Патент на изобретение №2152951
|
||||||||||||||||||||||||||
(54) 9-О-ОКСИМОВЫЕ ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА А И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ
(57) Реферат: Описываются новые 9-O-оксимовые производные эритромицина А общей формулы (I), где А, R1, R2, М, n, m и r имеют значения, указанные в п. 1 формулы, и фармацевтические композиции, содержащие их в качестве действующих веществ. Соединения формулы (I) пригодны для лечения инфекционных заболеваний. 2 с. и 3 з.п. ф-лы, 3 табл. 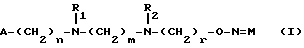
Изобретение относится к производным эритромицина A, обладающим антибиотической активностью, пригодным для лечения инфекционных заболеваний, в частности изобретение относится к производным 9-[O-(аминоалкил)оксима]эритромицина A, обладающим антибиотической активностью по отношению к грамположительным и грамотрицательным микроорганизмам. Эритромицин A [The Merck Index, XI изд., N 3626] представляет собой хорошо известный, встречающийся в естественных условиях макролид, который обладает антибиотической активностью и который имеет следующую структуру 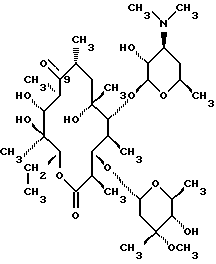 Помимо активности по отношению к некоторым микроорганизмам, не относящимся к бактериям, таким, как риккетсии и микоплазмы, эритромицин A обладает антибактериальной активностью в основном по отношению к грамположительным микроорганизмам, таким, как стрептококки, стафилококки и пневмококки, но он также проявляет эффективность по отношению к некоторым грамотрицательным микроорганизмам, таким, как, например, Haemophilus influenzae, Neisseria gonorrhoeae и Bordetella pertussis. Помимо хорошо известной активности по отношению к вышеуказанным прокариотам по появившимся недавно литературным данным эритромицин A и другие антибиотики из класса макролидов обладают активностью по отношению к эукариотичным паразитам [Р.А. Lartey и др., Advances in Pharmacology, 28, 307-343 (1994)]. По отношению к эритромицину A, равно как и по отношению к другим антибактериальным лекарствам, у некоторых штаммов бактерий наблюдали явление устойчивости. В частности это явление наблюдали при продолжительном применении эритромицина A при лечении инфекций, вызванных стафилококками [A. Kucers и N McK. Bennett, The use of antibiotics, A comprehensive Review with Clinical Emphasis, William Heinemann Medical, IV изд., (1987) 851-882]. Интерес к антибиотикам из класса макролидов обусловлен возможностью их применения в клинической терапии и ветеринарии при лечении некоторых инфекционных болезней, таких, как, например, инфекционные заболевания дыхательных путей, желудочно-кишечного тракта, мочеполовых путей и наружных органов, таких, как кожа, глаза и уши. Вследствие своей низкой стабильности в кислой среде эритромицин A необратимо превращается, например, в желудочно-кишечном тракте при оральном введении, в производное, лишенное антибиотической активности, что определяет плохую биологическую доступность действующего вещества [H.A. Kirst, Annual Reports in Medical Chemistry, 25, 119-128 (1989)]. Для преодоления вышеуказанных недостатков исследование было направлено на соединения, которые, сохраняя хорошие антибиотические свойства эритромицина A, характеризовались бы более высокой стабильностью по отношению к кислотам и лучшими фармакокинетическими свойствами, такими, как, например, более высокая биологическая доступность при оральном введении, концентрация в крови, способность проникать в ткани и период полураспада. В качестве примера в данной области техники можно привести карбаматы и карбонаты эритромицина A или 9-O-оксима эритромицина A, описанные в заявках на европейский патент 0216169 и 0284203 (обе на имя Beecham Group PLC), а также соединения, описанные в европейской заявке 0033255 (фирма Roussel-Uclaf). В европейской заявке 0033255, в частности, описаны 9-O-оксимовые производные эритромицина A формулы R-A-O-N=Ery, где Ery обозначает остаток эритромицина A, в котором оксимовая группа (-N= Ery) замещает карбонильную группу в положении 9 (O = Ery); А обозначает прямую или разветвленную алкильную группу с 1-6 атомами углерода; R обозначает, в частности, необязательно замещенную алкоксигруппу с 1-6 атомами углерода или группу [-N(R1)R2], в которой R1 и R2 имеют одинаковые или различные значения и обозначают атом водорода или необязательно замещенную алкильную группу с 1-6 атомами углерода. Соединения, описанные в заявке на европейский патент 0033255, такие, как, например, 9-[O-[(2-метоксиэтокси)метил]оксим] эритромицина A, известный под международным незапатентованным названием рокситромицин [The Merck Index, XI изд. , N 8253], 9-[O-[(2-диметиламино)этил]оксим]эритромицина A и 9-[O-[(2-диэтиламино)этил]оксим]эритромицина A, обладают спектром активности in vitro, сопоставимым с таковым эритромицина A, но обладают более высокой стабильностью по отношению к кислотам и лучшими фармакокинетическими свойствами. Указанные соединения, следовательно, проявляют антибиотическую активность по отношению к грамположительным микроорганизмам, таким, как стафилококки, стрептококки и пневмококки, и по отношению к некоторым грамотрицательным микроорганизмам, таким, как, например, Haemophilus influenzae и Haemophilus pertussis. Согласно настоящему изобретению были обнаружены соединения, являющиеся 9-O-оксимовыми производными эритромицина A, в частности соединения, представляющие собой производные 9-[О-(аминоалкил)оксима] эритромицина A, частично подпадающие под объем, но не приведенные в качестве примера в европейской заявке 0033255, которые обладают более широким спектром антибактериальной активности по отношению к грамположительным микроорганизмам, прежде всего по отношению к грамотрицательным микроорганизмам, и улучшенными фармакокинетическими свойствами, такими, как, например, большая продолжительность действия и больший период полураспада при элиминации в тканях по сравнению с соединениями, описанными в вышеуказанной европейской заявке. Таким образом, предметом настоящего изобретения являются соединения формулы 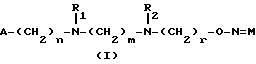 где A обозначает фенильную группу или гетероцикл с 5 или 6 членами, содержащий один или несколько гетероатомов, выбранных из азота, кислорода и серы, необязательно замещенный 1-3 одинаковыми или различными группами, выбранными из прямых или разветвленных (C1-C4)алкильных или алкоксигрупп, (C1-C2)алкилендиоксигрупп, (C1-C4)алкилсульфонильных групп, фенильной, фенокси-, гидрокси-, карбокси-, нитрогруппы, галогена и трифторметильной группы; R1 и R2 имеют одинаковые или различные значения и обозначают атом водорода или прямые, либо разветвленные (C1-C4)алкильные группы; n = 1 или 2; m = 1 – 8; r = 2 – 6; М обозначает группу формулы 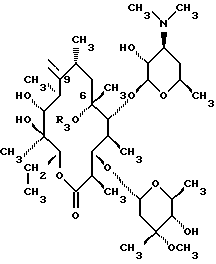 где R3 обозначает атом водорода или метильную группу; и их фармацевтически приемлемые соли. Оксимы формулы (I) могут иметь Z- или E-конфигурацию. Таким образом, предметом настоящего изобретения являются соединения формулы (I), имеющие Z- или E-конфигурацию, причем предпочтительны последние. Соединения формулы (I) проявляют антибиотическую активность и характеризуются высокой стабильностью по отношению к кислотам и хорошими фармакокинетическими свойствами, что позволяет применять их в терапии человека или в ветеринарии для лечения некоторых инфекционных заболеваний, таких, как, например, заболевания центральной нервной системы, верхних и нижних дыхательных путей, желудочно-кишечного тракта, мочеполовых путей, зубной ткани и наружных органов, таких, как кожа, глаза и уши. В настоящем описании, если не указано иное, под понятием (C1-C4)алкильная группа понимают прямую или разветвленную (C1-C4)алкильную группу, такую, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил; под понятием (C1-C4)алкоксигруппа понимают прямую или разветвленную (C1-C4)алкоксигруппу, такую, как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси; под понятием (C1-C2)алкилендиоксигруппа понимают метилендиокси- или этилендиоксигруппу. Под понятием гетероцикл с 5 или 6 членами, содержащий один или несколько гетероатомов, выбранных из азота, кислорода и серы, понимают гетероцикл, предпочтительно выбранный из пиридина, пиррола, пирролидина, фурана, тетрагидрофурана и тиофена. Предпочтительными соединениями являются соединения формулы (I), в которых A обозначает фенильную группу или гетероцикл, выбранный из пиридина и фурана, необязательно замещенный 1-3 группами, выбранными из гидрокси-, метокси-, метилендиокси-, н-бутокси-, феноксигрупп, фенила, метилсульфонила, нитрогруппы, галогена и трифторметила; R1 и R2 имеют одинаковые значения и обозначают атом водорода или метильную группу; R3 обозначает атом водорода. Еще более предпочтительными соединениями являются соединения формулы (I), в которых A обозначает фенильную группу, необязательно замещенную группой, выбранной из феноксигруппы, нитрогруппы и трифторметила; R1 