Патент на изобретение №2250906
|
||||||||||||||||||||||||||||
(54) СПОСОБЫ ПОЛУЧЕНИЯ ГЕМИНПЕПТИДОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ НУКЛЕОЛИТИЧЕСКИХ АГЕНТОВ
(57) Реферат:
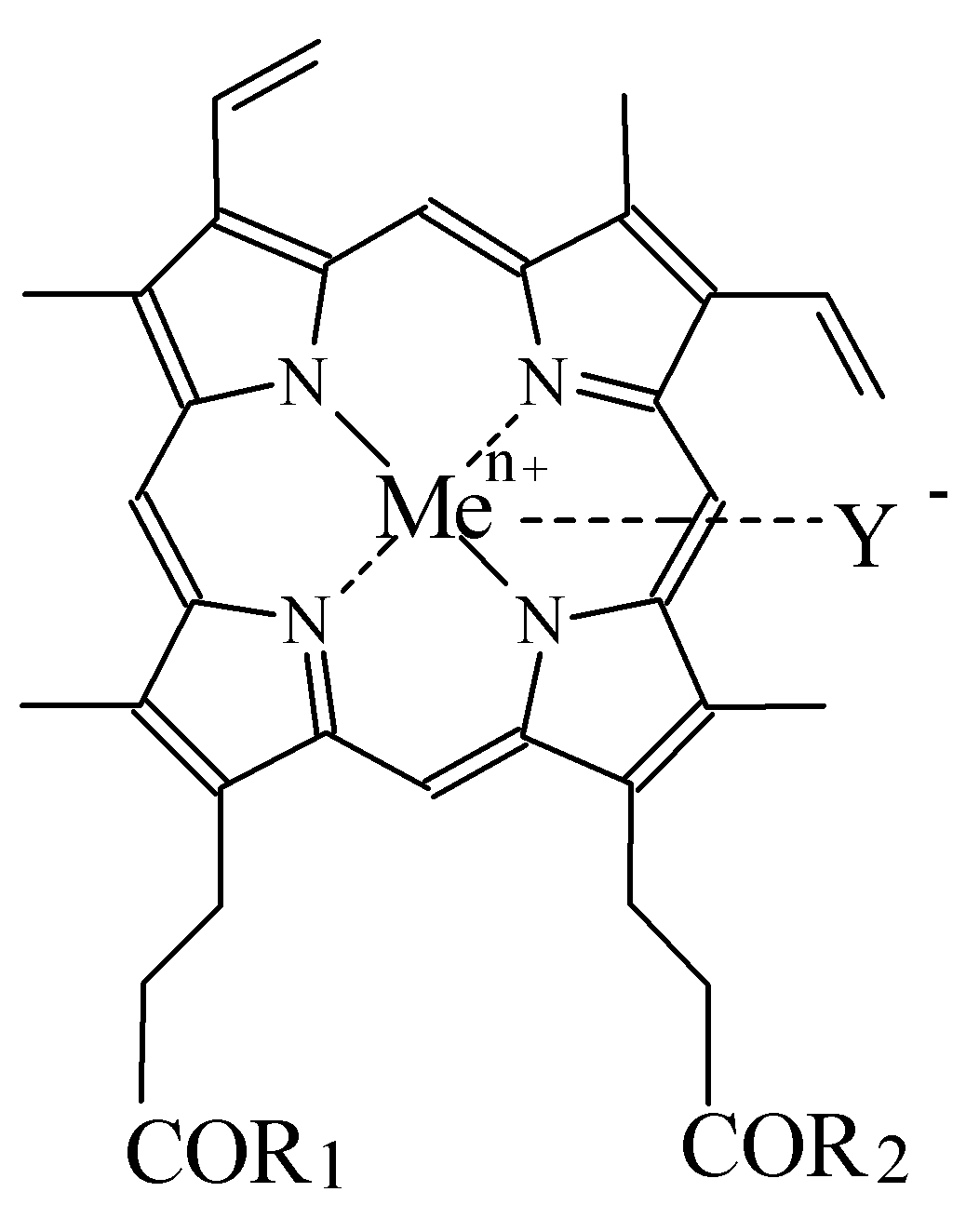
Изобретение относится к способам получения порфиринопептидов формулы I где R1 и R2 – заместители, которые могут представлять собой аминокислоты или пептиды, состоящие из 2-15 аминокислотных остатков, при этом
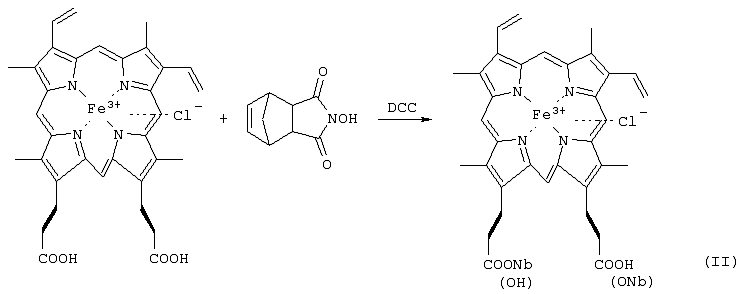
Изобретение относится к химии и медицине, а именно к новому способу синтеза конъюгатов порфиринов, в частности гемина с олигопептидами и аминокислотами, и их применению в качестве нуклеолитических агентов, общей формулы I: где R1 и R2 – заместители, которые могут представлять собой аминокислоты или пептиды, состоящие из 2-15 аминокислотных остатков, при этом Y– представляет собой Сl–; Me представляет собой Zn, Cu, Fe, Mn. Учитывая вышеуказанное, геминпептиды могут представлять собой более совершенные реагенты для расщепления ДНК (РНК). Твердофазный метод синтеза геминпептидов позволяет целенаправленно с высоким выходом получать монопептидные производные гемина и имеет преимущества перед классическим синтезом в растворе при получении геминпептидов, содержащих достаточно длинные пептиды со сложной аминокислотной последовательностью. Однако синтез твердофазным методом геминпептидов, содержащих остатки аминокислот или коротких пептидов, неоправдано сложен и трудоемок по сравнению с синтезом в растворе. Последний представляет собой смесь моно- и бис-эфиров с гемином при соотношении приблизительно 2:1:1, который вводится в реакцию с аминокомпонентом без выделения. Вследствие этого для выделения моно-аминоацильных и моно-пептидных производных гемина оказалось необходимым применение трудоемкой двухстадийной очистки, предусматривающей использование колоночной и препаративной тонкослойной хроматографии. В результате выходы геминпептидов (III-V) составили соответственно 17, 47 и 41%. Вышеуказанные недостатки делают известные способы малопригодными для препаративного синтеза низкомолекулярных геминпептидов, соответствующих общей формуле I. Задачей заявленного изобретения является упрощение процесса, повышение выхода и качества целевого продукта. Активацию карбоксильной группы гемина осуществляют путем превращения в N-окси-5-норборнен-2,3-дикарбоксиимидный эфир DCC-методом (схема 1) с применением эффекта концентрационного разбавления. Последний заключается в медленном прибавлении в течение 5-6 часов раствора 1 экв. DCC в диметилформамиде к раствору протогемина IX и N-гидрокси-5-норборнен-2,3-дикарбоксиимида (1:1) при 0° С и при интенсивном перемешивании. В результате соотношение гемин:моно-активированный эфир:бис-активированный эфир в реакционной смеси составляет приблизительно 2:4:1 соответственно. Таким образом, количество моно-активированного эфира в