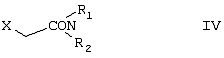Патент на изобретение №2248974
|
||||||||||||||||||||||||||
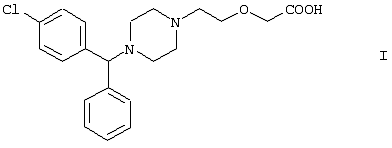
(54) СПОСОБ ПОЛУЧЕНИЯ {2-[4-(АЛЬФА-ФЕНИЛ-П-ХЛОРБЕНЗИЛ)ПИПЕРАЗИН-1-ИЛ]-ЭТОКСИ}-УКСУСНОЙ КИСЛОТЫ И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
(57) Реферат:
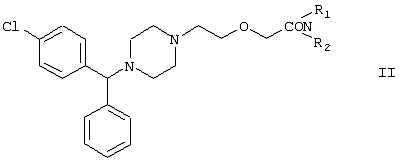
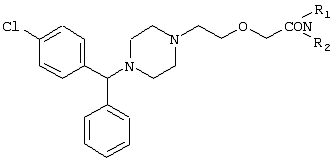
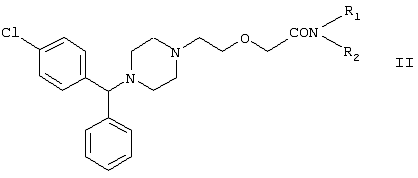
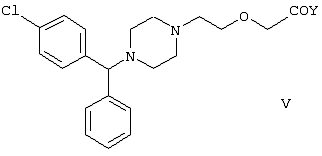
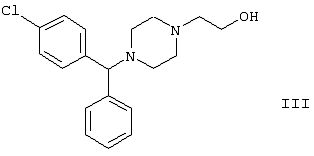
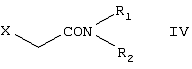
Настоящее изобретение относится к способу получения {2-[4-( заключающемуся в том, что соединение формулы II где R1 и R2 независимо представляют собой C1-4алкил, который может быть замещен фенилом, C2-4алкенил или циклогексил, или R1 и R2 образуют вместе с соседним атомом азота морфолиновую группу, гидролизуют, при необходимости в присутствии межфазного катализатора. Цетиризин используется как ингредиент противоаллергических фармацевтических композиций. Описан также способ получения промежуточных продуктов формулы II. 4 с. и 1 з.п. ф-лы.
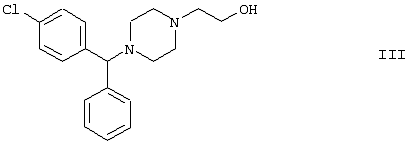
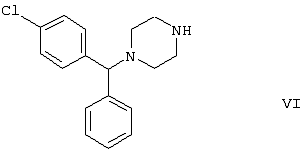
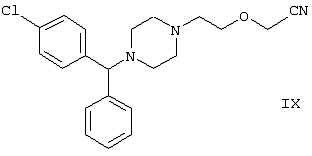
Область изобретения Изобретение касается нового способа получения {2-[4-( {2-[4-( Изобретение также касается новых {2-[4-( где R1 и R2 независимо представляют собой С1-4алкил, который может быть замещен фенилом, С2-4алкенил или циклогексил, или R1 и R2 образуют вместе с соседним атомом азота морфолиновую группу, и их кислотных аддитивных солей, которые являются полезными промежуточными соединениями при синтезе цетиризина. Предпосылки изобретения Известно несколько способов получения цетиризина формулы I. Согласно Европейской патентной заявке №58146 (Chem. Abstr., 98, 34599r), эфир или амид формулы где Y представляет собой группу формулы -OR1 или аминогруппу, где R1 означает алкил, гидролизуют для получения соединения формулы I. Используя в качестве исходного вещества метиловый эфир формулы V, при гидролизе с гидроксидом калия получают калиевую соль цетиризина с выходом 59%. Из калиевой соли образуется соответствующая кислота с выходом 81%, а выход ожидаемого дигидрохлорида цетиризина составляет 80%. Таким образом, общий выход при получении по известному способу составляет 38,2%. Существенным недостатком известного способа является то, что получение эфиров формулы V, которые используются в качестве исходных соединений, сопряжено с большими трудностями и с маленьким выходом. Так, 1-[(4-хлорфенил)фенилметил]пиперазин формулы VI реагирует с метил-2-хлорэтоксиацетатом формулы VII с образованием метилового эфира формулы V с выходом до 10,6% после хроматографии. В уже упоминавшейся Европейской патентной заявке говорится о том, что цетиризин можно получать также из ацетамидов формулы V, где Y представляет собой аминогруппу, путем гидролиза. Однако гидролиз ацетамида формулы V не подтверждается ни примерами, ни данными выхода. Согласно более позднему сообщению (Synthesis, 1995. 766), ацетамид формулы V гидролизуют в соляной кислоте при 50°С для получения цетиризина с выходом 71%. Для получения ацетамида формулы V известны еще три способа. Например, 1-[(4-хлорфенил)фенилметил]пиперазин формулы VI реагирует с 2-хлорэтоксиацетамидом формулы: в присутствии связывающего кислоту агента с образованием ацетамида формулы V с выходом 47%. Согласно второму способу {2-[4-( реагирует с гидридом натрия, давая натриевую соль, а соль реагирует с хлорацетамидом формулы где Х обозначает атом хлора, R1 и R2 обозначают атом водорода. Однако выход этой реакции всего 11%. Согласно третьему методу ацетамид формулы V может быть получен также из соответствующего метилового эфира формулы V, где Y обозначает метоксигруппу, путем реакции с аммиаком с выходом 54%. Из-за низкого выхода (27,8%) реакции получения метилового эфира общий выход составляет 15,0%. Следовательно, последний упомянутый способ синтеза цетиризина также неэкономичен. Способ, описанный в патенте UK No. 2225320 (Chem. Abstr., 113. 