и R2 имеют одинаковые значения и обозначают атом водорода или метильную группу; n = 1; m = 6; r = 2; R – атом водорода. Фармацевтически приемлемые соли соединений формулы (I) представляют собой соли органических или неорганических кислот, таких, как, например, соляная, бромистоводородная, йодистоводородная, азотная, серная, фосфорная, уксусная, винная, лимонная, бензойная, янтарная и глутаровая кислота. Конкретными примерами предпочтительных соединений формулы (I) являются: (Е)-9-[О-[2-[6-(фенилметиламино)гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[2-(фенилметиламино)этиламино]этил]оксим]эритромицина A; (Е)-9-[О-[6-[6-(фенилметиламино)гексиламино]гексил]оксим]эритромицина A; (Е)-9-[О-[6-[3-(фенилметиламино)пропиламино]гексил]оксим]эритромицина A; (Е)-9-[О-[6-[5-(фенилметиламино)пентиламино]гексил]оксим]эритромицина A; (Е)-9-[О-[2-[8-(фенилметиламино)октиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[5-(фенилметиламино)пентиламино]этил]оксим]эритромицина A; (Е)-9-[О-[5-[6-(фенилметиламино)гексиламино]пентил]оксим]эритромицина A; (Е)-9-[О-[3-[6-(фенилметиламино)гексиламино]пропил]оксим]эритромицина A; (Е)-9-[О-[3-[4-(фенилметиламино)бутиламино]пропил]оксим]эритромицина A; (Е)-9-[О-[2-[N-метил-6-(N’-метил-N’-фенилметиламино)гексиламино] этил] оксим]эритромицина A; (Е)-9-[О-[2-[6-[(бифенил-4-ил)метиламино] гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(3-феноксифенил)метиламино] гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(4-феноксифенил)метиламино] гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-(2- фурилметиламино)гексиламино] этил]оксим]эритромицина A; (E)-9-[O-[2-[6-(3-пиридилметиламино)гексиламино] этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(4-метоксифенил)метиламино]гексиламино]этил] оксим]эритромицина A; (Е)-9-[O-[2-[6-[(4-н-бутоксифенил)метиламино] гексиламино] этил] оксим] эритромицина A; (Е)-9-[О-[2-[6-[(3,4-метилендиоксифенил)метиламино] гексиламино] этил] оксим]эритромицина A; (Е)-9-[О-[2-[6-[(4-метилсульфонилфенил)метиламино] гексиламино] этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(4-фторфенил)метиламино]гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(2-трифторметилфенил)метиламино] гексиламино]этил]оксим] эритромицина A; (Е)-9-[О-[2-[6-[(3-трифторметилфенил)метиламино] гексиламино]этил]оксим] эритромицина A; (Е)-9-[О-[2-[6-[(4-трифторметилфенил)метиламино] гексиламино]этил]оксим] эритромицина A; (Е)-9-[О-[2-[6-[(2-гидроксифенил)метиламино]гексиламино]этил]оксим]эритромицина A; (Е)-9-[O-[2-[6-[(3-гидроксифенил)метиламино]гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(4-гидроксифенил)метиламино]гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(3,5-дихлор-2-гидроксифенил)метиламино]гексиламино]этил] оксим]эритромицина A; (Е)-9-[О-[2-[6-[(2-нитрофенил)метиламино] гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(3-нитрофенил)метиламино] гексиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[6-[(4-нитрофенил)метиламино] гексиламино]этил]оксим]эритромицина A; (E)-9-[O-[2-[6-[(4-гидрокси-3-нитрофенил)метиламино] гексиламино] этил] оксим]эритромицина A; (Е)-9-[О-[2-[6-[(3-гидрокси-4-нитрофенил)метиламино] гексиламино] этил] оксим]эритромицина A; (E)-9-[О-[2-[N-метил-6-[N’-метил-N’-(4-трифторметилфенил)метиламино] гексиламино]этил]оксим]эритромицина A. Получение соединений формулы (I), являющихся предметом настоящего изобретения, может быть осуществлено в соответствии с описанным ниже способом синтеза. Способ включает, во-первых, реакцию конденсации между соответствующей кислотой формулы 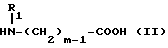 R1 и m имеют указанные выше значения, с хлористым ацилом формулы A-(CH2)n-1-COCl (III) где A и n имеют указанные выше значения. Реакцию конденсации осуществляют в соответствии со стандартными методами в инертном растворителе и в присутствии основания, такого, как, например, гидроксид щелочного металла, с получением соединений формулы 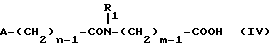 где A, R1, n и m имеют указанные выше значения. Полученные таким образом N-ациламинокислоты формулы (IV) далее подвергают конденсации в соответствии со стандартными методами с аминоэфирами формулы 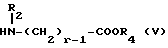 где R2 и r имеют указанные выше значения, a R4 обозначает метильную или этильную группу, с получением соединений формулы 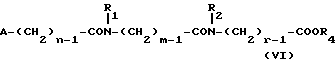 где A, R1, R2, R4, n, m и r имеют указанные выше значения. В соответствии со стандартными методами соединения формулы (VI) далее восстанавливают, например, с помощью борогидрида натрия в присутствии кислот, алюмогидрида лития, диметилсульфидборана или путем каталитического гидрирования, до соответствующих аминоспиртов формулы 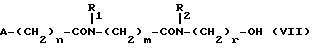 где A, R1, R2, n, m и r имеют указанные выше значения. Аминоспирты формулы (VII) затем превращают в соответствующие сульфонильные производные формулы (VIII), например, с помощью метансульфонилхлорида или пара-толуолсульфонилхлорида, и далее конденсируют с 9-O-оксимом эритромицина A или с 9-O-оксимом 6-O-метилэритромицина A, причем оба соединения могут быть представлены формулой (IX), с получением соединений формулы (I) 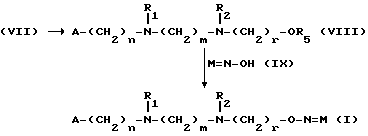 где A, R1, R2, M, n, m и r имеют указанные выше значения, a R5 обозначает мезильную или тозильную группу. Реакцию между соединениями формулы (VIII) и оксимами формулы (IX) осуществляют в инертном органическом растворителе, таком, как, например, тетрагидрофуран, этиловый эфир или 1,2-диметоксиэтан, в присутствии трет-бутилата калия и 18-краун-6-эфира в качестве комплексообразующего агента. Для специалиста в данной области техники очевидно, что, если реакцию сульфонилирования осуществляют с использованием соединений формулы (VII), в которых один или оба заместителя R1 и R2 обозначают атом водорода, может оказаться необходимым защитить атом или атомы азота перед осуществлением реакции сульфонилирования. В этом случае конденсацию полученных таким образом N-защищенных сульфонильных производных с оксимами формулы (IX) осуществляют аналогично тому, как это описано выше, а последующее удаление защитной группы осуществляют в соответствии с принятыми методами, что дает возможность получить соединения формулы (I), в которых один или оба заместителя R1 и R2 обозначают атом водорода. Более подробно защита аминов описана в литературе, например, см. T. W. Greene и P. G.M. Wuts, Protective groups in organic synthesis, John Wiley & Sons, Inc., 2-е изд., (1991), 309-405. Соединения формул (II), (III) и (V) являются известными или легко могут быть получены в соответствии с известными методами. Оксимы формулы (IX) также являются известными соединениями и могут быть получены в соответствии со стандартными методами, включающими, например, взаимодействие эритромицина A или 6-О-метилэритромицина A с гидрохлоридом гидроксиламина. Сложные эфиры формулы (VI) необязательно могут быть получены в соответствии с альтернативным методом синтеза, включающим, во-первых, конденсацию соответствующей аминокислоты формулы (II) с аминоэфиром формулы (V) c получением соединений формулы 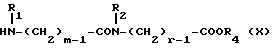 где R1, R2, R4, m и r имеют указанные выше значения. Специалисту в данной области техники очевидно, что перед осуществлением реакции конденсации между аминокислотой формулы (II) и аминоэфиром формулы (V) может оказаться необходимым соответствующим образом защитить аминогруппу аминокислоты формулы (II) аналогично тому, как это уже было описано для реакции сульфонилирования. Последующую конденсацию соединений формулы (X) с соединением формулы (III) осуществляют в соответствии со стандартными методами, а необязательное удаление защитной группы затем позволяет получить соединения формулы (VI). Получение соединений формулы (I), в которых по крайней мере один из двух заместителей R1 и R2 обозначает группу, выбранную из этила, н-пропила, н-бутила и изобутила, может быть осуществлено в соответствии с альтернативным способом синтеза, описанным ниже. Этот способ включает, во-первых, ацилирование атома или атомов азота аминоспиртов формулы (VII), где один или оба заместителя R1 и R2 обозначают атом водорода. Например, при использовании соединения формулы (VII), в котором оба заместителя R1 и R2 обозначают атом водорода, в соответствии со стандартными методами в присутствии пригодного ацилхлорида (R’COCl) можно получить соединения формулы 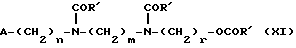 где A, n, m и r имеют указанные выше значения, a R’ обозначает прямую или разветвленную (C1-C3)алкильную группу. Восстановление соединений формулы (XI), осуществляемое в соответствии со стандартными методами, позволяет получить соединения формулы 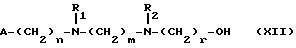 где A, n, m и r имеют указанные выше значения, a R1 и R2 обозначают этил, н-пропил, н-бутил или изобутил, из которых после превращения в соответствующие