реакционной смеси увеличивается в два раза, а количество бис-активированного эфира соответственно уменьшается по сравнению с подобной реакцией с N-оксисукцинимидом. Схема 1 Синтез 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира протогемина IX Полученный моно-ONb-эфир гемина (II) без выделения вводят в реакцию с аминокомпонентами H-ArgOMe или H-Leu-HisOMe или H-Leu-Leu-Val-PheOMe. Реакции протекают с более высокой скоростью по сравнению с аналогичными реакциями с участием N-оксисукцинимидного эфира гемина. При этом наблюдается полная конверсия моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира и аминокомпонентов с преимущественным образованием моно-аминоацильного или монопептидного производных гемина; соотношение образующихся моно- и бис-аминоацильных или пептидных производных гемина в реакционной смеси составляет приблизительно 4:1. Выходы геминпептидов (III, IV, V), полученных с использованием N-окси-5-норборнен-2,3-дикарбоксиимидных эфиров гемина, были несколько выше, чем в случае N-оксисукцинимидных эфиров гемина, и составляют соответственно 36, 60 и 51%. Кроме того, поставленная задача заявленного изобретения, заключающаяся в повышении выхода и качества моно-производных протогемина IX и упрощения процесса их выделения, решается и другим, альтернативным методом с применением дифенилфосфорилазида (DPPA) для активации СООН-группы гемина. Вышеуказанные факторы и отсутствие устойчивых активированных производных гемина после завершения реакции позволяют упростить процесс выделения и повысить выходы целевых моно-производных гемина. Для их очистки оказалось достаточно однократной колоночной хроматографии на силикагеле и выходы аминоацильного производного гемина и геминпептидов составили соответственно 63, 66 и 62%. Кроме того, трифторацетат может выступать в качестве противоиона по иону металла вместо Сl– в составе порфиринового остатка. Вышеуказанное может изменять и ухудшать качество и выход геминпептидов, в том числе их устойчивость при хранении. Так, геминпептиды (IV, V), полученные с использование трифторуксусной кислоты, неустойчивы в растворах DMSO, что подтверждается методами ТСХ, электронной и масс-спектрометрии, что может быть причиной плохой воспроизводимости результатов биологических и биохимических испытаний, проводимых в растворах, содержащих DMSO. Поставленная в заявке на изобретение задача повышения качества и выходов геминпептидов решается путем замены трифторуксусной кислоты на минеральную, например соляную, при отщеплении N-защиты, например Воc, от N-защищенных пептидов – аминокомпонентов при синтезе геминпептидов формулы I. Полученные таким способом геминпептиды устойчивы в растворах, содержащих DMSO. Заключение Таким образом, способ активации СООН-групп гемина с применением моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира и DCC или DPPA в присутствии основания и использование