191395s), направлен на устранение недостатков упомянутых выше известных способов путем получения цетиризина из {2-[4-( Последний из известных способов имеет еще один недостаток. Реакцию производного этанола формулы III с хлорацетатом натрия можно осуществить с относительно приемлемым выходом, если добавлять реагенты в раствор производного этанола формулы III несколькими порциями. Добавление реагентов нужно программировать на основе постоянного мониторинга состава реакционной смеси. Эта технология очень неудобна, особенно в промышленном масштабе. Последний из известных способов пытаются улучшить с помощью способа, описанного в патенте PL No. 163415 (Chem. Abstr., 123. 55923s), согласно которому реакцию проводят в системе, состоящей из двух фаз, в присутствии нейтрального органического растворителя и водного раствора гидроксида натрия, получая дигидрохлорид цетиризина с выходом 60%. Хотя этот способ менее сложен, чем способ, опубликованный в патенте UK No.2225320, общий выход составляет всего 54%, как рассчитано для производного этанола формулы III, которое может быть получено более подходящим способом. Другой способ получения цетиризина известен из патента UK No. 2225321 (Chem. Abstr., 113, 191396t), согласно которому 1-[(4-хлорфенил)фенилметил]пиперазин формулы VI подвергают реакции с хлорэтоксиацетонитрилом, получая производное нитрила, имеющее следующую формулу: которое гидролизуют в кислой или щелочной среде до цетиризина. При таком способе дигидрохлорид цетиризина получают из производного нитрила формулы IX с общим выходом 60,5% в случае кислотного гидролиза и 65,5% в случае щелочного гидролиза. Однако реакция 1-[(4-хлорфенил)фенилметил]пиперазина формулы VI с хлорэтоксиацетонитрилом дает производное нитрила с выходом 86,4%, уменьшая общий выход в процессе синтеза до 52,2% и 56,6%, соответственно. Следует принимать во внимание также, что согласно литературе (E.J. Salmi, R. Leimu and H. Kallio, Suomen Kemistilehti, 17B, 17-19 (1944)), хлорэтоксиацетонитрил может быть получен из хлоргидрина этилена в две стадии с использованием очень ядовитого цианида меди (I) с общим выходом 58%. Наконец, простой способ получения возможен благодаря способу, известному из опубликованной Европейской патентной заявки No. 801064, согласно которой цетиризин получают из 1-[(4-хлорфенил)фенилметил]пиперазина формулы VI путем реакции с 2-хлорэтоксиуксусной кислотой в нейтральном растворителе в присутствии связывающего кислоту агента. В данной публикации приводится только один Пример, который не содержит ни данных выхода, ни качественных данных о продукте. К тому же, большим недостатком этого способа является недоступность 2-хлорэтоксиуксусной кислоты в промышленных масштабах. Целью данного изобретения является создание экономичного способа получения цетиризина, который удовлетворял бы самым строгим требованиям, предъявляемым к качеству вещества. Краткое описание изобретения Было обнаружено, что описанная выше цель достигается благодаря созданию способа получения {2-[4-( Изобретение включает {2-[4-( Описание предпочтительных воплощений В способе изобретения исходным соединением является ацетамид формулы II или его кислотная аддитивная соль, образующаяся с органической или неорганической кислотой, и исходное соединение подвергают щелочному или кислотному гидролизу способом, известным per se, и при необходимости в присутствии