сульфонильные производные и конденсации с оксимами формулы (IX) аналогично тому, как это описано выше, можно получить соединения формулы 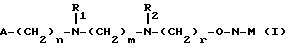 где A, M, n, m и r имеют указанные выше значения, a R1 и R2 обозначают этил, н-пропил, н-бутил или изобутил. Ниже описан процесс синтеза, альтернативный по отношению к тому, который описан ранее для получения соединений формулы (I), являющихся предметом настоящего изобретения. Этот процесс включает, во-первых, окисление соответствующего N-защищенного аминоспирта, такого, как, например, N-бензилоксикарбониламиноспирт формулы (XIII), в присутствии гипохлорита натрия и свободного радикала 2,2,6,6-тетраметилпиперидинокси (ТЕМПО) в инертном органическом растворителе с получением соединений формулы (XIV) 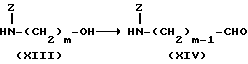 где m имеет указанные выше значения, а Z обозначает защитную группу. Примерами инертных органических растворителей, пригодных для реакции окисления, являются, например, метиленхлорид, хлороформ, черыреххлористый углерод, 1,2-дихлорэтан, этилацетат, бензол и толуол. Аминирование полученного таким образом альдегида в присутствии пригодного аминоспирта формулы (XV) и восстановление полученного промежуточного продукта, например, в присутствии боргидрида натрия, позволяет получить аминоспирты формулы (XVI) 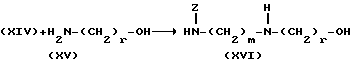 где Z, m и r имеют указанные выше значения. Последующая защита азота аминогруппы соединений формулы (XVI) и тем самым превращение в соответствующие сульфонильные производные, конденсация с оксимами формулы (IX) и удаление защитной группы атомов азота аналогично тому, как это описано выше, позволяют получить соединения формулы 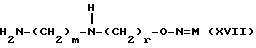 где М, m и r имеют указанные выше значения. Промежуточные оксимы формулы (XVII), сконденсированные с соответствующим альдегидом формулы (XVIII) и восстановленные, например, каталитическим гидрированием, позволяют получить соединения формулы (I) 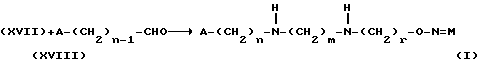 где A, M, m и r имеют указанные выше значения. Соединения формул (XIII), (XV) и (XVIII) являются известными или легко могут быть получены в соответствии с известными методами. Соединения формулы (I), в которых один или оба заместителя R1 и R2 обозначают атом водорода, полученные в соответствии с одним из описанных выше способов, необязательно могут быть алкилированы по атому или атомам азота диаминофрагмента в соответствии со стандартными методами, включающими, например, конденсацию с соответствующим альдегидом и восстановление полученного промежуточного продукта. Таким путем получают соединения формулы (I), в которых R1 и R2 имеют одинаковые или различные значения и обозначают прямую или разветвленную (C1-C4)алкильную группу. Получение соединений формулы (I), имеющих Z- или E-конфигурацию, осуществляют в соответствии с одной из схем синтеза, описанных выше, используя оксим формулы (IX) в требуемой конфигурации [J.C. Casс и др., The Journal of Antibiotics, 44, 313-330, (1991)]. Было установлено, что соединения формулы (I) проявляют антибактериальную активность по отношению к некоторым грамположительным и грамотрицательным микроорганизмам и пригодны для применения в клинической терапии и в ветеринарии при лечении некоторых инфекционных заболеваний, таких, как, например, заболевания центральной нервной системы, верхних и нижних дыхательных путей, желудочно-кишечного тракта, мочеполовых путей, зубной ткани и наружных органов, таких, как кожа, глаза и уши. Кроме того, эти соединения обладают активностью по отношению к некоторым представляющим клинический интерес грамположительным микроорганизмам, устойчивым к эритромицину A или в более общем случае к антибиотикам из класса макролидов, характеризующихся наличием 14- или 15-членных макролактонов. Антибактериальную активность соединений формулы (I) по отношению к грамположительным микроорганизмам, таким, как Streptococcus pyogenes, Streptococcus pneumoniae, Enterococcus faecalis и Staphylococcus aureus, и грамотрицательным микроорганизмам, таким, как Escherichia coli и Klebsiella pneumoniae, оценивали с помощью опытов in vitro, пригодных для оценки минимальной концентрации антибиотика, позволяющей ингибировать рост бактерий (МИК) (пример 23). В качестве эталонов применяли рокситромицин и кларитромицин [The Merck Index, XI изд., N 8253 и N 2340 соответственно]. Было установлено, что антибактериальная активность соединений формулы (I) по отношению к грамположительным микроорганизмам практически сопоставима с таковой рокситромицина и кларитромицина, т.е. двух макролидов, характеризующихся в опытах in vitro высокой антибактериальной активностью (таблица 1). Было установлено также, что по отношению к грамотрицательным микроорганизмам, в частности по отношению к энтеробактериям, таким, как Escherichia coli и Klebsiella pneumoniae, соединения формулы (I) являются существенно более активными по сравнению с обоими соединениями, используемыми в качестве эталонов (таблица 2). В этой связи интересно отметить, что соединения формулы (I), являющиеся предметом настоящего изобретения, оказались более активными, чем рокситромицин, описанный в вышеуказанной европейской заявке 0033255 и выбранный в качестве предпочтительного соединения по отношению к нескольким другим производным, таким, как, например, 9-[О-[(2-диметиламино)этил]оксим]эритромицина A [J.C. Casс и др., The Journal of Antibiotics, 44, 313-330, (1991)]. Кроме того, было установлено, что соединения формулы (I) проявляют активность в опытах in vivo (таблица 3). Антибактериальную активность соединений формулы (I) в опытах in vivo, выраженную в виде средней защищающей дозы PD50 (мг/кг), оценивали с помощью экспериментальной легочной инфекции, вызванной у мышей с помощью Streptococcus pyogenes (пример 23). При анализе данных об активности, обнаруженной в опытах in vivo, становится очевидным, что соединения формулы (I) отличаются пролонгированным действием и длительным периодом полураспада при элиминации из ткани. Действительно, после внутрибрюшинного введения мыши соединения формулы (I) быстро и повсеместно распределялись по всему организму, а уровни в тканях оказывались выше, чем в плазме. Эти результаты становятся особенно очевидными при анализе значений PD50 для соединений формулы (I), введенных за 24 часа до или через 1 час после заражения. Действительно, эти величины оказались практически неизмененными после введения за 24 часа до или через 1 час после заражения. В случае экспериментальной легочной инфекции, вызванной у мышей с помощью Streptococcus pyogenes, т.е. патогена, обычно вызывающего респираторные заболевания, эффективные концентрации соединений формулы (I), введенные внутрибрюшинно, сохранялись в области легких в течение 24-48 часов после введения. В отличие от этого рокситромицин и кларитромицин, т.е. используемые в качестве эталонов соединения, будучи введенными за 24 часа до заражения, оказались неактивными. Следовательно, соединения формулы (I) также обладают избирательной активностью по отношению к легким и могут успешно применяться для лечения инфекций дыхательных путей. В дополнение к вышеуказанной активности по отношению к бактериальным микроорганизмам соединения формулы (I), являющиеся предметом настоящего изобретения, являются активными по отношению к эукариотным патогенам, в частности, они обладают выраженной активностью по отношению к простейшим, таким, как Plasmodium falciparum, который является хорошо известным возбудителем малярии. Следовательно, соединения формулы (I) также могут успешно применяться для лечения заболеваний малярией. Наряду с тем, что они характеризуются широким спектром антибиотической активности по отношению к грамположительным и грамотрицательным микроорганизмам и простейшим, хорошей стабильностью по отношению к кислотам и хорошими фармакокинетическими свойствами, острая токсичность для мышей соединений формулы (I) сопоставима с таковой рокситромицина. Следовательно, отличаясь высокой безопасностью при применении, они могут успешно использоваться в терапии человека и в ветеринарии. Соединения формулы (I) предпочтительно следует использовать в соответствующей фармацевтической форме, пригодной для орального, парентерального введения, в виде суппозитория или для местного применения. Таким образом, предметом настоящего изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество одного или нескольких соединений формулы (I) в смеси с фармацевтически приемлемым носителем. Эти фармацевтические формы включают таблетки, капсулы, сиропы, растворы для инъекций, готовые к применению или получаемые перед применением путем разбавления лиофилизированной формы, суппозитории, растворы, кремы, мази и глазные примочки. Для ветеринарных целей в дополнение к вышеуказанным композициям возможно изготовление твердых или жидких концентратов, подлежащих разбавлению в пище или в воде для