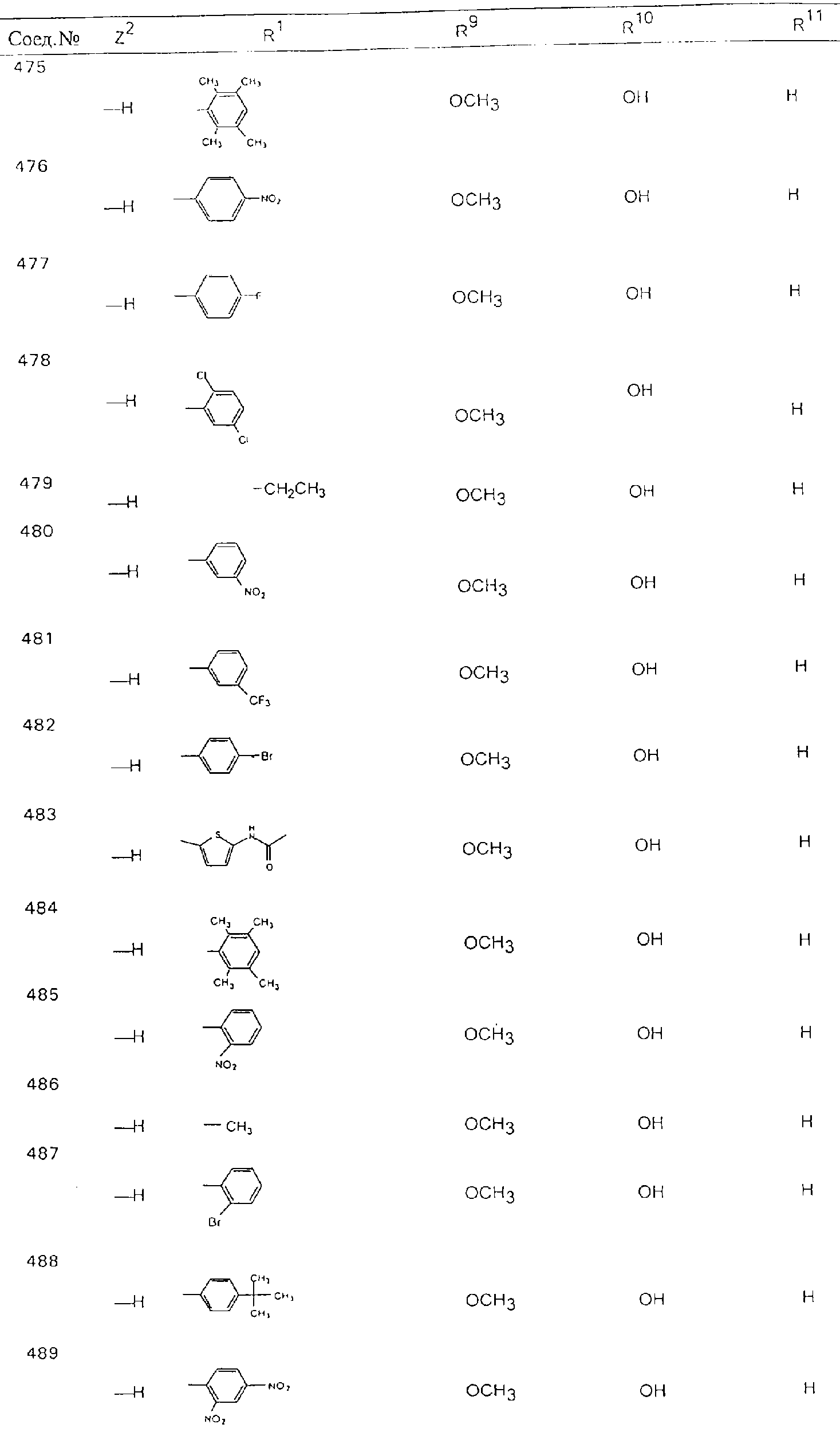
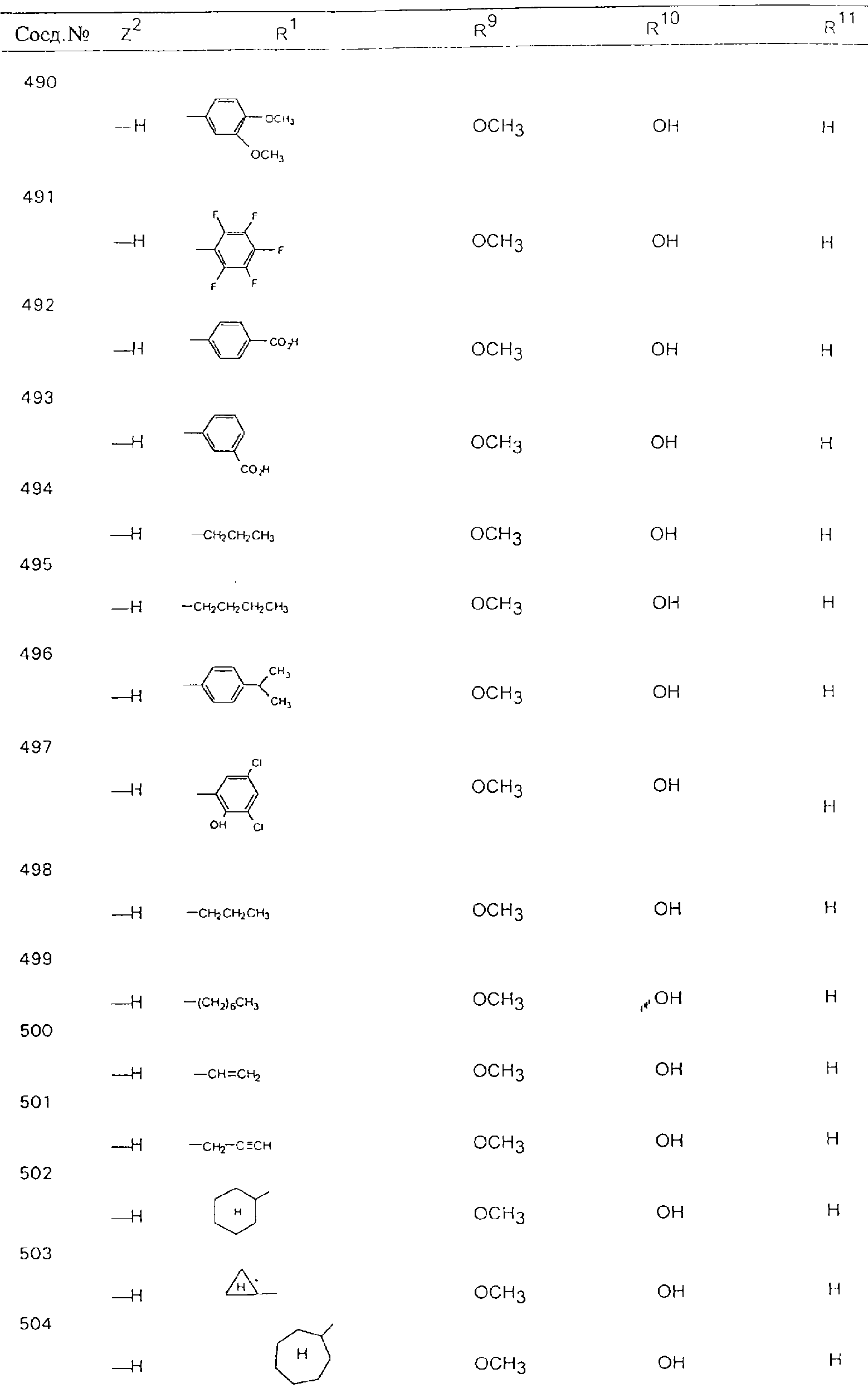
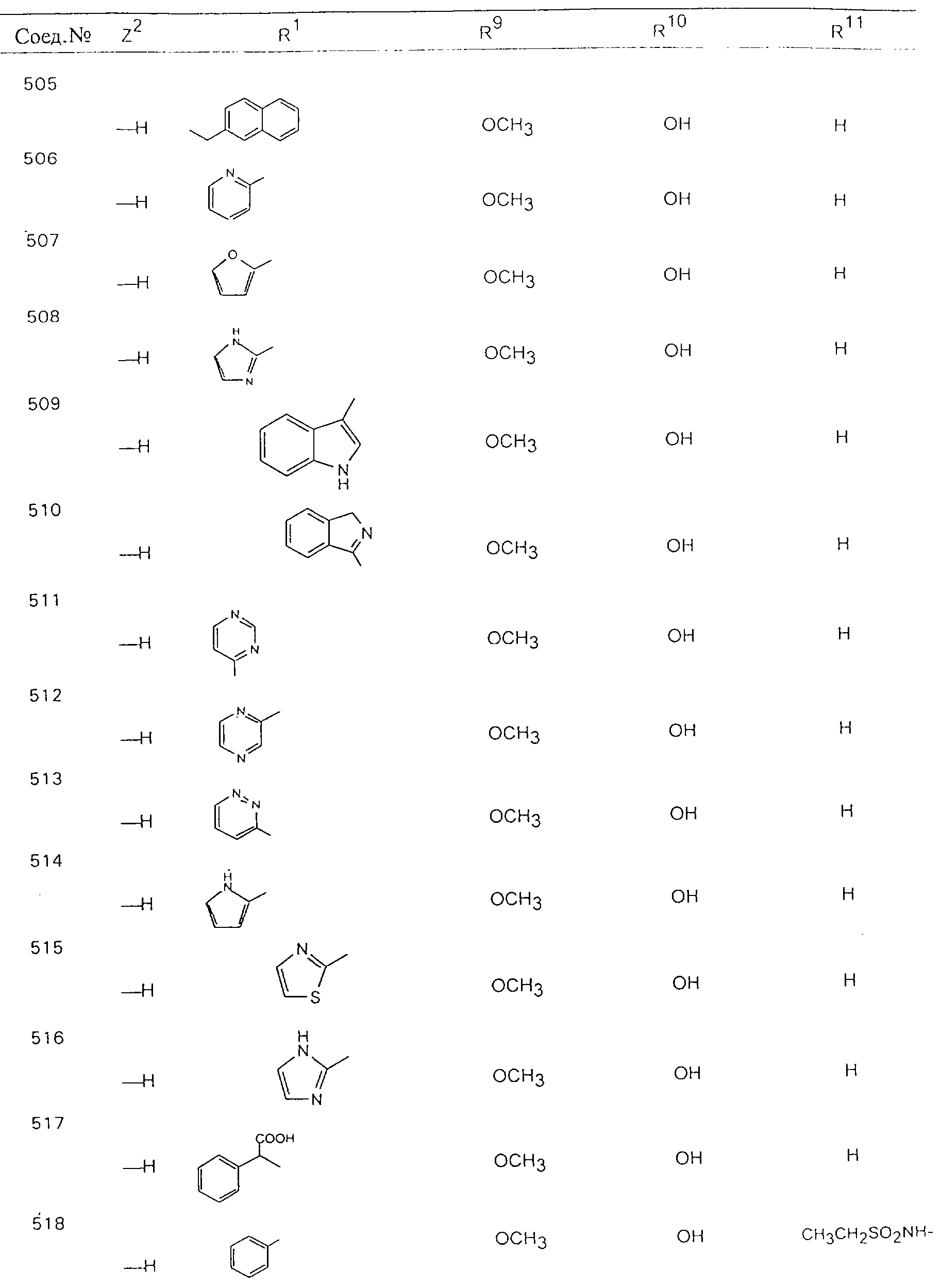
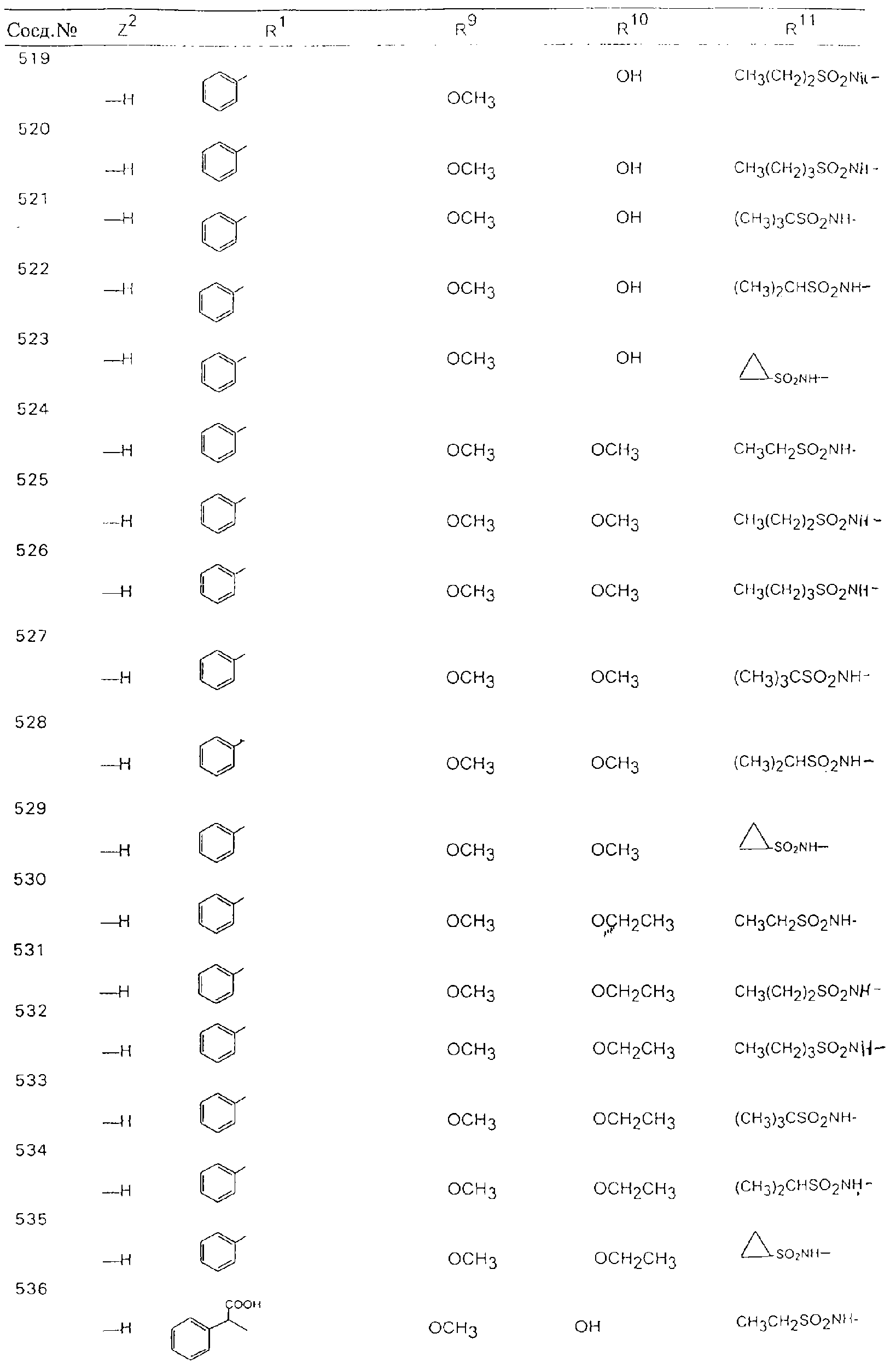
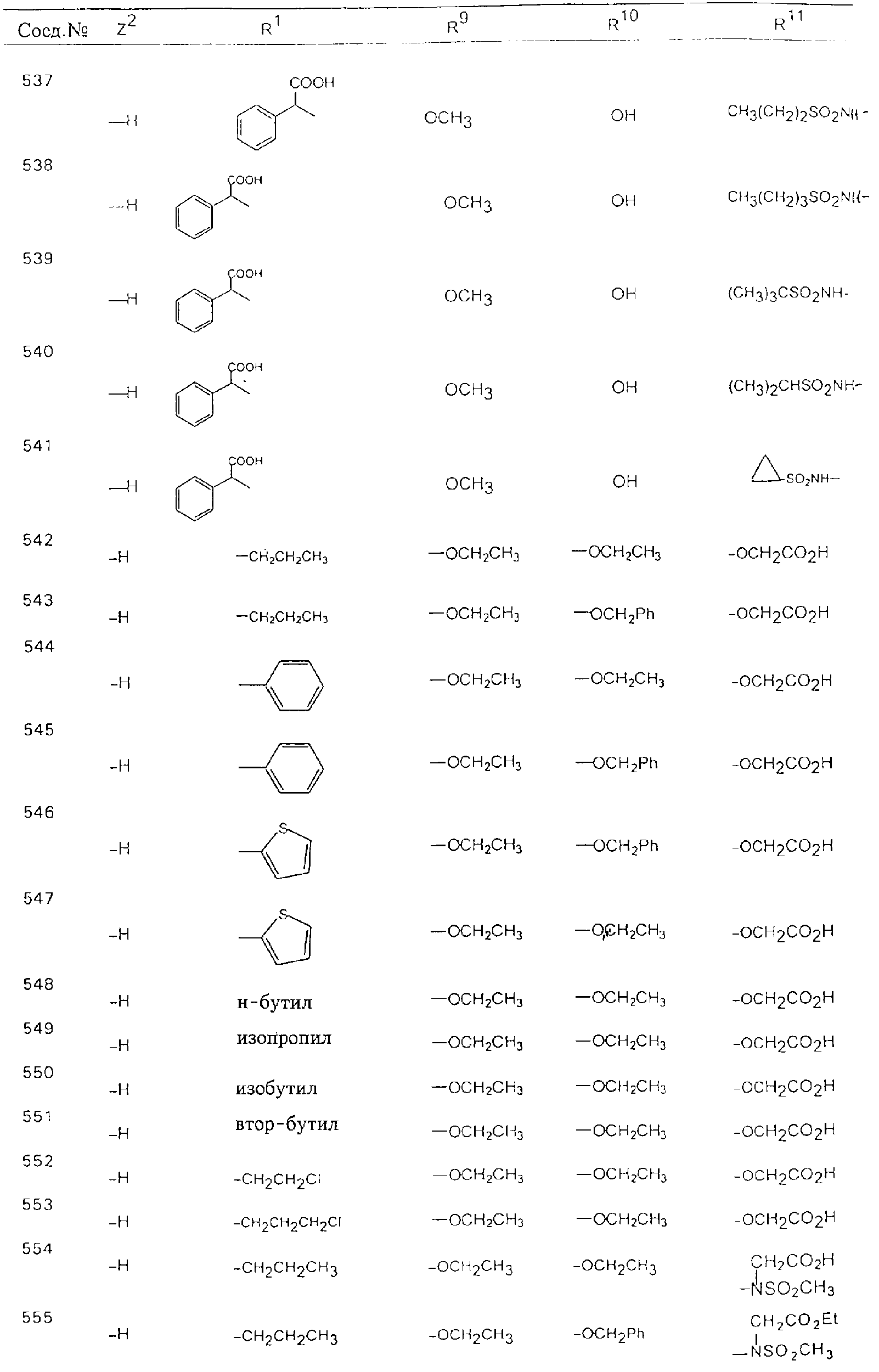