межфазного катализатора. При желании полученный продукт превращают в фармацевтически приемлемую кислотную аддитивную соль или соль металла, и/или основание высвобождают из кислотной аддитивной соли способом, известным per se. Гидролиз {2-[4-( Эксперта может удивить то, что гидролиз {2-[4-( В качестве ацетамида формулы II предпочтительней использовать N,N-диметил-{2-[4-( Согласно предпочтительному способу изобретения гидролиз {2-[4-( После гидролиза и доведения рН до 3,8-4,0 возможна экстракция раствора цетиризинового основания в дихлорметане предпочтительней с помощью 5-30% соляной кислоты, водный раствор концентрируют при пониженном давлении для уменьшения содержания воды до 20-25 мас.%, и затем следуют процедуре, описанной выше для получения дигидрохлорида цетиризина. Похожий на мед осадок, полученный после упаривания раствора цетиризинового основания в дихлорметане, может быть растворен в смешивающемся с водой органическом растворителе, предпочтительней в ацетоне, метилэтилкетоне или тетрагидрофуране, и полученный таким образом раствор цетиризинового основания в органическом растворителе затем обрабатывают соляной кислотой. Кислый раствор подвергают любой из вышеописанных процедур для получения дигидрохлорида цетиризина. Согласно еще одному предпочтительному способу изобретения гидролиз ацетамида формулы II осуществляют в водном растворе неорганической кислоты, предпочтительней соляной или серной кислоты, при 40-110°С, предпочтительней при 70-110°С, более предпочтительней при точке кипения раствора. Обычно используют 2-25 мас.%, предпочтительней 10-20 мас.%, более предпочтительней 15 мас.% раствор неорганической кислоты. Для отделения продукта полученный кислотный гидролизат разбавляют водой, доводят рН до значения 3,8-4,0 путем добавления водного раствора гидроксида натрия, и затем следуют процедуре, описанной для щелочного гидролиза. И щелочной, и кислотный гидролиз можно проводить в присутствии межфазного катализатора и при необходимости также сорастворителя. В качестве межфазного катализатора может быть использован галид тетраалкиламмония, предпочтительней хлорид тетрабутиламмония, хлорид триэтилбензиламмония или хлорид триоктиметиламмония (Aliquat 336) или краун-эфир, предпочтительней 15-краун-5 или 18-краун-6. Сорастворителем может быть смешивающийся с водой органический растворитель, как, например, этанол, бутанол, этиленгликоль или диоксан. Если в способе изобретения в качестве исходного соединения используется кислотная аддитивная соль ацетамида формулы II, полученная с органической кислотой, эта органическая кислота кристаллизуется из реакционной смеси после осаждения продукта кислотой. В этом случае выпавшую органическую кислоту можно отделить путем фильтрации перед экстракцией продукта дихлорметаном, для того чтобы добиться более легкого разделения фаз. Однако с реакционной смесью можно работать и без отделения органической кислоты. Таким образом, способ изобретения экономичен, может быть легко выполнен и дает цетиризин или его кислотную аддитивную соль в очень чистом виде. Второй аспект данного изобретения касается новых промежуточных соединений формулы II, где R1 и R2 определены выше, и их кислотных аддитивных солей, образуемых при взаимодействии с органическими и неорганическими кислотами. Изобретение включает изомеры {2-[4-( В формуле II, при определении R1 и R2, С1-4алкил, который может быть замещен фенилом, может иметь прямую или разветвленную цепь, например метил, этил, изопропил, трет-бутил или бензил и так далее. С2-4алкенил может быть, например, аллилом или металлилом. Предпочтительным соединением формулы II является (RS)-N,N-диметил-{2-[4-( {2-[4-( Реакцию проводят при температуре не выше 120°С, предпочтительней