питья. В соответствии с типом композиции кроме терапевтически эффективного количества одного или нескольких соединений формулы (I) эти композиции должны содержать твердые или жидкие эксципиенты или растворители для фармацевтической или ветеринарной целей и необязательно другие добавки, обычно используемые в технике изготовления препаративных форм, такие, как загущающие агенты, агрегирующие агенты, замасливатели, агенты, способствующие дезинтеграции, корригенты и красители. Для лечения конкретных инфекций соединение формулы (I) может быть объединено с эффективным количеством другого действующего вещества. Эффективное количество соединения формулы (I) может варьироваться в зависимости от различных факторов, таких, как серьезность и фаза заболевания, орган или система, подверженная заболеванию, характеристики видов хозяев, чувствительность бактериальных видов, обусловливающих инфекцию, и выбранный путь введения. Терапевтическая доза обычно составляет от 0,5 до 100 мг/кг веса тела в день и может применяться в виде однократной дозы или нескольких суточных доз. Ниже изобретение проиллюстрировано на примерах, не ограничивающих его объем. Строение соединений формулы (I) и промежуточных продуктов синтеза при их получении подтверждали с помощью 1H-ЯМР- или 16C-ЯМР-спектроскопии. Величины значимых сигналов наиболее важных промежуточных продуктов и соединений формулы (I) приведены ниже в описании. Пример 1 Получение N-бензоил-6-аминокапроновой кислоты Раствор бензоилхлорида (0,18 моля) в этиловом эфире (160 мл) и раствор 1 н. гидроксида натрия (180 мл) одновременно добавляли к смеси 6-аминокапроновой кислоты (0,15 моля) в этиловом эфире (150 мл) и воды (200 мл), содержащей гидроксид натрия (0,15 моля), и перемешивали при температуре от 0 до 5oC. По завершении добавления соединений температуру реакционной смеси доводили до комнатной и перемешивали в течение еще 4 часов. После разделения фаз водную фазу промывали этиловым эфиром (200 мл) и подкисляли концентрированной соляной кислотой с использованием реактивной бумаги конго. После экстракции этилацетатом (3х200 мл) объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (200 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Таким путем получали N-бензоил-6-аминокапроновую кислоту, которую непосредственно применяли в последующих реакциях. По аналогичной методике получали следующие соединения: N-бензоил-3-аминопропановую кислоту; N-бензоилглицин; N-бензоил-8-аминооктановую кислоту; N-бензоил-4-аминобутановую кислоту; N-фенилацетил-6-аминокапроновую кислоту; N-фенилацетилглицин; N-бензоил-N-изопропил-4-аминобутановую кислоту; N-бензоил-N-изопропил-6-аминокапроновую кислоту. Пример 2 Получение этилового эфира N-[6-(бензоиламино)гексаноил]глицина Раствор дициклогексилкарбодиимида (112 ммолей) в безводном тетрагидрофуране (44 мл) постепенно добавляли к суспензии, содержащей N-бензоил-6-аминокапроновую кислоту (93,5 ммоля), полученную согласно примеру 1, гидрохлорид этилового эфира глицина (112 ммолей), триэтиламин (112 ммолей) и безводный 1-гидроксибензотриазол (112 ммолей) в тетрагидрофуране (330 мл) и перемешивали при температуре 0oC. Далее температуру реакционной смесь доводили до комнатной и перемешивали в течение 16 часов. В результате получали осадок, который удаляли фильтрацией, а полученный таким образом фильтрат упаривали при пониженном давлении. Остаток объединяли с этилацетатом (300 мл) и последовательно промывали раствором 5%-ной соляной кислоты (2х100 мл), насыщенным раствором хлорида натрия (100 мл), раствором 5%-ного бикарбоната натрия (2х100 мл) и в завершение насыщенным раствором хлорида натрия (100 мл). Органическую фазу сушили над сульфатом натрия и упаривали досуха при пониженном давлении, получая таким образом этиловый эфир N-[6-(бензоиламино)гексаноил]глицина, который непосредственно применяли в последующих реакциях. По аналогичной методике получали следующие соединения: этиловый эфир N-1(бензоиламино)ацетил] глицина; этиловый эфир N-[6-(фенилацетиламино)гексаноил]глицина; этиловый эфир N-[(фенилацетиламино)ацетил] глицина; этил-6-[6-(бензоиламино)гексаноиламино] гексаноат; метиловый эфир N-[5-(бензоиламино)пентаноил]глицина; метил-6-[5-(бензоиламино)пентаноиламино]гексаноат; метиловый эфир N-[7-(бензоиламино)гептаноил] глицина; метил-5-[6-(бензоиламино)гексаноиламино] пентаноат; метил-6-[(бензоиламино)ацетиламино] гексаноат; метил-3-[6-(бензоиламино)гексаноиламино]пропионоат; этил-6-[N-изопропил(фенилацетиламино)ацетиламино]гексаноат; метил-6-[4-(бензоиламино)бутаноиламино] гексаноат; метил-4-[N-изопропил-4-(N’-изопропил-N’-бензоиламино)бутаноиламино]бутаноат. Пример 3 Получение этилового эфира N-(6-аминоногексаноил)глицина а) К раствору гидроксида натрия (33,54 г; 0,831 моля) в воде (840 мл) и метаноле (400 мл) добавляли 6-аминокапроновую кислоту (100 г; 0,762 моля) и постепенно добавляли раствор ди-трет-бутилдикарбоната (168 г; 0,762 моля) в метаноле (140 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После этого вновь добавляли твердый ди-трет-бутилдикарбонат (17,5 г; 0,08 моля) и реакционную смесь перемешивали еще в течение 16 часов. Затем реакционную смесь промывали гексаном (2х400 мл), подкисляли раствором бисульфата калия до pH 1,5 и экстрагировали этилацетатом (3х450 мл). Объединенные органические фазы сушили над сульфатом натрия и упаривали досуха, получая при этом 6-(трет-бутоксикарбониламино)капроновую кислоту (163 г) в виде масла. б) Аналогично методике, описанной в примере 2, 6-(трет-бутоксикарбониламино) капроновую кислоту (163 г) непосредственно конденсировали с гидрохлоридом этилового эфира глицина (118 г; 0,845 моля), получая таким образом этиловый эфир N-[6-(трет-бутоксикарбониламино)гексаноил]глицина (285 г) в виде неочищенного продукта, который непосредственно применяли в последующей реакции, tпл 76-77oC (изопропиловый эфир). в) Раствор 6н. соляной кислоты (150 мл) в этилацетате (150 мл) добавляли к раствору этилового эфира N-[6-(трет-бутоксикарбониламино)гексаноил]глицина (285 г) в этилацетате (500 мл) и перемешивали при комнатной температуре. Через 24 часа образовывался осадок, который отфильтровывали, промывали этилацетатом и этиловым эфиром и сушили в термостате (50oC) под вакуумом. Таким путем получали этиловый эфир N-(6-аминоногексаноил)глицина (93 г) в виде неочищенного продукта, который непосредственно применяли в последующих реакциях. ТСХ (метиленхлорид:метанол:аммиак = 10:2:1) Rf=0,2. Пример 4 Получение этилового эфира N-[6-[(4-фторбензоил)амино]гексаноил]глицина Раствор 4-фторбензоилхлорида (47,4 ммоля) в метиленхлориде (30 мл) постепенно добавляли к суспензии, содержащей этиловый эфир N-(6-аминоногексаноил)глицина (39,5 ммоля), полученный согласно примеру 3, и триэтиламин (87 ммолей) в метиленхлориде (150 мл) и перемешивали при 0oC. Температуру полученной таким образом смеси, к которой затем добавляли триэтиламин (2 мл), доводили до комнатной и перемешивали. После выдержки в течение 1 часа в этих условиях реакционную смесь промывали раствором 5%-ной соляной кислоты (2х100 мл) и насыщенным раствором хлорида натрия (3х100 мл). Отделенную органическую фазу сушили над сульфатом натрия и упаривали досуха под вакуумом. Таким путем получали этиловый эфир N-[6-[(4-фторбензоил)амино]гексаноил]глицина в виде неочищенного продукта, который непосредственно применяли в последующих реакциях, tпл 121-122oC (этилацетат); ТСХ (этилацетат) Rf=0,3. По аналогичной методике получали следующее соединение: этиловый эфир N-[6-(2-фуроиламино)гексаноил] глицина, tпл 104-106oC (ацетонитрил/изопропиловый эфир); ТСХ (метиленхлорид:метанол = 95:5) Rf=0,3. Пример 5 Получение этилового эфира N-[6-[(4-метоксибензоил)амино]гексаноил]глицина Аналогично примеру 2 с использованием 4-метоксибензойной кислоты (33 ммоля) и этилового эфира N-(6-аминоногексаноил)глицина (39,5 ммоля), полученного согласно примеру 3, получали этиловый эфир N-[6-[(4-метоксибензоил)амино] гексаноил] глицина в виде неочищенного продукта, который непосредственно применяли в последующих реакциях, tпл 106-107oC. ТСХ (метиленхлорид:метанол = 90:10) Rf=0,46. По аналогичной методике получали следующие соединения: этиловый эфир N-[6-[(3,4-метилендиоксибензоил)амино]гексаноил]глицина, ТСХ (метиленхлорид:метанол = 90:10) Rf= 0,39; этиловый эфир N-[6-[(4-метилсульфонилбензоил)амино]гексаноил]глицина, tпл 124-126oC; ТСХ (метиленхлорид: метанол = 96:4) Rf=0,31; этиловый эфир N-[6-[(3-трифторметилбензоил)амино] гексаноил] глицина, tпл 102-104oC; ТСХ (метиленхлорид:метанол = 95:5) Rf= 0,38. Пример 6 Получение 2-[6-(фенилметиламино)гексиламино]этанола Серную кислоту (6 н.) в этиловом эфире (40,9 мл; 700 ммолей), полученную смешением 96%-ной серной кислоты (33 мл) и этилового эфира (100 мл), постепенно добавляли к суспензии, содержащей этиловый эфир N-[6-(бензоиламино)гексаноил] глицина (46,8 ммоля), полученный согласно примеру 2, и борогидрид натрия (700 ммолей) в