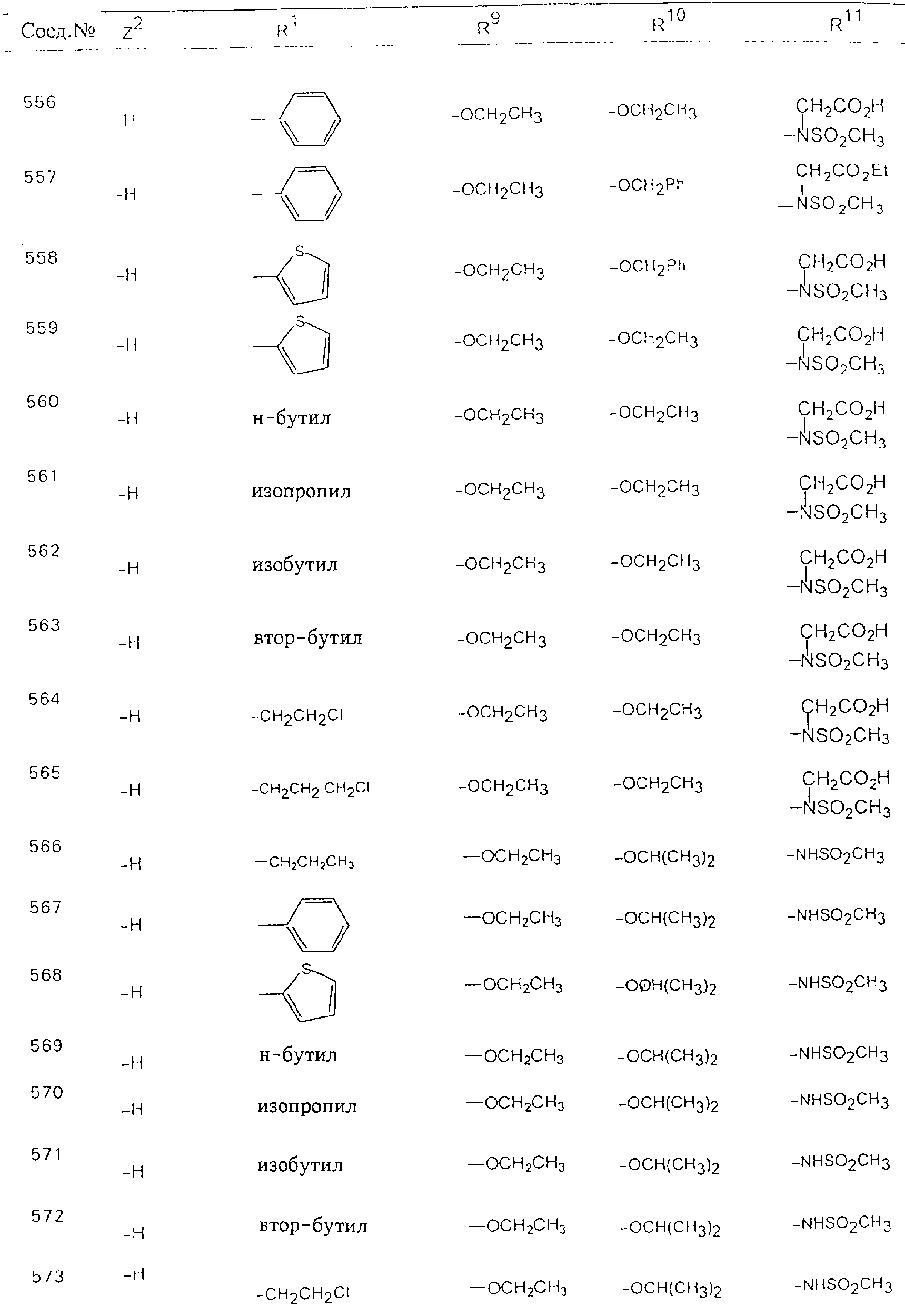
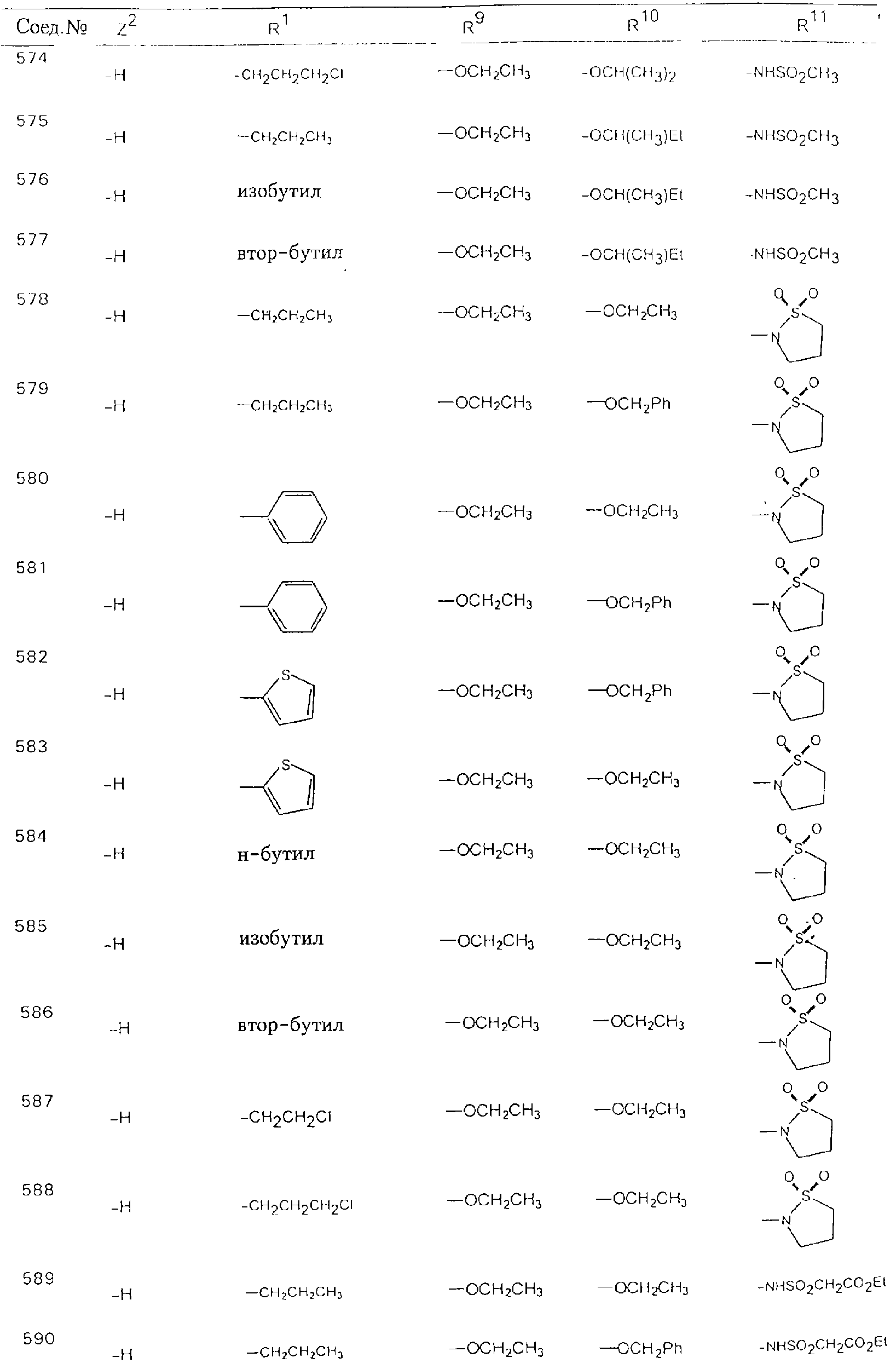
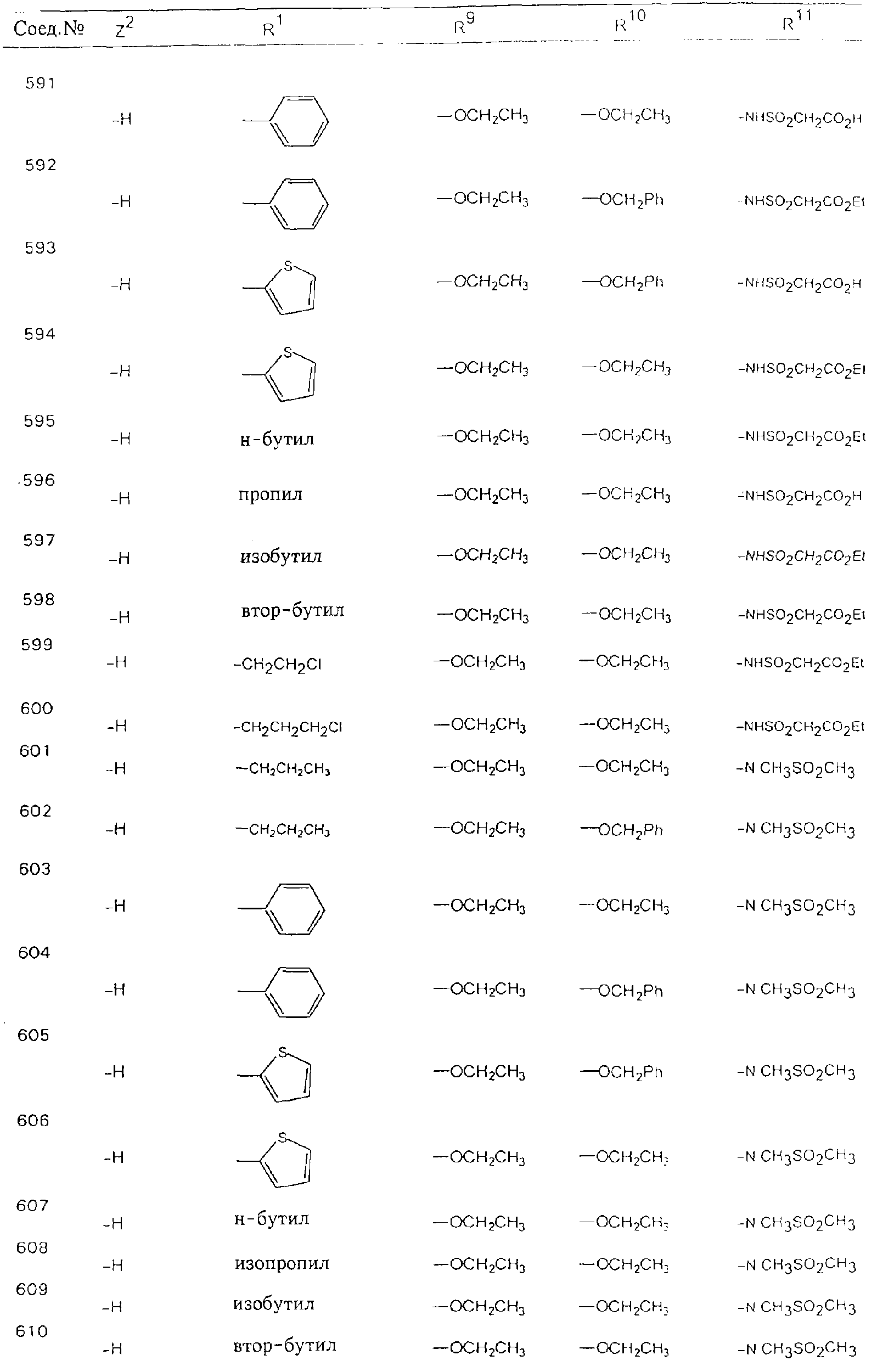
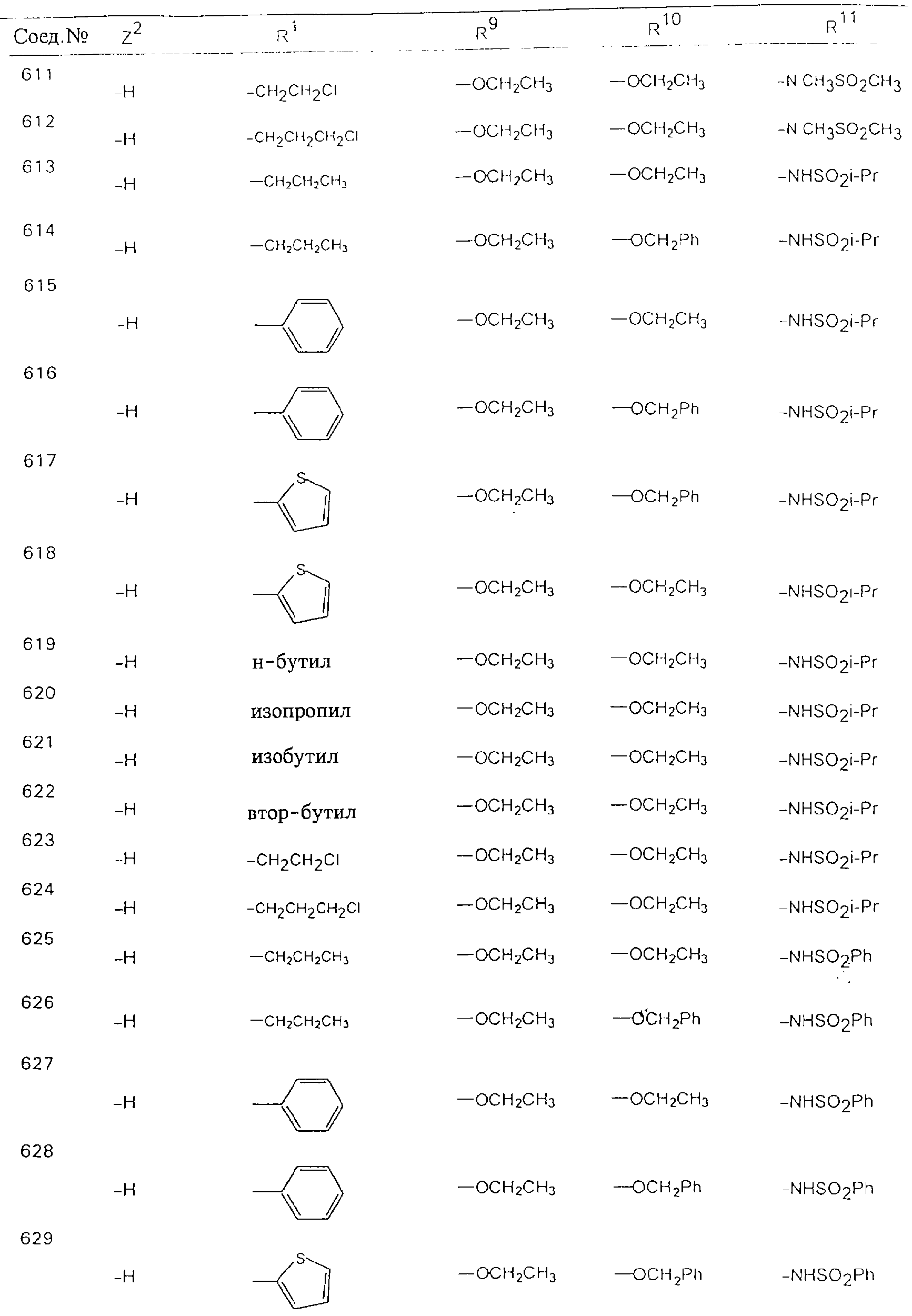
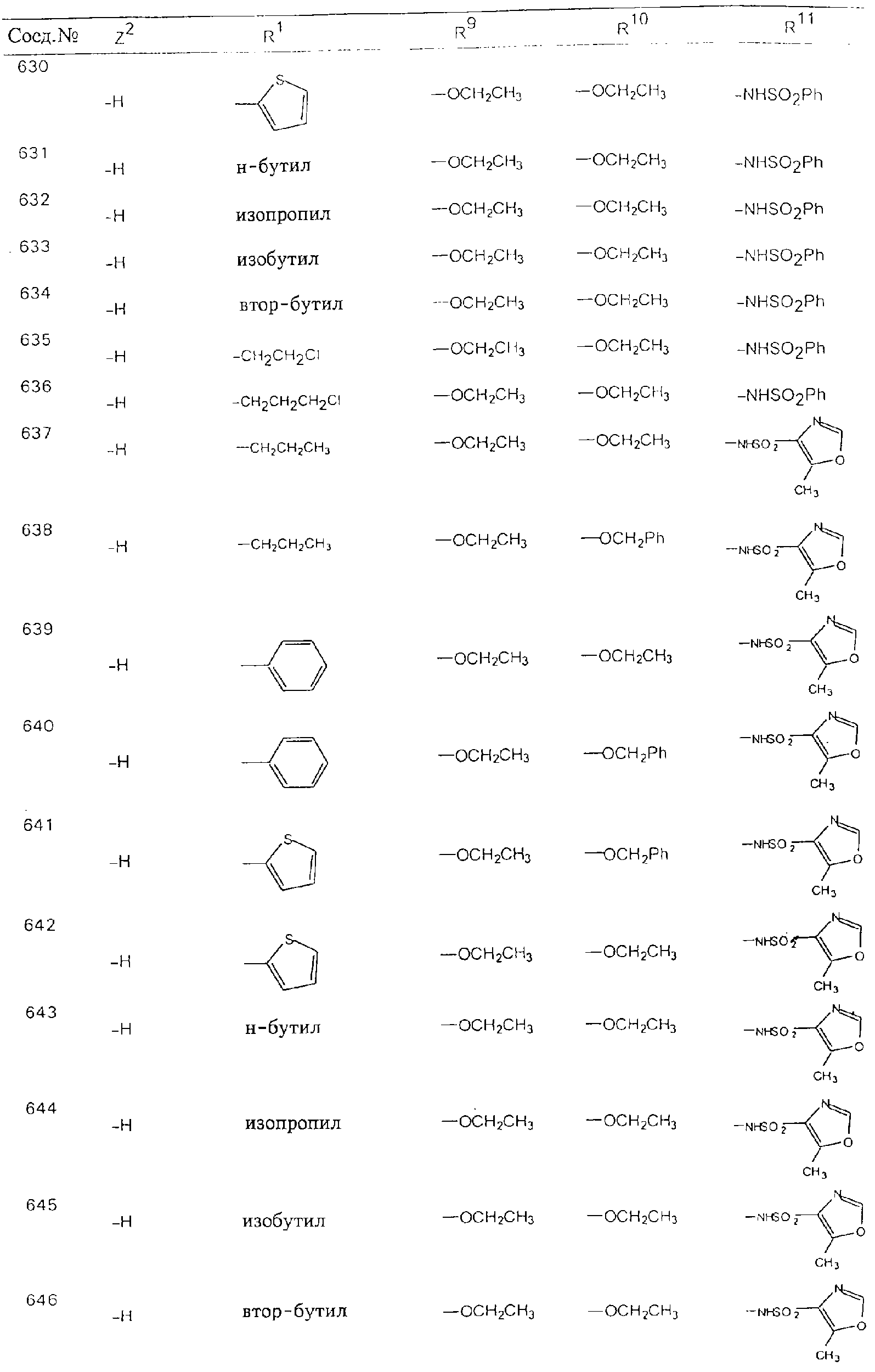
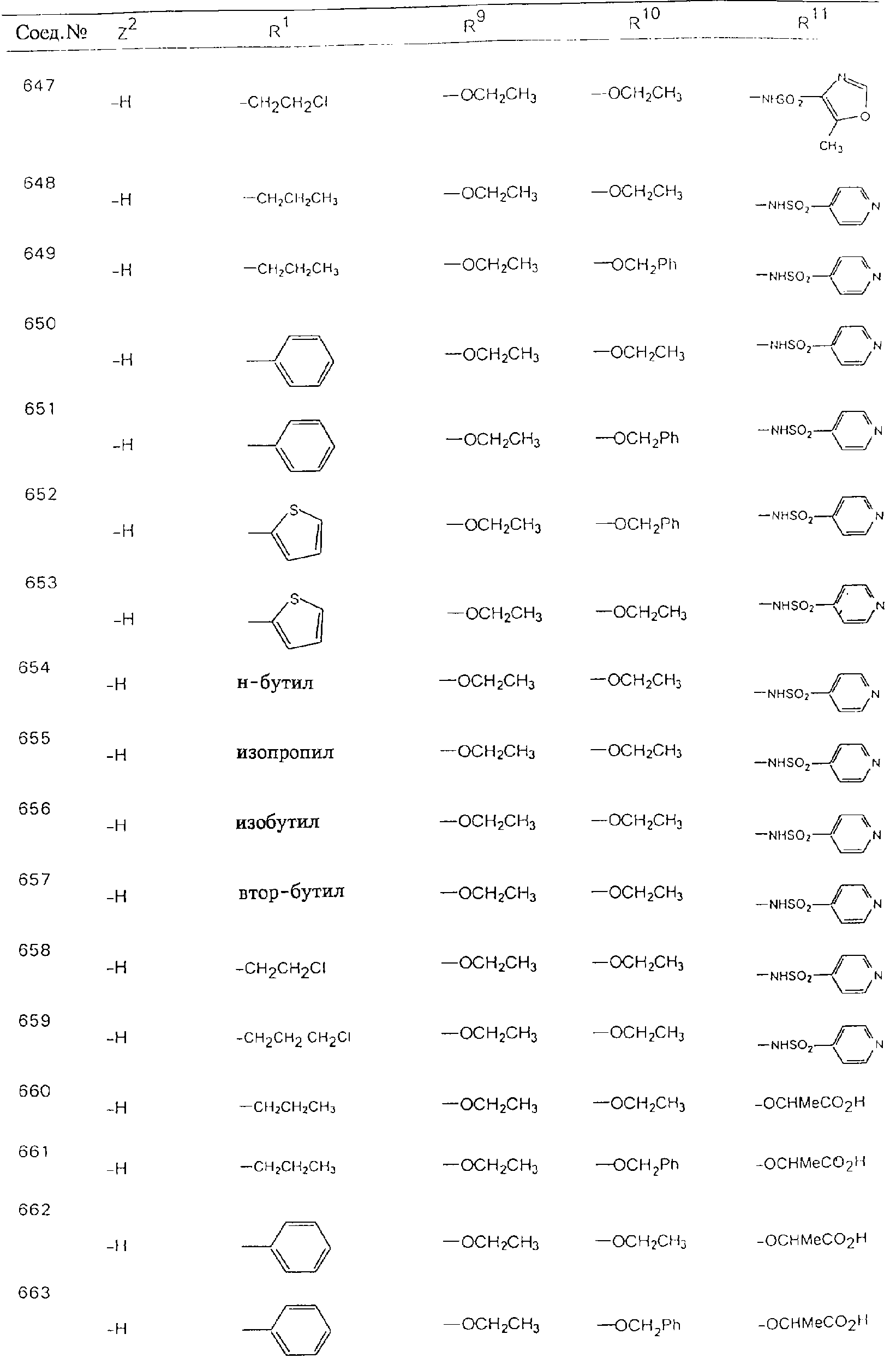