при 60-90°С. Соль щелочного металла {2-[4-( Новые {2-[4-( Согласно особенно предпочтительному способу разделения, во время описанного выше разделения соединения формулы II между водой и несмешивающимся с водой органическим растворителем, примеси удаляют из органического раствора путем доведения рН раствора до 6,2-6,7, предпочтительней до 6,4, с помощью водного раствора неорганической или органической кислоты, предпочтительней соляной кислоты, во время выполнения экстракции и разделения фаз. Затем к органическому раствору добавляют еще одну порцию воды, доводят рН приблизительно до 4 добавлением водного раствора кислоты, таким образом превращая продукт в его кислотную аддитивную соль, которая разлагается в водной фазе. После разделения фаз основание высвобождают щелочью, предпочтительней водным раствором гидроксида натрия, и снова экстрагируют несмешивающимся с водой органическим растворителем, предпочтительней дихлорметаном. После упаривания раствора остаток растворяют в более низком алканоле, предпочтительней изопропаноле, и полученный раствор обрабатывают алканолом, содержащим хлористый водород, предпочтительней изопропанолом, содержащим хлористый водород, для получения дигидрохлорида, который осаждают в кристаллической форме добавлением растворителя, который не растворяет продукт, предпочтительней ди(низший алкил)кетона, более предпочтительней ацетона. Согласно другому предпочтительному способу выделения дигидрохлорида ацетамида формулы II после упаривания раствора основания в органическом растворителе осадок растворяют в холодной соляной кислоте, упаривают воду при пониженном давлении и к осадку добавляют ди(низший алкил)кетон, более предпочтительней ацетон для кристаллизации дигидрохлорида. Таким образом, ацетамид формулы II получают в очень чистом виде, что более предпочтительно для получения чистого цетиризина. {2-(4-( 2-Галогенацетамиды формулы IV также известны из литературы [W. E.Weaver and W. M. Whaley, J. Amer. Chem. Soc., 69, 516 (1947); J. Kasprzyk et al., J. Heterocycl. Chem. 30, 119 (1993)] и могут быть легко получены способами, описанными в указанных выше ссылках. Изобретение далее поясняется следующими Примерами. Получение {2-[4-( Пример 1 (RS)-N,N-Диметил-{2-[4-( К раствору 99,3 г (0,3 моль) (RS)-{2-[4-( Таким образом получают 120 г (81,1%) (RS)-N,N-диметил-{2-[4-( Спектральные данные основания: ИК(КВr); ПМР (CDCl3): Пример 2 (RS)-N,N-Диметил-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что похожий на мед осадок основания растворяют в 100 мл 20% соляной кислоты, полученный раствор концентрируют при 10-20°С при пониженном давлении для уменьшения содержания воды приблизительно до 5%, осадок растворяют в 100 мл изопропанола, к полученному раствору прибавляют 1000 мл ацетона и затем снова выполняют процедуру, описанную в Примере 1.Таким образом получают 110 г (76,4%) названного соединения, т.пл.: 184-189°С. Пример 3 (RS)-N,N-Диметил-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что похожий на мед осадок растворяют в 30 мл метанола и к полученному раствору прибавляют 80 мл метанола, содержащего 27% хлористого водорода, затем отделяют дигидрохлорид, как описано в Примере 1. Таким образом получают 108,9 г (75,6%) названного соединения, т.пл.: 185-190°С. Пример 4 (RS)-N,N-Диметил-{2-[4-( 126,8 г (0,3 моль) (RS)-{2-[4-( Таким образом получают 129,2 г (88,1%) названного соединения, т.