аминокомпонентов – пептидов в виде солей с минеральной кислотой, например хлоргидратов, с последующей нейтрализацией основанием обеспечивают упрощение процесса, повышение выходов и качества геминпептидов. Список сокращений Воc – трет-бутилоксикарбонил DCC – N,N’-дициклогексилкарбодиимид DCU – N,N’-дициклогексилмочевина DMF – N,N’-диметилформамид DMSO – диметилсульфоксид DPPA – дифенилфосфорилазид МеОН – метиловый спирт ОМе – метиловый эфир ONb – норборнендикарбоксиимидоокси Трис-НСl – (гидроксиметил)аминометан-гидрохлорид ТСХ – хроматография в тонком слое ЭДТА – этилендиаминтетраацетат Экспериментальная часть Синтез порфиринопептидов отражен в примерах 2-7. Предлагаемые порфиринопептиды получают с высокими выходами и высокой степенью чистоты и характеризуют УФ-, ИК-спектроскопией, масс-спектрометрией, ТСХ. Индивидуальность полученных соединений проверяют методом ТСХ на пластинах Kieselgel 60 F254 (фирмы “Merck”, Германия) в системах: хлороформ – метанол 9:1 (1), хлороформ – метанол – уксусная кислота 5:3:1 (2), хлороформ – метанол 4:1 (3), на пластинах Alufol (нейтр., фирмы “Merck”, Германия) – в метаноле (4), в системе хлороформ – метанол 9:1 (5). Хроматограммы проявляют нингидрином, хлор-толидиновым реактивом и по свечению в УФ-свете, имидазолсодержащие производные – реактивом Паули. Масс-спектры высокого разрешения получают на времяпролетном масс-спектрометре REFLEX ИК-спектры регистрируют на приборах: Perkin ELMER FT-IR Spectrometer 1725X (Швейцария) и Magna 750 (Nicolet, США). Электронные спектры снимают на приборе Jasco model UV/VS 7800 Spectrophotometer (Япония). Пример 1. Синтез 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира протогемина IX (II) К раствору 0.7 г (1.08 ммоль) протогемина IX в 7 мл DMF и 0.20 г (1.08 ммоль) N-гидрокси-5-норборнен-2,3-дикарбоксиимида при охлаждении до -10° С и перемешивании добавляют по каплям 0.22 г (1.08 ммоль) DCC в 3 мл DMF в течение 6 ч. Раствор оставляют на 48 ч при 0° С. DCU отделяют. Дополнительную очистку от DCU проводят переосаждением из DMF эфиром. В осадке получают смесь соединений с Rfocн. (II) 0.27(1), Rfминорн. 0.5(1), Rfгемина 0.15 (1). Пример 2. N К раствору 0.16 мл (1.2 ммоль) Еt3N в 2 мл DMF прибавляют 0.16 г (0.6 ммоль) гидрохлорида метилового эфира аргинина, оставляют на 15 мин при 0° С. К раствору полученного аминокомпонента добавляют раствор 20.8 мл (0.6 ммоль) 6(7)-моно-N-гидрокси-5-норборнен-2,3-дикарбоксиимидного эфира протогемина (IX) в DMF, полученного по вышеуказанной методике. Реакционную смесь перемешивают 5 ч до полного исчезновения моно-активированного эфира гемина с Rf 0.27(1). Раствор концентрируют в вакууме до 2 мл и прибавляют 6 мл эфира. Осадок отделяют от раствора декантированием и сушат над безводным CaCl2. Вещество очищают на колонке (24× 1.2 см) с Kieselgel 60, целевой продукт элюируют смесью хлороформ-метанол (7:3). Фракции, содержащие