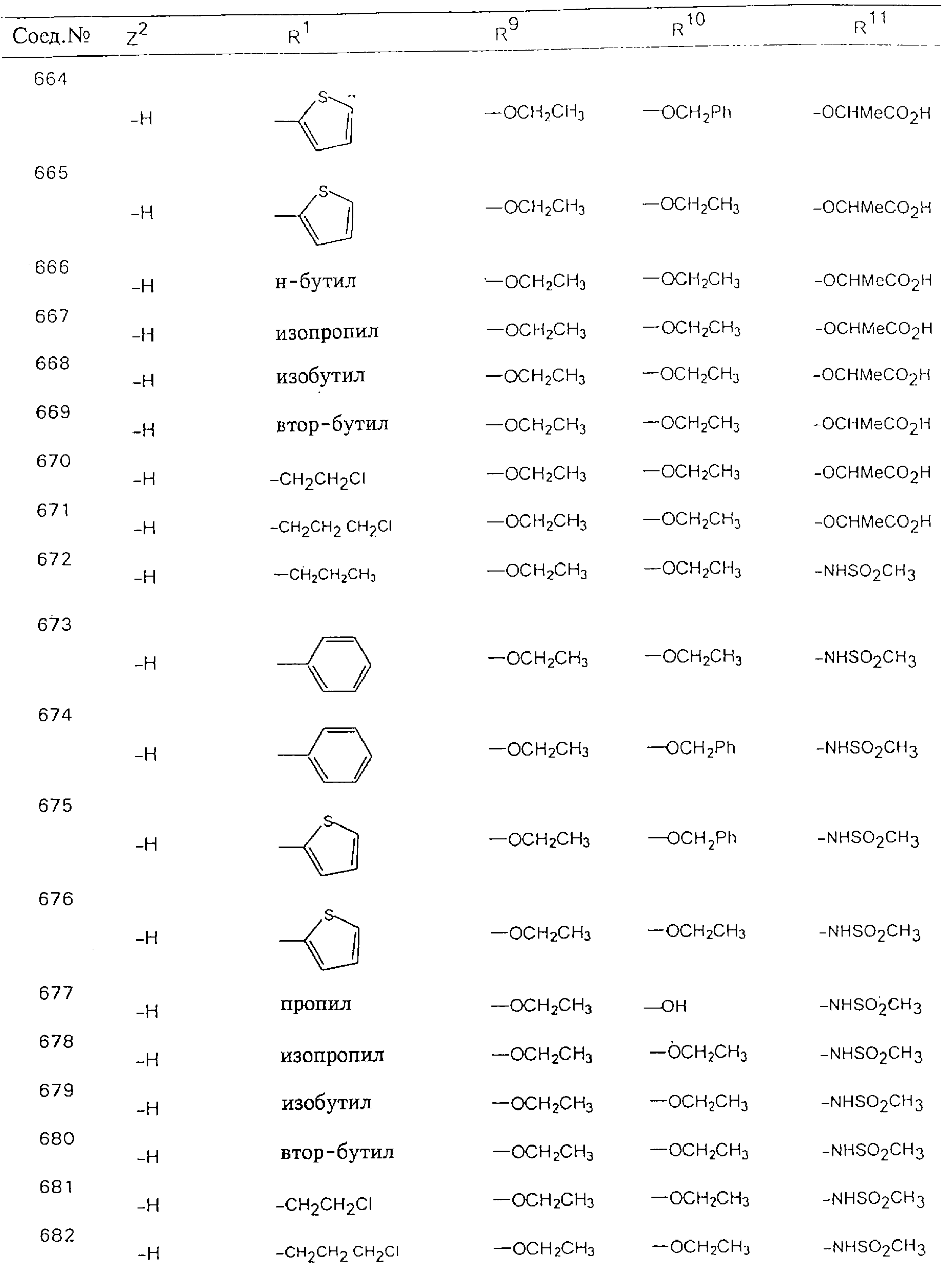
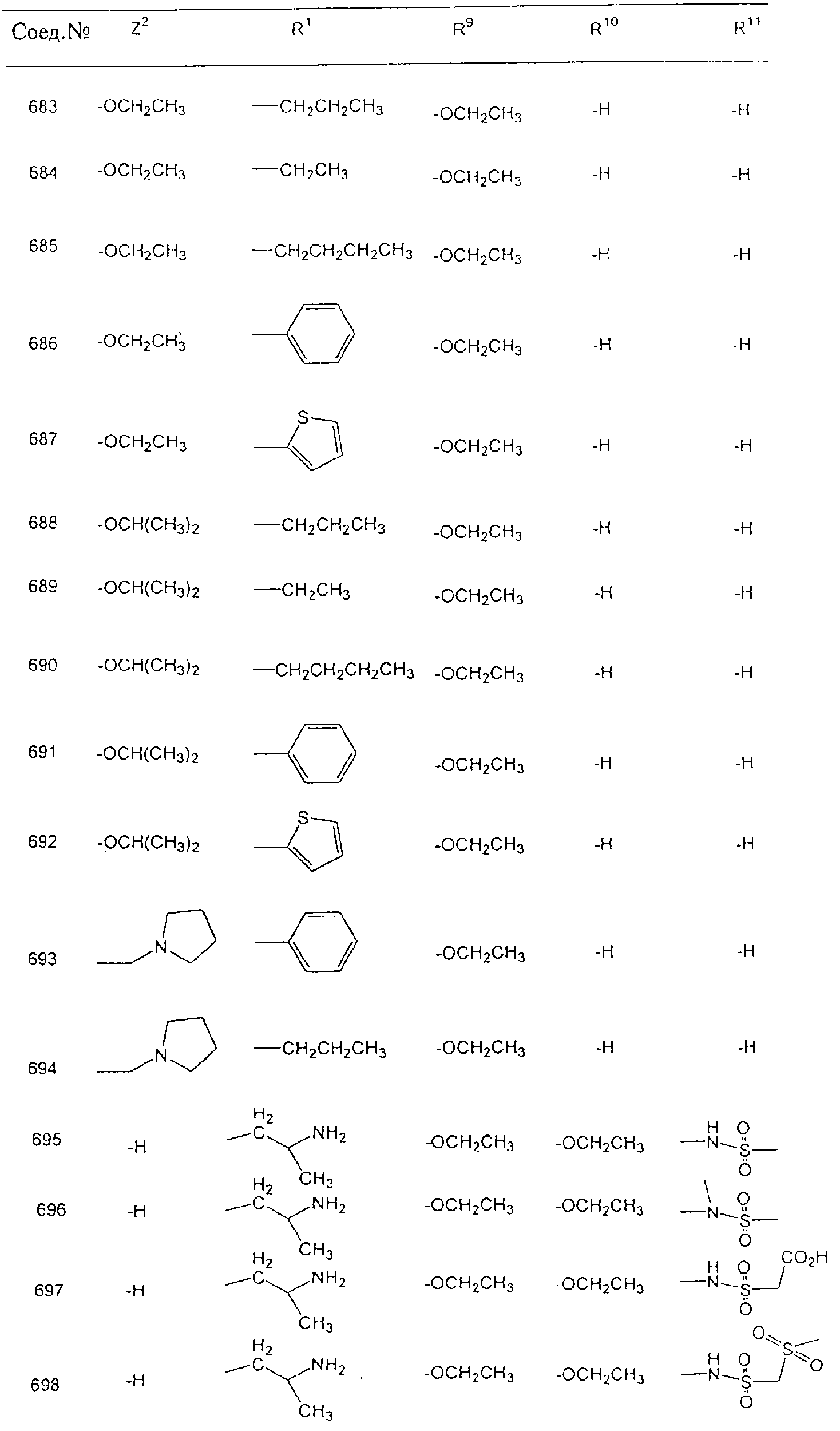
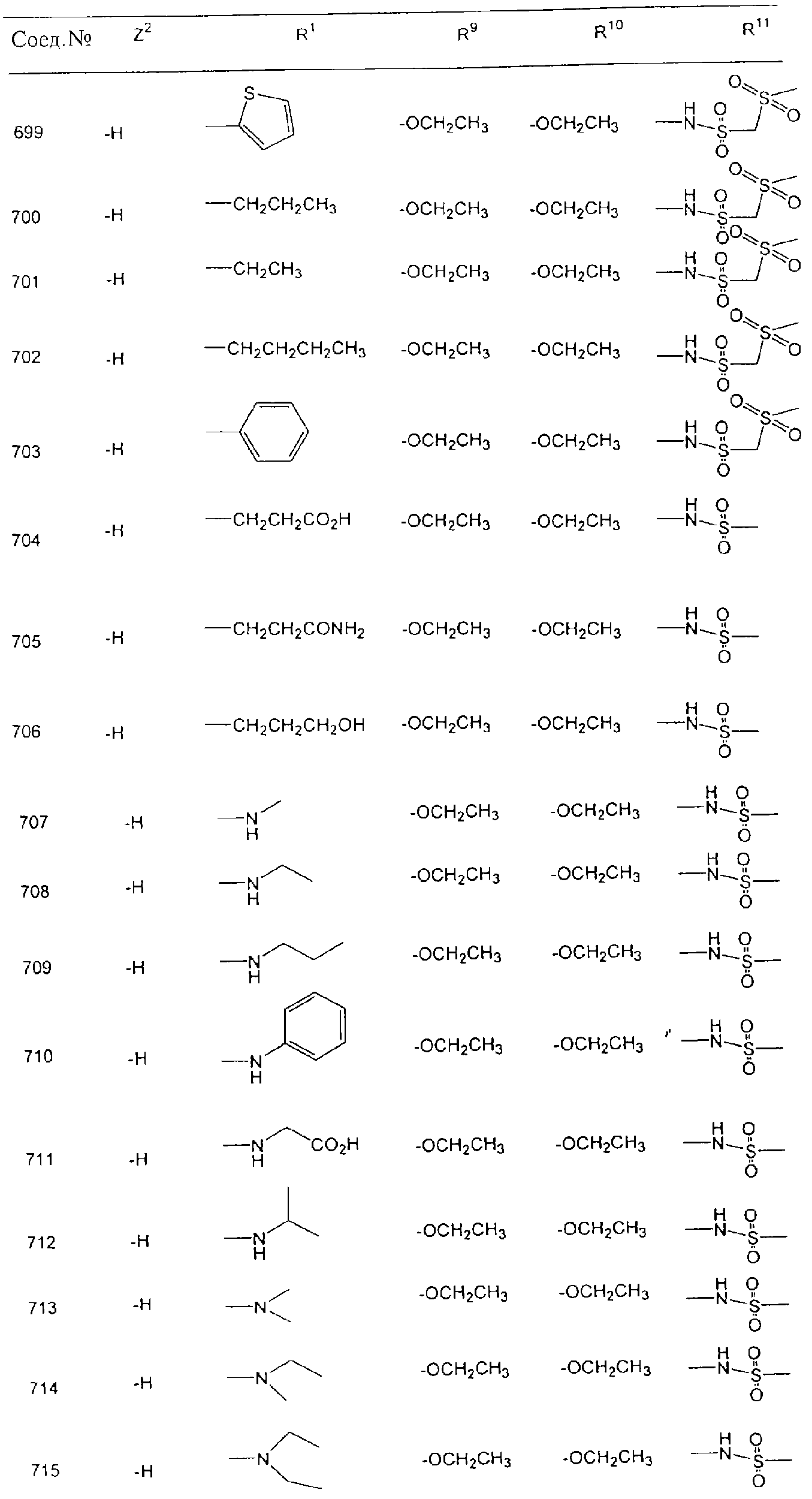
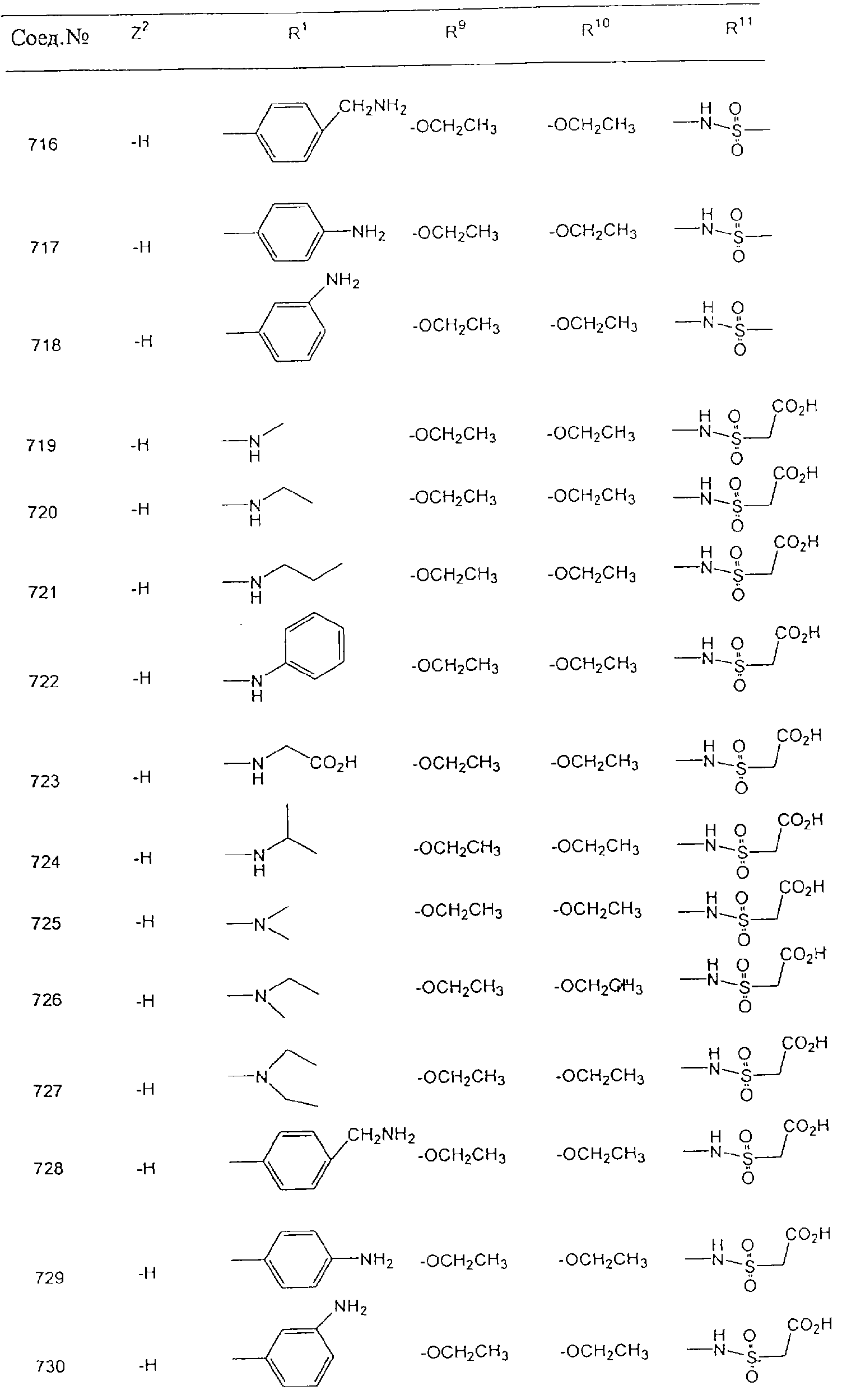
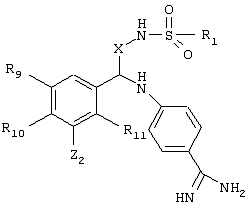