пл.: 185-190°С. Чистота: больше 99,6%, как определено ВЭЖХ. Пример 5 (RS)-N,N-Диметил-{2-[4-(-фенил-п-хлорбензил)пиперазин-1-ил]этокси}ацетамида фумарат Выполняют процедуру, описанную в Примере 1, с той разницей, что реакцию проводят с 0,025 моль, похожее на мед основание (10,3 г, 98%) растворяют в 10 мл изопропанола, к полученному раствору прибавляют 2,9 г (0,024 моль) фумаровой кислоты и смесь нагревают до растворения. К теплому раствору добавляют 40 мл этилацетата для последующей кристаллизации продукта. После охлаждения кристаллы отфильтровывают и промывают этилацетатом. Таким образом получают 11,2 г (84,0%) названного соединения, т.пл.: 138-141°С. Пример 6 (RS)-N,N-Диэтил-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что реакцию проводят с 0,025 моль, и вместо N,N-диметил-2-хлорацетамида используют 4,11 г (0,0275 моль) N,N-диэтил-2-хлорацетамида. В процессе обработки реакционной смеси похожее на мед основание (9,98 г, 89,9%) не превращают в дигидрохлорид, а очищают посредством хроматографии на колонке с силикагелем, которую подвергают элюции хлороформом, содержащим 1-10% метанола, при постоянном увеличении полярности смеси. Таким образом получают 9,16 г (82,5%) названного соединения в виде бледно-желтого, похожего на мед продукта. Rf=0,45 (пластина Kieselgel Merck, хлороформ-метанол=9:1). ИК(КВr); ПМР (CDCl3): Пример 7 (RS)-N,N-Диаллил-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что вместо N,N-диэтил-2-хлорацетамида используют 4,77 г (0,0275 моль) N,N-диаллил-2-хлорацетамида. Похожее на мед основание (10,5 г, 89%), полученное после обработки реакционной смеси, очищают посредством хроматографии на колонке с силикагелем, используя в качестве элюента смесь хлороформа и 2% метанола. Таким образом получают 9,45 г (80,1%) названного соединения в виде бледно-желтого, похожего на мед продукта. Rf=0,5 (пластина Kieselgel Merck, хлороформ-метанол=9:1). ИК(КВr); ПМР (CDCl3): Пример 8 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что реакцию проводят с 0,05 моль, и вместо N,N-диметил-2-хлорацетамида используют 8,9 г (0,05 моль) хлорацетилморфолида. Похожее на мед основание (10,4 г, 45,4%), полученное после обработки реакционной смеси, очищают посредством хроматографии на колонке с силикагелем, используя в качестве элюента смесь этилацетата и 10% метанола. Таким образом получают 8,3 г (36,2%) названного соединения в виде маслянистого продукта. Rf=0,35 (пластина Kieselgel Merck, хлороформ-метанол=9:1). ИК(КВr); ПМР (CDCl3): MC(Cl):(M+1)+=458 Пример 9 (RS)-N,N-Дициклогексил-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что реакцию проводят с 0,02 моль, и вместо N,N-диметил-2-хлорацетамида используют 5,2 г (0,02 моль) N,N-дихлоргексил-2-хлорацетамида. Таким образом получают 9,8 г (88,9%) названного соединения в виде похожего на мед продукта. Чистота: больше 99%, как определено ВЭЖХ. ИК(КВr); ПМР (CDCI3): Пример 10 (RS)-N,N-Дициклогексил-{2-[4-( 1,1 г похожего на мед основания, полученного по способу, описанному в Примере 9, растворяют в 3 мл изопропанола, к полученному раствору при комнатной температуре прибавляют раствор 0,23 г малеиновой кислоты в 2 мл изопропанола и реакционную смесь перемешивают при комнатной температуре в течение 14 часов. Выпавшие кристаллы отфильтровывают и промывают некоторым количеством изопропанола. Таким образом получают 9,8 г (88,9%) названного соединения. Чистота: 99,6%, как определено ВЭЖХ. Пример 11 (RS)-N,N-Дибензил-{2-[4-( Выполняют процедуру, описанную в Примере 1, с той разницей, что реакцию проводят с 0,05 моль, и вместо N,N-диметил-2-хлорацетамида используют 13,7 г (0,05 моль) N,N-дибензил-2-хлорацетамида. Похожее на мед основание (9,98 г, 89,9%), полученное после обработки реакционной смеси, очищают посредством хроматографии на колонке с силикагелем, которую подвергают элюции смесью этилацетата и н-гексана при постоянном увеличении полярности смеси. Таким образом получают 17,0 г (57%) названного соединения в виде похожего на мед продукта. Rf=0,7 (пластина Kieselgel Merck, этилацетат-метанол=1:1). ИК(КВr); ПМР (СDСl3): Пример 12 (RS)-N,N-Диметил-{2-[4-( К раствору 99,3 г (0,3 моль) (RS)-{2-[4-( Таким образом получают 120 г (81,8%) (RS)-N,N-{2-[4-( Получение {2-[4-( Пример 13 (RS)-{2-[4-( К раствору 97,8 г (0,2 моль) (RS)-{2-[4-( Таким образом получают 74,3 г (80,5%) чистого названного соединения, т.