вещество с Rf 0.78(2), объединяют. Растворитель удаляют в вакууме. Вещество растворяют в 8 мл бутанола и промывают водой (3× 3 мл). Растворитель из органического слоя удаляют в вакууме. Для введения противоиона Сl– геминпептид (III) в 10 мл смеси хлороформ-метанол (9:1) встряхивают с 6 мл 0.5 н соляной кислоты, 6 мл 2% водного раствора гидрокарбоната натрия и водой до нейтральной реакции. Органический слой сушат над безводным Na2SO4, растворитель удаляют в вакууме, остаток сушат над безводным CaCl2 в вакууме. Выход 0.14 г (36%). Rf 0.78 (2). Электронный спектр, Пример 3. N Раствор 0.14 г (0.36 ммоль) Boc-Leu-His-OMe в 3.25 н НСl в смеси диоксана и метанола выдерживают 25 мин при 20° С. Кислоту и растворитель удаляют в вакууме, остаток растирают с эфиром. Осадок отделяют центрифугированием и сушат в вакууме над безводным NaOH. К раствору дихлоргидрата метилового эфира лейцилгистидина в 3.0 мл DMF при перемешивании и охлаждении до 0° С добавляют 0.1 мл (0.72 ммоль) Et3N. Оставляют на 15 мин при 0° С. К раствору полученного аминокомпонента добавляют раствор 8 мл (0.36 ммоль) 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира протогемина IX (I), полученного ранее. Реакционную смесь перемешивают в течение 4 ч до исчезновения аминокомпонента. Раствор концентрируют в вакууме до 1.5 мл и добавляют 6 мл эфира. Осадок отделяют от растворителя декантированием и сушат над безводным CaCl2. Вещество очищают на колонке (19× 3.5 см) с Kieselgel 60, целевой продукт элюируют смесью хлороформ-метанол (7:3). Фракцию, содержащую смесь моно- и бис-(метилового эфира N Пример 4. N Соединение (V) получают аналогично соединению (IV) из 0.105 г (0.18 ммоль) Boc-Leu-Leu-Val-Phe-OMe и 4.5 мл (0.18 ммоль) 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира протогемина IX (II), полученного ранее. Целевое вещество выделяют на колонке (35× 1.2 см) с Kieselgel 60, элюируя смесью растворителей хлороформ-метанол (9:1). Для введения противоиона Сl– геминпептид (V) обрабатывают в соответствии с методикой, описанной для соединения (III). Выход 0.190 г (51%). Rf 0.5 (1), Rf 0 (5). Аминокислотный анализ: Leu 1.9 (2), Val 1.07 (1), Phe 1.02 (1). ИК-спектр, Пример 5. N К раствору 0.05 г (0.20 ммоль) H-ArgOMex2HCl и 0.13 г (0.20 ммоль) протогемина IX в 2 мл DMF при охлаждении до -10° С и перемешивании прибавляют 0.044 мл (0.20 ммоль) DPPA и 0.084 мл (0.6 ммоль) Et3N. Реакционную смесь перемешивают в течение 2 ч при -10° С и 15 ч при 20° С. Раствор концентрируют в вакууме до 1.5 мл и добавляют 6 мл эфира. Осадок отделяют от растворителя декантированием и сушат над безводным СаСl2. Соединение очищают на колонке с Kieselgel 60 F254 (12× 31.5 см) (Merck). Целевое вещество с Rf 0.78 (2) элюируют смесью хлороформ-метанол (7:3). Растворитель удаляют в вакууме. Для введения противоиона Сl– геминпептид (IIIa) обрабатывают в соответствии с методикой, описанной