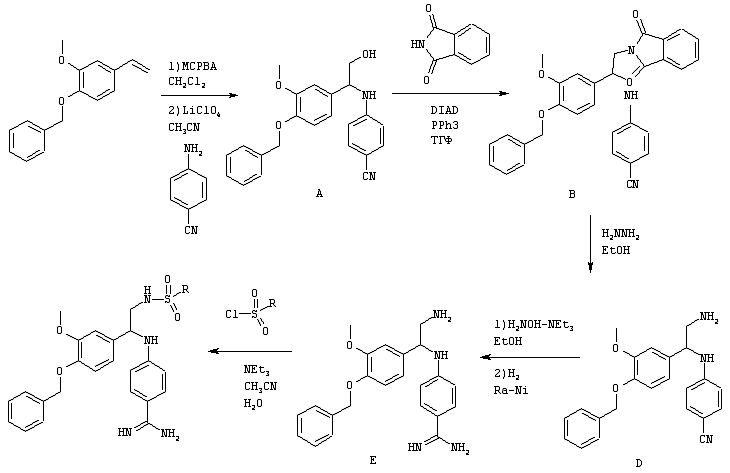
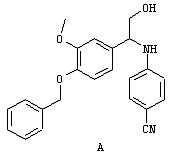
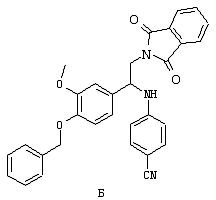
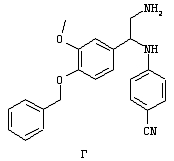
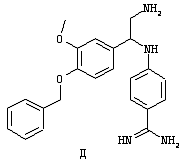
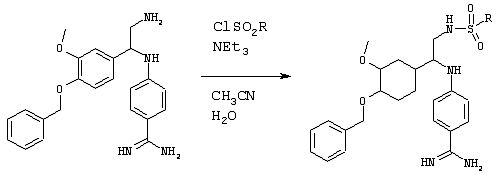