безводном тетрагидрофуране (200 мл), и перемешивали при температуре от 15oC до 20oC. Реакционную смесь доводили до температуры кипения в течение 24 часов и затем охлаждали до 0oC. Затем при перемешивании добавляли метанол (150 мл). Растворитель выпаривали при пониженном давлении и остаток объединяли с раствором 6 н. гидроксида натрия (200 мл), выдерживая полученную смесь при температуре кипения в течение 24 часов. Затем реакционную смесь, охлажденную до комнатной температуры, экстрагировали тетрагидрофураном (2х100 мл) и органическую фазу упаривали досуха, объединяли с этилацетатом и сушили над сульфатом натрия. Путем подкисления эфирным раствором соляной кислоты получали осадок, представляющий собой 2-[6-(фенилметиламино)гексиламино]этанол в виде гидрохлорида. Полученный таким образом неочищенный продукт непосредственно применяли в последующих реакциях. По аналогичной методике получали следующие соединения: 2-[2-(фенилметиламино)этиламино]этанол; 2-[6-(2-фенилэтиламино)гексиламино] этанол; 6-[6-(фенилметиламино)гексиламино]гексанол; 2-[5-(фенилметиламино)пентиламино] этанол; 2-[8-(фенилметиламино)октиламино]этанол; 5-[6-(фенилметиламино)гексиламино]пентанол; 6-[3-(фенилметиламино)пропиламино] гексанол; 3-[6-(фенилметиламино)гексиламино] пропанол; 3-[4-(фенилметиламино)бутиламино]пропанол; 6-[2-(фенилметиламино)этиламино]гексанол; 6-[N-изопропил-4-(фенилметиламино)бутиламино]гексанол; 2-[6-[(4-фторфенил)метиламино]гексиламино] этанол; 2-[6-[(4-метоксифенил)метиламино] гексиламино]этанол; 2-[6-[(3,4-метилендиоксифенил)метиламино] гексиламино] этанол; 2-[6-[(3-трифторметилфенил)метиламино] гексиламино] этанол; 2-[6-[(4-метилсульфонилфенил)метиламино] гексиламино] этанол; 4-[N-изопропил-4-(N’-изопропил-N’-фенилметиламино)бутиламино]бутанол. Пример 7 Получение 6-[N-ацетил-6-(N’-ацетил-N-фенилметиламино)гексиламино]гексилацетата Триэтиламин (1,95 мл; 14 ммолей) и раствор ацетилхлорида (0,62 мл; 8,69 ммоля) в метиленхлориде (5 мл) постепенно добавляли к суспензии, содержащей 6-[6-(фенилметиламино)гексиламино]гексанол (1 г; 2,6 ммоля), полученный согласно примеру 6, в метиленхлориде (15 мл), и перемешивали при 0oC. После перемешивания в течение 1 часа при 0oC температуру реакционной смеси доводили до комнатной и перемешивали еще в течение 16 часов. Затем реакционную смесь промывали раствором 10%-ной соляной кислоты (10 мл) и насыщенным раствором хлорида натрия. После разделения фаз органическую фазу сушили над сульфатом натрия и упаривали досуха под вакуумом, получая таким образом 6-[N-ацетил-6-(N’-ацетил-N’-фенилметиламино) гексиламино]гексилацетат (1,18 г) в виде масла, который непосредственно применяли в последующих реакциях. По аналогичной методике получали следующее соединение: 2-[N-ацетил-6-[N’-ацетил-N’-(2-фенилэтил)амино]гексиламино] этилацетат. Пример 8 Получение 6-[N-этил-6-(N’-этил-N’-фенилметиламино)гексиламино]гексанола Аналогично примеру 6 с использованием 6-[N-ацетил-6-(N’-ацетил-N’- фенилметиламино)гексиламино]гексилацетата, полученного согласно примеру 7, получали 6-[N-этил-6-(N’-этил-N’-фенилметиламино)гексиламино]гексанол. По аналогичной методике получали следующее соединение: 2-[N-этил-6-[N’-этил-N’-(2-фенилэтил)амино]гексиламино]этанол. Пример 9 Получение 2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’- фенилметиламино)гексиламино]этанола Раствор 1 н. гидроксида натрия (44,5 мл) и толуоловый раствор 50%-ного бензилхлорформиата (44,5 ммоля) в этилацетате (33 мл) постепенно и одновременно добавляли к раствору дигидрохлорида 2-[6-(фенилметиламино)гексиламино] этанола (18,5 ммоля), полученного согласно примеру 6, в растворе 1 н. гидроксида натрия (37,1 мл) и этилацетата (40 мл), и перемешивали при температуре 0oC. По завершении добавлений температуру реакционной смеси доводили до комнатной и перемешивали в течение 24 часов. После разделения фаз водную фазу промывали этилацетатом (2х50 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (50 мл), сушили над сульфатом натрия и упаривали досуха под вакуумом. Таким путем получали 2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’-фенилметиламино)гексиламино] этанол в виде масла, который непосредственно применяли в последующих реакциях. ТСХ (этилацетат:гексан = 50:50) Rf=0,20. По аналогичной методике получали следующие соединения: 2-[N-бензилоксикарбонил-2-(N’-бензилоксикарбонил-N’-фенилметиламино) этиламино]этанол, ТСХ (этилацетат:гексан = 60:40) Rf=0,25; 6-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’-фенилметиламино)гексиламино]гексанол, ТСХ (этилацетат:гексан = 50:50) Rf=0,27; 6-[N-бензилоксикарбонил-5-(N’-бензилоксикарбонил-N’-фенилметиламино) пентиламино]гексанол; 2-[N-бензилоксикарбонил-5-(N’-бензилоксикарбонил-N’-фенилметиламино) пентиламино]этанол; 2-[N-бензилоксикарбонил-8-(N’-бензилоксикарбонил-N’-фенилметиламино) октиламино]этанол; 5-[N-бензилоксикарбонил-6-(N’-бензилoкcикapбoнил-N’-фенилметилaмино) гексиламино]пентанол; 6-[N-бензилоксикарбонил-3-(N’-бензилоксикарбонил-N’-фенилметиламино) пропиламино]гексанол; 3-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’-фенилметиламино) гексиламино]пропанол; 3-[N-бензилоксикарбонил-4-(N’- бензилоксикарбонил-N’-фенилметиламино) бутиламино]пропанол; 6-[N-изопропил-2-[N’-бензилоксикарбонил-N’-(2-фенилэтил)амино]этиламино] гексанол, ТСХ (метиленхлорид:метанол:аммиак = 95:5:0,5) Rf=0,33; 6-[N-бензилоксикарбонил-4-(N’-изопропил-N’-фенилметиламино)бутиламино] гексанол, ТСХ (метиленхлорид:метанол:аммиак = 95:5:0,5) Rf=0,42; 2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’-[(4-фторфенил)метил] амино]гексиламино]этанол, ТСХ (этилацетат:гексан = 60:40) Rf=0,35; 2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’-[(4-метоксифенил) метил]амино]гексиламино]этанол, ТСХ (этилацетат:гексан = 50:50) Rf=0,2; 2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’- [(3,4-метилендиоксифенил)метил] амино] гексиламино] этанол, ТСХ (этилацетат:гексан = 60:40) Rf=0,26; 2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’- [(3-трифторметилфенил)метил] амино] гексиламино] этанол, ТСХ (этилацетат:гексан = 50:50) Rf= 0,25; 2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’-[(4- метилсульфонилфенил)метил] амино] гексиламино]этанол, ТСХ (этилацетат:гексан = 90:10) Rf= 0,36. Пример 10 Получение 2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’-фенилметиламино) гексиламино]этилметансульфоната Раствор метансульфонилхлорида (3,16 ммоля) в метиленхлориде (5 мл) постепенно добавляли к раствору 2-[N-бензилоксикарбонил-6-(N‘-бензилоксикарбонил-N’- фенилметиламино)гексиламино]этанола (2,6 ммоля), полученного согласно примеру 9, в метиленхлориде (15 мл), содержащем триэтиламин (0,44 мл; 3,16 ммоля), и перемешивали при температуре 0oC. Реакционную смесь, температуру которой доводили до комнатной и перемешивали в течение 5 часов, добавляли к раствору 5%-ной соляной кислоты (20 мл). После разделения фаз органическую фазу промывали 5%-ной соляной кислотой (10 мл) и насыщенным раствором хлорида натрия (3х10 мл). Затем органическую фазу сушили над сульфатом натрия и упаривали досуха, получая таким образом 2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’-фенилметиламино)гексиламино]этилметансульфонат, который непосредственно применяли в реакции, приведенной в следующем примере. Пример 11 Получение (Е)-9-[О-[2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’-фенилметиламино) гексиламино]этил]оксима]эритромицина A (Е)-9-O-оксим эритромицина A (627 мг; 0,84 ммоля), 18-краун-6-эфир (220 мг; 0,84 ммоля) и раствор 2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’- фенилметиламино)гексиламино]этилметансульфоната (0,84 ммоля), полученного согласно примеру 10, в безводном тетрагидрофуране (5 мл) добавляли в указанном порядке к суспензии трет-бутилата калия (103 мг; 0,92 ммоля) в безводном тетрагидрофуране (5 мл) и выдерживали при комнатной температуре и при перемешивании в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов и затем упаривали при пониженном давлении. Остаток объединяли с этилацетатом (10 мл) и полученную таким образом смесь промывали насыщенным раствором хлорида натрия (10 мл). Водную фазу экстрагировали этилацетатом (2х10 мл) и объединенные органические фазы сушили над сульфатом натрия и упаривали досуха. Таким путем получали (Е)-9-[О-[2-[N-бензилоксикарбонил-6-(N‘бензилоксикарбонил-N’- фенилметиламино)гексиламино] этил]оксим] эритромицина A и применяли его непосредственно в последующих реакциях. ТСХ (метиленхлорид:метанол:аммиак = 90:9:1) Rf=0,58; МС (C. I. ) (М+Н)+ = 1250; 1H-ЯМР (200 МГц, CDCl3):  (част./млн) 7,38-7,10 (m, 15Н, (ароматические соединения); 5,18-5,10 (m, 4H, (част./млн) 7,38-7,10 (m, 15Н, (ароматические соединения); 5,18-5,10 (m, 4H, 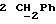 ); 3,30 (s, 3Н, OCH3); 2,26 (s, 6Н, 2NCH3); 0,81 (t, 3Н ); 3,30 (s, 3Н, OCH3); 2,26 (s, 6Н, 2NCH3); 0,81 (t, 3Н 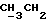 ).