пл.: 226-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997, 1084. Пример 14 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что вместо (RS)-N,N-диметил-{2-[4-( Пример 15 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что вместо (RS)-N,N-диметил-{2-[4-( Пример 16 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 14, с той разницей, что вместо (RS)-N,N-диметил-{2-[4-( Пример 17 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 14, с той разницей, что вместо (RS)-N,N-диметил-{2-[4-( Пример 18 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 14, с той разницей, что вместо (RS)-N,N-диметил-{2-[4-( Пример 19 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что после гидролиза реакционную смесь разбавляют 3500 мл воды, экстрагируют 600 мл этилацетата и 200 мл диизопропилового эфира, остатки органических растворителей удаляют из водного раствора при пониженном давлении и продолжают выполнять процедуру из Примера 13. Таким образом получают 68,1 г (73,8%) названного соединения, т.пл.: 225-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084. Пример 20 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что после гидролиза реакционную смесь разбавляют 3500 мл воды, охлаждают до 0°С, выпавшую кристаллическую натриевую соль цетиризина осаждают в течение 1 часа, фильтруют, затем растворяют в 1000 мл воды, доводят рН до 3,8 добавлением соляной кислоты, затем продолжают выполнять процедуру из Примера 13. Таким образом получают 65,7 г (71,2%) названного соединения, т.пл.: 225-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084. Пример 21 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что гидролиз проводят в присутствии 170 мл (2,0 моль) концентрированной соляной кислоты, и к концу гидролиза рН реакционной смеси доводят до нужного уровня 3,8 с помощью приблизительно 270 мл 40% водного раствора гидроксида натрия. Таким образом получают 70,6 г (76,5%) названного соединения, т.пл.: 225-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084. Пример 22 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что гидролиз проводят в присутствии 170 мл (0,9 моль) 50% серной кислоты, и к концу гидролиза рН реакционной смеси доводят до нужного уровня 3,8 с помощью приблизительно 260 мл 40% водного раствора гидроксида натрия. Таким образом получают 67,3 г (72,9%) названного соединения, т.пл.: 225-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084. Пример 23 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что к реакционной смеси кроме водного раствора гидроксида натрия прибавляют 2 г Aliquat 336 (хлорид триоктиметиламмония). Таким образом получают 74,9 г (81,2%) названного соединения, т.пл.: 226-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084. Пример 24 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что к реакционной смеси кроме водного раствора гидроксида натрия прибавляют 1 г 15-краун-5 (1,4,7,10,13-пентаоксациклопентадекан). Таким образом получают 76,1 г (82,5%) названного соединения, т.пл.: 226-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084. Пример 25 (RS)-{2-[4-( Выполняют процедуру, описанную в Примере 13, с той разницей, что гидролиз проводят в присутствии 280 мл 40% водного раствора гидроксида калия. Таким образом получают 75,0 г (81,3%) названного соединения, т.пл.: 226-228°С. Чистота продукта соответствует требованиям Европейской Фармакопеи, 3, 1997,1084.