для соединения (III). Выход 0.100 г (63%). Rf 0.78 (2). Электронный спектр, Пример 6. N Раствор 0.10 г (0.26 ммоль) Boc-Leu-His-OMe в 3.25 н НСl в смеси диоксана и метанола выдерживают 25 мин при 20° С. Кислоту и растворитель удаляют в вакууме, остаток растирают с эфиром. Осадок отделяют центрифугированием и сушат в вакууме над безводным NaOH. К раствору полученного аминокомпонента и 0,17 г (0.26 ммоль) протогемина IX в 3 мл DMF, при охлаждении до -10° С и перемешивании прибавляют 0.056 мл (0.26 ммоль) DPPA и 0.11 мл (0.8 ммоль) Et3N. Реакционную смесь перемешивают в течение 2 ч при -10 С и 15 ч при 20° С. Раствор концентрируют в вакууме до 1.5 мл и добавляют 6 мл эфира. Осадок отделяют от растворителя декантированием и сушат над безводным CaCl2. Соединение (IVa) очищают на колонке с Kieselgel 60 F254 (35× 1.2 см) (Merck). Растворитель удаляют в вакууме. Для введения противоиона Сl– геминпептид (IVa) обрабатывают в соответствии с методикой, описанной для соединения (III). Выход 0.156 г (65.6%). Rf 0.38 (3), Rf 0 (4). Аминокислотный анализ: Leu 1.12 (1), His 1.07 (1). ИК-спектр, Пример 7. N Деблокирование аминокомпонента и синтез геминпептида (Va) осуществляют в соответствии с условиями, описанными для соединения (IVa), исходя из 0.075 г (0.12 ммоль) Boc-Leu-Leu-Val-Phe-OMe, 0.078 г (0.12 ммоль) протогемина IX в 3 мл DMF, 0.03 мл (0.12 ммоль) DPPA и 0.033 мл (0.24 ммоль) Еt3N. Целевое вещество выделяют на колонке (40× 1.2 см) с Kieselgel 60, элюируя смесью растворителей хлороформ-метанол (9:0.5). Для введения противоиона Сl– геминпептид (Va) обрабатывают в соответствии с методикой, описанной для соединения (III). Выход 0.084 г (62%). Rf 0.5 (1), Rf 0 (5). Аминокислотный анализ: Leu 2.01 (2), Val 1.06 (1), Phe 0.9 (1). ИК-спектр, Исследование нуклеолитической активности геминпептидов Другим объектом изобретения является применение геминпептидов общей формулы I в качестве нуклеолитических агентов. Исследование нуклеолитической активности геминпептидов (III, IV, V) проводят по типичной методике [Maniatis Т., Fritish E., Sambrook J. Molecular Cloning (A Laboratory Manual) Cold Spring Harbor; N.-Y.: Cold Spring Harbor Cloning (A Laboratory Manual) Cold Spring Harbor; N.-Y.: Cold Spring Harbor Laboratory Press, 1982] с применением ДНК плазмиды pGem, а также ДНК плазмиды pUKB244, несущей полноразмерный вирусный ген Gag-P24 (ВИЧ), выделенных из E.coli. Геминпептиды в концентрации 10-4-10-8 М инкубируют с 0.5 мкг плазмидной ДНК в течение 1 ч при 37° С в 10 мкл реакционной смеси. Продукты расщепления анализируют с помощью электрофореза в 1% агарозном геле в ТАЕ-буфере (0.04 М Трис-ацетат (рН 8.0), 0.02 М ЭДТА) при напряжении 10 В/см. Продукты реакции визуализируют путем окрашивания их флуоресцирующим красителем бромистым этидием. Полосы ДНК детектируют с помощью источника УФ-света длинноволновой области с Активность