По аналогичной методике получали следующие соединения: ).
По аналогичной методике получали следующие соединения:(Е)-9-[О-[2-[N-бензилоксикарбонил-2-(N’-бензилоксикарбонил-N’- фенилметиламино)этиламино]этил]оксим]эритромицина A, ТСХ (метиленхлорид:метанол:аммиак = 90:10:1) Rf=0,5; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,11-6,97 (m, 15Н, ароматические соединения); 5,18-4,97 (m, 4Н, 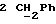 ; 3,30 (s, 3Н, ОCH3); 2,25 (s, 6H, 2NCH3); 0,82 (t, 3Н, ; 3,30 (s, 3Н, ОCH3); 2,25 (s, 6H, 2NCH3); 0,82 (t, 3Н, 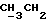 ); );(Е)-9-[О-[6-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’- фенилметиламино)гексиламино] гексил] оксим]эритромицина A, ТСХ (метиленхлорид:метанол: аммиак = 90:10:1) Rf=0,6; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,27-6,96 (m, 15Н, ароматические соединения); 5,05-4,92 (m, 4Н, 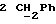 ); 3,17 (s, 3Н, OCH3); 2,13 (s, 6Н, 2NCH3); 0,70 (t, 3Н, ); 3,17 (s, 3Н, OCH3); 2,13 (s, 6Н, 2NCH3); 0,70 (t, 3Н, 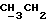 ); );(E)-9-[O-[6-[N-бензилоксикарбонил-3-(N’-бензилоксикарбонил-N‘– фенилметиламино)пропиламино] гексил] оксим]эритромицина A, ТСХ (метиленхлорид:метанол: аммиак = 90: 10:1) Rf=0,65; МС (C.I.) (М+Н)+ = 1194; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,39-7,01 (m, 15Н, ароматические соединения); 5,17-5,02 (m, 4H, 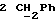 ); 3,30 (s, 3Н, OCH3); 2,27 (s, 6H, 2NCH3); 0,82 (t, 3Н, ); 3,30 (s, 3Н, OCH3); 2,27 (s, 6H, 2NCH3); 0,82 (t, 3Н, 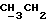 ); );(E)-9-[O-[6-[N-бензилоксикарбонил-5-(N’-бензилоксикарбонил-N’- фенилметиламино)пентиламино]гексил]оксим]эритромицина A; (Е)-9-[О-[2-[N-бензилоксикарбонил-8-(N’-бензилоксикарбонил-N’- фенилметиламино)октиламино]этил]оксим]эритромицина A; (Е)-9-[О-[2-[N-бензилоксикарбонил-5-(N’-бензилоксикарбонил-N’- фенилметиламино)пентиламино]этил]оксим]эритромицина A; (E)-9-[O-[5-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’- фенилметиламино)гексиламино]пентил]оксим]эритромицина A; (Е)-9-[О-[3-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил-N’- фенилметиламино)гексиламино]пропил]оксим]эритромицина A; (E)-9-[O-[3-[N-бензилоксикарбонил-4-(N’-бензилоксикарбонил-N’- фенилметиламино)бутиламино]пропил]оксим]эритромицина A; (Е)-9-[О-[6-[N-бензилоксикарбонил-2-[N’-бензилоксикарбонил-N’-(2- фенилэтил)амино] этиламино]гексил]оксим]эритромицина A, tпл 74-76oC; МС (C.I.) (М+Н)+ = 1172; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,38-7,03 (m, 10H, ароматические соединения); 5,13-5,03 (m, 2Н, 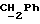 ); 3,29 (s, 3Н, OCH3); 2,25 (s, 6Н, 2NCH3); ); 3,29 (s, 3Н, OCH3); 2,25 (s, 6Н, 2NCH3);(Е)-9-[О-[6-[N-этил-6-(N’-этил-N’-фенилметиламино)гексиламино] гексил] оксим] эритромицина A (соединение 1), tпл 80-82oC (ацетонитрил); МС (C.I.) (М+Н)+ = 1094; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 175,20; 171,35; 140,06; 128,86; 128,07; 126,62; 102,96; 96,27; 53,54;(Е)-9-[O-[2-[N-этил-6-[N’-этил-N’-(2-фенилэтил)амино] гексиламино] этил] оксим]эритромицина A (соединение 2), ТСХ (хлороформ:гексан:триэтиламин = 45: 45: 10) Rf= 0,2; МС (C.I.) (М+Н)+ = 1052; 1H-ЯМР (200 МГц, CDCl3): (част. /млн) 7,26-7,04 (m, 5Н, ароматические соединения); 3,22 (s, 3Н, OCH3); 2,20 (s, 6Н, 2NCH3); 0,79 (t, 3Н, 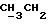 ); );(Е)-9-[О-[6-[N-бензилоксикарбонил-4-(N’-изопропил-N’- фенилметиламино)бутиламино] гексил] оксим] эритромицина A, tпл 75-77oC; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,47-7,12 (m, 10H, ароматические соединения); 5,18-4,97 (m, 4Н, 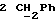 );3,30 (s, 3Н, OCH3); 2,25 (s, 6Н, 2NCH3); 0,82 (t, 3Н, );3,30 (s, 3Н, OCH3); 2,25 (s, 6Н, 2NCH3); 0,82 (t, 3Н, 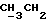 ); );(Е)-9-[O-[2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’- [(4-фторфенил)метил]амино]гексиламино]этил]оксим]эритромицина A, ТСХ (метиленхлорид: метанол:аммиак = 90:10:1) Rf= 0,62; 1H-ЯМР (200 МГц, CDCl3): (част. /млн) 7,38-6,88 (m, 15H, ароматические соединения); 5,17-5,03 (m, 2Н, 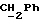 ); 3,29 (s, 3Н, OCH3); 2,26 (s, 6H, 2NCH3); 0,81 (t, 3Н, ); 3,29 (s, 3Н, OCH3); 2,26 (s, 6H, 2NCH3); 0,81 (t, 3Н, 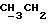 ); );(E)-9-[O-[2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’-[(4- метоксифенил)метил]амино]гексиламино]этил]оксим]эритромицина A, ТСХ (метиленхлорид:метанол:аммиак = 45:45:10) Rf=0,3; 1H-ЯМР (200 МГц, CDCl3): (част. /млн) 7,40-7,23 (m, 10Н,  ); 7,20-6,75 (m, 4Н, ); 7,20-6,75 (m, 4Н, 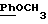 ); 5,52-5,17 (m, 4Н, ); 5,52-5,17 (m, 4Н, 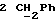 ); 3,77 (s, 3Н, ); 3,77 (s, 3Н,  ); 3,29 (s, 3Н, OCH3); 2,25 (s, 6H, 2NCH3); 0,82 (t, 3H, ); 3,29 (s, 3Н, OCH3); 2,25 (s, 6H, 2NCH3); 0,82 (t, 3H, 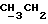 ); );(E)-9-[O-[2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’- [(3,4-метилендиоксифенил)метил] амино] гексиламино] этил]оксим]эритромицина A, ТСХ (метиленхлорид: метанол:аммиак = 95:5:0,5) Rf=0,31; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,38-7,22 (m, 10Н,  ); 6,78-6,55 (m, 3Н, ароматические соединения); 5,90 (s, 2Н, OCH2O); 5,15-5,02 (m, 4H, ); 6,78-6,55 (m, 3Н, ароматические соединения); 5,90 (s, 2Н, OCH2O); 5,15-5,02 (m, 4H, 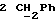 ); 3,29 (s, 3Н, OCH3); 2,26 (s, 6Н, 2NCH3); 0,82 (t, 3Н, ); 3,29 (s, 3Н, OCH3); 2,26 (s, 6Н, 2NCH3); 0,82 (t, 3Н, 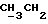 ); );(E)-9-[O-[2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-[(3- трифторметилфенил)метил]амино]гексиламино]этил]оксим]эритромицина A, ТСХ (метиленхлорид:метанол:аммиак = 90:10:1) Rf=0,65; 1H-ЯМР (200 МГц, CDCl3): (част. /млн) 7,54-7,15 (m, 14H, ароматические соединения); 5,20-5,03 (m, 4H, 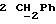 ); 3,30 (s, 3Н, OCH3); 2,26 (s, 6Н, 2NCH3); 0,82 (t, 3H, ); 3,30 (s, 3Н, OCH3); 2,26 (s, 6Н, 2NCH3); 0,82 (t, 3H, 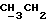 ); );(E)-9-[O-[2-[N-бензилоксикарбонил-6-[N’-бензилоксикарбонил-N’- [(4-метилсульфонилфенил)метил] амино] гексиламино] этил] оксим]эритромицина A, ТСХ (метиленхлорид:метанол:аммиак = 95:5:0,5) Rf=0,5; 1H-ЯМР (200 МГц, CDCl3): (част. /млн) 7,90-7,79 (m, 4H, 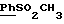 ); 7,48-7,15 (m, 10H, ); 7,48-7,15 (m, 10H,  ); 5,19-5,03 (m, 4H, ); 5,19-5,03 (m, 4H, 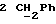 ); 3,30 (s, 3Н, OCH3); 3,02 (s, 3Н, CH3SO2); 2,27 (s, 6Н, 2NCH3); 0,82 (t, 3Н, ); 3,30 (s, 3Н, OCH3); 3,02 (s, 3Н, CH3SO2); 2,27 (s, 6Н, 2NCH3); 0,82 (t, 3Н, 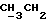 ); );(E)-9-[О-[4-[N-изопропил-4-(N’-изопропил-N’-фенилметиламино) бутиламино] бутил] оксим] эритромицина A (соединение 3), tпл 83-85oC (гексан); MC (C.I.) (М+Н)+ = 1066; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,37-7,10 (m, 5H, ароматические соединения); 3,50 (s, 2Н, 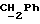 ); 3,30 (s, 3Н, OCH3); 2,26 (s, 6H, 2NCH3); 0,82 (t, 3Н, ); 3,30 (s, 3Н, OCH3); 2,26 (s, 6H, 2NCH3); 0,82 (t, 3Н, 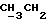 ).