Формула изобретения
1. Способ получения {2-[4-( или ее фармацевтически приемлемой кислотной аддитивной соли или соли металла путем гидролиза соответствующего ацетамида в щелочной или кислой среде и, при необходимости, превращения полученного продукта в кислотную аддитивную соль или соль металла и/или высвобождения основания из кислотной аддитивной соли или соли металла, отличающийся тем, что {2-[4-( где R1 и R2 независимо представляют собой C1-4алкил, который может быть замещен фенилом, C2-4алкенил или циклогексил, или R1 и R2 образуют вместе с соседним атомом азота морфолиновую группу, или его кислотную аддитивную соль используют в качестве ацетамида, и, при необходимости, проводят гидролиз в присутствии межфазного катализатора. 2. Способ по п.1, отличающийся тем, что {2-[4-( 3. {2-[4-( 4. (RS)-N,N-Диметил-{2-[4-( 5. Способ получения {2-[4-( подвергают реакции с 2-галогенацетамидом формулы где R1 и R2 определены выше, Х обозначает атом галогена, предпочтительней атом хлора, и, при необходимости, полученное основание формулы II превращают в кислотную аддитивную соль с неорганической или органической кислотой, или основание высвобождают из его кислотной аддитивной соли.
|
||||||||||||||||||||||||||
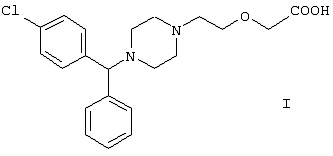
 -фенил-п-хлорбензил)пиперазин-1-ил]этокси}уксусной кислоты (цетиризина) формулы I
-фенил-п-хлорбензил)пиперазин-1-ил]этокси}уксусной кислоты (цетиризина) формулы I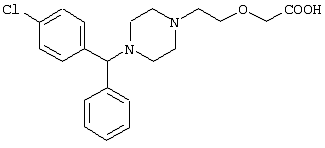
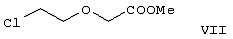
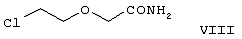
 С=O 1658 см-1
С=O 1658 см-1 м.д. 2,41 (уш.с, 4Н, пиперазин-3,5-NСН2), 2,53 (т. J=5,8 Гц, 4Н, пиперазин-2,6-NCH2), 2,62 (т, J=7,0 Гц, 2Н, этокси-NСН2), 2,92 и 2,98 (двойной с, 2×3H, NCH3), 3,63 (т, J=7,0 Гц, 2Н этокси-ОСН2), 4,14 (с, 2Н, СОСН2), 4,20 (с, 1Н, СН), 7,15-7,37 (м, 9Н, АrН).
м.д. 2,41 (уш.с, 4Н, пиперазин-3,5-NСН2), 2,53 (т. J=5,8 Гц, 4Н, пиперазин-2,6-NCH2), 2,62 (т, J=7,0 Гц, 2Н, этокси-NСН2), 2,92 и 2,98 (двойной с, 2×3H, NCH3), 3,63 (т, J=7,0 Гц, 2Н этокси-ОСН2), 4,14 (с, 2Н, СОСН2), 4,20 (с, 1Н, СН), 7,15-7,37 (м, 9Н, АrН).