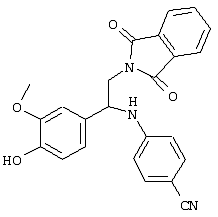
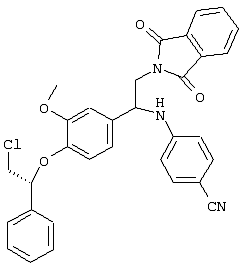
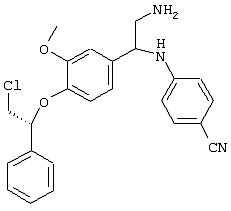
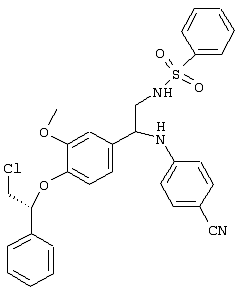
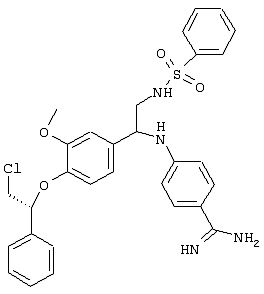
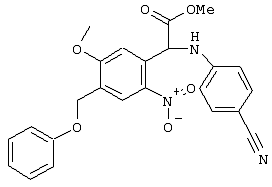
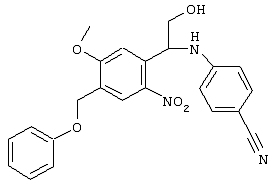
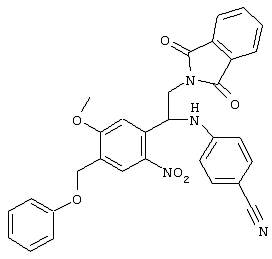
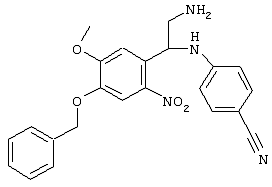
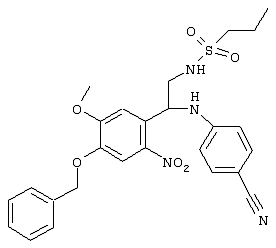