Пример 12 ).
Пример 12Получение (Е)-9-[O-[2-[6-(фенилметиламино)гексиламино]этил]оксима] эритромицина A (соединение 4) К раствору (Е)-9-[О-[2-[N-бензилоксикарбонил-6-(N’-бензилоксикарбонил- N’-фенилметиламино)гексиламино]этил]оксима]эритромицина A (5,9 ммоля), полученного согласно примеру 11, в этаноле (150 мл) добавляли 10%-ный палладий на угле (750 мг). Полученную таким образом смесь помещали в гидрогенизатор Парра, загруженный водородом (1 бар), и перемешивали при комнатной температуре. Через 7 часов катализатор отфильтровывали и спиртовой раствор упаривали досуха. Таким путем после очистки с помощью хроматографии на силикагеле (элюент метиленхлорид: метанол: аммиак = 90:10:1) получали (Е)-9-[О-[2-[6-(фенилметиламино)гексиламино] этил]оксим] эритромицина A. МС (C.I.) (М+Н)+ = 982; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 140,48; 128,39; 128,11; 126,88.
По аналогичной методике получали следующие соединения:(Е)-9-[О-[2-[2-(фенилметиламино)этиламино] этил] оксим] эритромицина A (соединение 5), 13C-ЯМР (50 МГц, CDCl3): (част./млн) 176,51; 172,36; 140,96; 129,08; 128,95; 127,67; 103,84; 96,86; 53,35;(Е)-9-[O-[6-[6-(фенилметиламино)гексиламино]гексил]оксим] эритромицина A (соединение 6), МС (C.I.) (M+H)+ = 1038; 13C-ЯМР (50 МГц, CDCl3): (част. /млн) 175,24; 171,31; 140,33; 128,37; 128,13; 126,89; 102,92; 96,27; 54,01;(Е) 9-[O-[6-[3-(фенилметиламино)пропиламино]гексил]оксим] эритромицина A (соединение 7), МС (C.I.) (М+Н)+ = 995; 13C-ЯМР (50 МГц, CDCl3): (част. /млн) 175,15; 171,37; 140,41; 128,38; 128,09; 126,89; 102,92; 96,27; 54,04;(Е)-9-[O-[6-[5-(фенилметиламино)пентиламино]гексил]оксим] эритромицина A (соединение 8), 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,35-7,15 (m, 5H, ароматические соединения); 3,75 (s, 2Н, CH2Ph); 2,25 (s, 6Н, 2NCH3); 0,81 (t, 3Н, 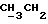 ); );(Е)-9-[O-[2-[8-(фенилметиламино)октиламино] этил] оксим] эритромицина A (соединение 9), 1H-ЯМР (200 МГц, CDCl3): (част./млн)7,40-7,15 (m, 5H, ароматические соединения); 3,75 (s, 2H, 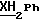 ), 3,29 (s, 3Н, OCH2); 2,25 (s, 6H, 2NHC3); 0,82 (t, 3Н, ), 3,29 (s, 3Н, OCH2); 2,25 (s, 6H, 2NHC3); 0,82 (t, 3Н, 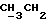 ); );(Е)-9-[О-[2-[5-(фенилметиламино)пентиламино] этил] оксим] эритромицина A (соединение 10), 13C-ЯМР (50 МГц, CDCl3): (част./млн) 174,96; 172,00; 140,31; 128,39; 128,14; 126,93; 103,16; 96,20; 53,98;(E)-9-[O-[5-[6-(фенилметиламино)гексиламино]пентил]оксим] эритромицина A (соединение 11), 13C-ЯМР (50 МГц, CDCl3): (част./млн) 175,24; 171,24; 140,41; 128,38; 128,13; 126,88; 102,97; 96,28; 54,06;(E)-9-[O-[3-[6-(фенилметиламино)гексиламино]пропил]оксим] эритромицина A (соединение 12), 13C-ЯМР (50 МГц, CDCl3): (част./млн) 175,23; 171,46; 140,45; 128,39; 128,13; 126,88; 102,99; 96,29; 50,06;(E)-9-[O-[3-[4-(фенилметиламино)бутиламино] пропил]оксим] эритромицина A (соединение 13), 13C-ЯМР (50 МГц, CDCl3): (част./млн) 175,22; 171,45; 140,35; 128,39; 128,13; 126,90; 102,98; 96,26; 53,94;(Е)-9-[O-[6-[N-изопропил-2-(2-фенилэтиламино)этиламино] гексил] оксим] эритромицина A (соединение 14), tпл 93-95oC; МС (C.I.) (М+Н)+ = 1038; 13C-ЯМР (50 МГц, CDCl3): (част. /млн) 174,92; 170,96; 139,71; 128,42; 128,10; 125,79; 102,62; 95,94; 50,93;(E)-9-[O-[6-[4-(N-изопропилфенилметиламино)бутиламино] гексил] оксим] эритромицина A (соединение 15), tпл 78-80oC; МС (C.I.) (М+Н)+ = 1052; 13C-ЯМР (50 МГц, CDCl3): част. /млн) 175,47; 171,30; 140,95; 128,61; 128,02; 126,70; 116,87; 102,94; 53,94;(E)-9-[O-[2-[6-[(4-фторфенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 16), МС (C. I.) (М+Н)+ = 999,5; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 161,88; 136,06; 129,65; 115,14;(E)-9-[O-[2-[6-[(4-метоксифенил)метиламино]гексиламино]этил]оксим] эритромицина A (соединение 17), МС (C. I. ) (М+Н)+ = 1011; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 158,57; 132,58; 129,31; 113,76;(E)-9-[O-[2-[6-[(3,4-метилендиоксифенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 18), МС (C.I.) (М+Н)+ = 1025; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 147,65; 146,44; 134,39; 121,18; 108,66; 108,06;(E)-9-[O-[2-[6-[(3-трифторметилфенил)метиламино] гексиламино]этил]оксим] эритромицина A (соединение 19), МС (C.I.) (М+Н)+ = 1050; 13C-ЯМР (50 МГц, CDCl3): (част. /млн) 141,57; 131,40; 130,63; 128,76; 124,22; 124,72; 123,56;(E)-9-[O-[2-[6-[(4-метилсульфонилфенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 20), МС (C.I.) (М+Н)+ = 1059; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 147,23; 138,97; 128,79; 127,49.