соединений исследуют при кислых и нейтральных значениях рН в следующих буферных растворах: буфер I:100 мМ AcONH4 (рН 4.5), 10 мМ MnCl2; буфер II: 100 мМ AcONH4 (pH 4.5), 20 мМ ZnOAC2; буфер III: 100 мМ AcONH4 (pH 4.5), 10 MM MgCl2; буфер IV: 20 мМ Трис-HCl (pH 7.5), 20 мМ МnСl2; буфер V: 20 мМ Трис-HCl (pH 7.5), 20 мМ ZnOAC2; буфер VI: 100 мМ Трис-HCl (pH 7.5), 10 мМ MgCl2; буфер VII: 20 мМ Трис-HCl (pH 7.5), 2 мМ FeSO4; буфер VIII: 10 мМ Трис-HCl (рН 8.5), 10 мМ MgCl2, 100 мМ КСl, 0.1 mg/ml BSA, 200 мМ ЭДТА; буфер IX: 10 мМ Трис-HCl (рН 7.5), 10 мМ MgCl2, 0.1 mg/ml BSA, 200 мМ ЭДТА. Геминпептиды прибавляют к реакционной смеси в виде раствора в DMSO, причем концентрация DMSO в конечной пробе составляет 10%. Исчезновение полос, соответствующих различным формам ДНК, свидетельствуют о расщеплении ДНК по окислительному типу. Результаты Введение в реакционную среду геминпептидов вызывает исчезновение полос (расщепление) ДНК в различных условиях (рН, ионы металлов и др.) (см. таблицу).
Заключение Порфиринопептиды, полученные заявленным способом, можно применять в качестве нуклеолитических агентов, например для биомедицинских исследований, или в качестве потенциальных лекарственных средств для химиотерапии вирусных и онкологических заболеваний.
Формула изобретения
1. Способы получения порфиринопептидов общей формулы I или их фармацевтически приемлемых солей (I) где R1 и R2 – заместители, которые могут представлять собой аминокислоты или пептиды, состоящие из 2÷ 15 аминокислотных остатков, при этом Y– представляет собой Cl–; Me представляет собой Zn, Cu, Fe, Mn; включающие активацию карбоксильной группы порфирина и последующее взаимодействие активированного производного порфирина с аминокислотой или пептидом, отличающиеся тем, что карбоксильную группу порфирина активируют действием N-окси-5-норборнен-2,3-дикарбоксиимида при молярном соотношении исходных реагентов 1:1 и процесс ведут в присутствии DCC, или действием DPPA при эквимолекулярном соотношении порфирин: DPPA в присутствии основания, затем активированный по карбоксильной группе порфирин вводят в реакцию с аминокомпонентом – аминокислотой или пептидом, находящимся в виде соли с минеральной кислотой, которую нейтрализуют основанием. 2. Способы получения порфиринопептидов по п.1, отличающиеся тем, что в соединениях общей формулы I остаток порфирина представляет собой гемин; остатки аминокислоты или пептида представляют собой -Arg-OMe, -Leu-HisOMe, -Leu-Leu-Val-PheOMe, а основание для активации СООН группы гемина действием DPPA и нейтрализации аминокомпонента, находящегося в виде хлористоводородной соли, представляет собой триэтиламин. 3. Применение порфиринопептидов общей формулы I в качестве нуклеолитических агентов. 4. Применение порфиринопептидов по п.3, отличающееся тем, что соединения общей формулы I проявляют нуклеолитическую активность по отношению к вирусной ДНК.