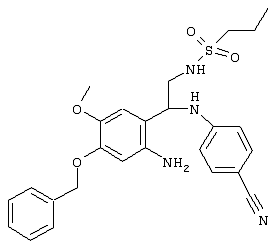
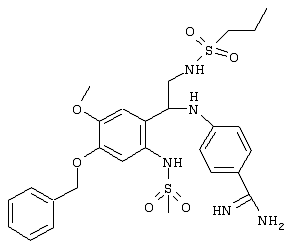
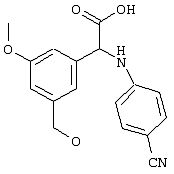
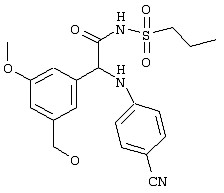
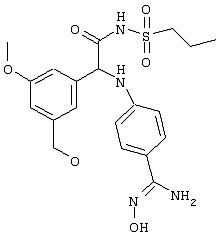
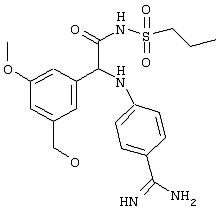
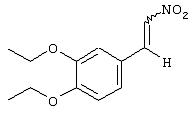
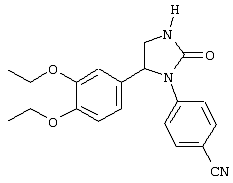
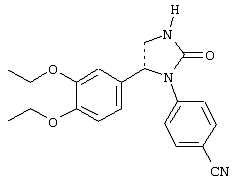
 ]Nа -55,0 (с 2,20, ацетон). МС (М+Н)=352.
]Nа -55,0 (с 2,20, ацетон). МС (М+Н)=352.