|
||||||||||||||||||||||||||||
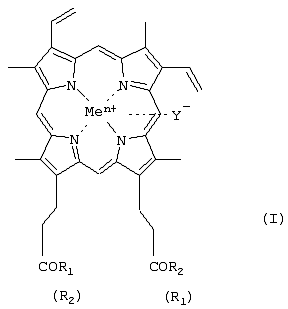
 -карбоксильные группы аминокислот или пептидов могут быть модифицированы С1-С8алкиловым эфиром, а боковые функции аминокислот или пептидов могут быть защищены, причем возможно, что R1=R2 или R1
-карбоксильные группы аминокислот или пептидов могут быть модифицированы С1-С8алкиловым эфиром, а боковые функции аминокислот или пептидов могут быть защищены, причем возможно, что R1=R2 или R1  R2, в частности R1=-ArgOMe, R2=-ОН (III); R1=-LeuHisOMe, R2=-OH (IV); R1=-LeuLeuValPheOMe, R2=-OH (V); карбоксильная группа порфирина может быть модифицирована метиловым или другим C1-C8 эфиром или физиологически приемлемой солью; Y– представляет собой Cl–; Me представляет собой Zn, Cu, Fe, Mn; путем активации карбоксильной группы порфирина действием N-окси-5-норборнен-2,3-дикарбоксиимидом при молярном соотношении исходных реагентов 1:1 и процесс ведут в присутствии N,N‘ – дициклогексилкарбодиимида, или действием дифенилфосфорилазида (DPPA) при эквимолярном соотношении порфирин:DPPA в присутствии основания, затем активированный по карбоксильной группе порфирин вводят в реакцию с аминокомпонентом – аминокислотой или пептидом, находящимся в виде соли с минеральной кислотой, которую нейтрализуют основанием; и к применению I в качестве нуклеолитических агентов. 2 н. и 2 з.п. ф-лы, 1 табл.
R2, в частности R1=-ArgOMe, R2=-ОН (III); R1=-LeuHisOMe, R2=-OH (IV); R1=-LeuLeuValPheOMe, R2=-OH (V); карбоксильная группа порфирина может быть модифицирована метиловым или другим C1-C8 эфиром или физиологически приемлемой солью; Y– представляет собой Cl–; Me представляет собой Zn, Cu, Fe, Mn; путем активации карбоксильной группы порфирина действием N-окси-5-норборнен-2,3-дикарбоксиимидом при молярном соотношении исходных реагентов 1:1 и процесс ведут в присутствии N,N‘ – дициклогексилкарбодиимида, или действием дифенилфосфорилазида (DPPA) при эквимолярном соотношении порфирин:DPPA в присутствии основания, затем активированный по карбоксильной группе порфирин вводят в реакцию с аминокомпонентом – аминокислотой или пептидом, находящимся в виде соли с минеральной кислотой, которую нейтрализуют основанием; и к применению I в качестве нуклеолитических агентов. 2 н. и 2 з.п. ф-лы, 1 табл.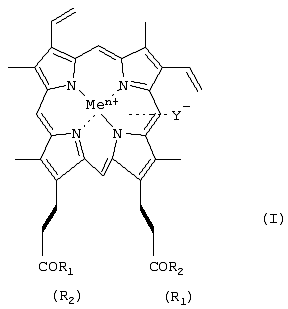
 III (BRUKER, Германия) методом матриксной лазернодесорбционной ионизации, в качестве матрицы использовалась 2,5-дигидрооксибензойная кислота.
III (BRUKER, Германия) методом матриксной лазернодесорбционной ионизации, в качестве матрицы использовалась 2,5-дигидрооксибензойная кислота. max, нм, СНСl3-МеОН (1:1), (
max, нм, СНСl3-МеОН (1:1), ( · 10-3): 397.4 (101.5), 500.80 (4.49), 540.40 (2.63). 626.0 (1.14). ИК-спектр,
· 10-3): 397.4 (101.5), 500.80 (4.49), 540.40 (2.63). 626.0 (1.14). ИК-спектр,  , см-1, вазелиновое масло: 1745 (СО cл.эф.), 1650 (амид I), 1514 (амид II). Масс-спектр, m/z: [M]+ 786.41.
, см-1, вазелиновое масло: 1745 (СО cл.эф.), 1650 (амид I), 1514 (амид II). Масс-спектр, m/z: [M]+ 786.41.