Пример 13Получение N-бензилоксикарбонил-6-аминогексанола Бензилхлорформиат (50%-ный в толуоле; 84,8 мл; 0,256 моля) в этилацетате (171 мл) и раствор 1 н. гидроксида натрия (256 мл) постепенно и одновременно добавляли к смеси 6-аминогексанола (25 г; 0,21 моля) в этилацетате (250 мл) и воды (200 мл) и перемешивали при 0oC. Температуру реакционной смеси (pH 9) доводили до комнатной и перемешивали в течение 5 часов. После разделения фаз водную фазу промывали этилацетатом (200 мл). 3атем объединенные органические фазы промывали насыщенным раствором хлорида натрия (150 мл), сушили над сульфатом натрия и упаривали досуха. Остаток объединяли с этиловым эфиром (300 мл) и образовавшийся осадок отфильтровывали и сушили под вакуумом при 50oC, получая таким образом N-бензилоксикарбонил-6-аминогексанол (44,5 г), tпл 80-82oC. Пример 14 Получение N-бензилоксикарбонил-6-аминогексанала Раствор бромида калия (1,89 г; 16 ммолей) в воде (31 мл) добавляли к раствору N-бензилоксикарбонил-6-аминогексанола (40 г; 0,159 моля), полученного согласно примеру 13, в метиленхлориде (600 мл), содержащему свободный радикал 2,2,6,6-тетраметилпиперидинокси (ТЕМПО) (0,248 г; 1,6 ммоля). К реакционной смеси для установления значения pH на 8,7 постепенно добавляли раствор гипохлорита натрия (215 мл), полученный путем смешения 7%-ного раствора гипохлорита натрия (240 мл) с бикарбонатом натрия (4,22 г) и 5%-ной соляной кислотой (5 мл), и перемешивали при 10oC. По окончании добавления и после разделения фаз органическую фазу промывали метиленхлоридом (2х200 мл), сушили над сульфатом натрия и упаривали досуха. Таким путем получали N-бензилоксикарбонил-6-аминогексанал (39,45 г) в виде масла. ТСХ (этилацетат: гексан = 1:1) Rf=0,41. Пример 15 Получение 2-[6-(бензилоксикарбониламино)гексиламино]этанола Смесь, содержащую N-бензилоксикарбонил-6-аминогексанал (35 г; 0,14 моля) и 2-аминоэтанол (51,3 г; 0,84 моля) в этаноле (250 мл), в присутствии молекулярных сит (3 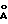 ) перемешивали при комнатной температуре в течение 2 часов. 3атем реакционную смесь фильтровали через целит и к образовавшемуся раствору добавляли борогидрид натрия (6,33 г; 0,168 моля). После перемешивания в течение 4 часов при комнатной температуре реакционный растворитель выпаривали под вакуумом и остаток объединяли с водой (500 мл) и этилацетатом (500 мл). После разделения фаз водную фазу дополнительно экстрагировали этилацетатом (200 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (250 мл), сушили над сульфатом натрия и упаривали досуха, получая таким образом 2-[6-(бензилоксикарбониламино)гексиламино] этанол (38,36 г). ТСХ (этилацетат:метанол:аммиак = 10:2:1) Rf=0,4.
Пример 16 ) перемешивали при комнатной температуре в течение 2 часов. 3атем реакционную смесь фильтровали через целит и к образовавшемуся раствору добавляли борогидрид натрия (6,33 г; 0,168 моля). После перемешивания в течение 4 часов при комнатной температуре реакционный растворитель выпаривали под вакуумом и остаток объединяли с водой (500 мл) и этилацетатом (500 мл). После разделения фаз водную фазу дополнительно экстрагировали этилацетатом (200 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (250 мл), сушили над сульфатом натрия и упаривали досуха, получая таким образом 2-[6-(бензилоксикарбониламино)гексиламино] этанол (38,36 г). ТСХ (этилацетат:метанол:аммиак = 10:2:1) Rf=0,4.
Пример 16Получение 2-[N-бензилоксикарбонил-6-(бензилоксикарбониламино) гексиламино]этанола Аналогично примеру 9 с использованием 2-[6-(бензилоксикарбониламино) гексиламино] этанола (38,3 г; 0,13 моля), полученного согласно примеру 15, получали 2-[N-бензилоксикарбонил-6- (бензилоксикарбониламино)гексиламино] этанол в виде масла. ТСХ (этилацетат:гексан = 65:35) Rf=0,45. Пример 17 Получение 2-[N-бензилоксикарбонил-6-(бензилоксикарбониламино) гексиламино]этилметансульфоната Аналогично примеру 10 с использованием 2-[N-бензилоксикарбонил-6- (бензилоксикарбониламино)гексиламино]этанола (20 г; 47,8 ммоля), полученного согласно примеру 16, получали 2-[N-бензилоксикарбонил-6- (бензилоксикарбониламино)гексиламино] этилметансульфонат (24,35 г), который использовали непосредственно в последующих реакциях. Пример 18 Получение (E)-9-[O-[2-[N-бензилоксикарбонил-6-(бензилоксикарбониламино) гексиламино]этил]оксима]эритромицина A Аналогично примеру 11 с использованием 2-[N-бензилоксикарбонил-6- (бензилоксикарбониламино)гексиламино] этилметансульфоната (24,25 г; 47,8 ммоля), полученного согласно примеру 17, после хроматографии на силикагеле (элюент метиленхлорид: метанол:аммиак = 95:5:0,5) получали (E)-9-[О-[2-[N-бензилоксикарбонил-6-(бензилоксикарбониламино) гексиламино] этил] оксим] эритромицина A (36,1 г). ТСХ (метиленхлорид:метанол:аммиак = 85:15:1,5) Rf= 0,5; 1H-ЯМР (200 МГц, CDCl3): (част./млн) 7,39-7,22 (m, 10H, ароматические соединения); 5,14-5,05 (m, 4H, 2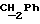 ); 3,29 (s, 3Н, OCH3); 2,25 (s, 6H, 2NCH2); 0,80 (t, 3Н, ); 3,29 (s, 3Н, OCH3); 2,25 (s, 6H, 2NCH2); 0,80 (t, 3Н, 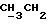 ).
Пример 19 ).
Пример 19Получение (Е)-9-[О-[2-(6-аминогексиламино)этил]оксима]эритромицина A Аналогично примеру 12 с использованием (E)-9-[О-[2-[N- бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламино] этил] оксима] эритромицина A, полученного согласно примеру 18, после хроматографии на силикагеле (элюент метиленхлорид: метанол: аммиак = 85:15:1,5) получали (Е)-9-[O-[2-(6-аминогексиламино)этил] оксим] эритромицина A. ТСХ (метиленхлорид:метанол:аммиак = 85: 15: 1,5) Rf=0,2; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 175,18; 171,26; 102,96; 96,28.
Пример 20Получение (Е)-9-[O-[2-[6-[(2-трифторметилфенил)метиламино] гексиламино] этил]оксима] эритромицина A (соединение 21) К раствору (Е)-9-[О-[2-(6-аминогексиламино)этил]оксима]эритромицина A (2 г; 2,24 ммоля), полученного согласно примеру 19, в этаноле (50 мл) добавляли 2-трифторметилбензальдегид (0,4 г) и молекулярные сита (4,5 г; 3  ) и перемешивали при комнатной температуре. Через 2 часа 30 молекулярные сита отфильтровывали и к образовавшемуся раствору добавляли 10%-ный палладий на угле (0,2 г). Реакционную смесь помещали в гидрогенизатор Парра, загруженный водородом (1 бар). Через 1 час по окончании реакции гидрирования катализатор отфильтровывали и растворитель выпаривали. Остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлорид:метанол:аммиак = 95:5:0,5), получая таким образом (Е)-9-[O-[2-[6-[(2-трифторметилфенил)метиламино] гексиламино]этил]оксим]эритромицина A (2 г). МС (C.I.) (М+Н)+ 1050; 13C-ЯМР (50 МГц, CDCl3): част./млн) 139,14; 131,88; 130,38; 127,58; 126,81; 125,82.
По аналогичной методике получали следующие соединения: ) и перемешивали при комнатной температуре. Через 2 часа 30 молекулярные сита отфильтровывали и к образовавшемуся раствору добавляли 10%-ный палладий на угле (0,2 г). Реакционную смесь помещали в гидрогенизатор Парра, загруженный водородом (1 бар). Через 1 час по окончании реакции гидрирования катализатор отфильтровывали и растворитель выпаривали. Остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлорид:метанол:аммиак = 95:5:0,5), получая таким образом (Е)-9-[O-[2-[6-[(2-трифторметилфенил)метиламино] гексиламино]этил]оксим]эритромицина A (2 г). МС (C.I.) (М+Н)+ 1050; 13C-ЯМР (50 МГц, CDCl3): част./млн) 139,14; 131,88; 130,38; 127,58; 126,81; 125,82.
По аналогичной методике получали следующие соединения:(Е)-9-[О-[2-[6-(3-пиридилметиламино)гексиламино]этил]оксим] эритромицина A (соединение 22), МС (C.I.) (М+Н)+ = 982; 13C-ЯМР (50 МГц, CDCl3): (част. /млн) 149,66; 148,39; 135,81; 123,40;(Е)-9-[O-[2-[6-[(4-трифторметилфенил)метиламино]гексиламино]этил] оксим] эритромицина A (соединение 23), МС (C.I.) (М+Н)+ = 1050; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 144,73; 128,23; 125,25; 124,26;(Е)-9-[О-[2-[6-[(2-гидроксифенил)метиламино]гексиламино]этил]оксим] эритромицина A (соединение 24), МС (C. I. ) (М+Н)+ = 997; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 158,37; 128,60; 128,19; 122,54; 118,88; 116,32;(Е)-9-[O-[2-[6-[(3-гидроксифенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 25), МС (C.I.) (М+Н)+ = 997; 13C-ЯМР (50 МГц, CDCl (част./млн) 157,28; 140,46; 129,56; 119,70; 115,55; 114,89;(Е)-9-[O-[2-[6-[(4-н-бутоксифенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 26), МС (C.I.) (М+Н)+ = 1053; 13C-ЯМР (50 МГц, CDCl (част./млн) 158,27; 131,65; 129,40; 114,40;(E)-9-[O-[2-[6-[(3-феноксифенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 27), МС (C.I.) (М+Н)+ = 1073; 13C-ЯМР (50 МГц, CDCl (част./млн) 157,32; 157,28; 142,69; 129,72; 129,64; 123,16; 122,91; 118,84; 118,52; 117,29;(Е)-9-[O-[2-[6-[(4-гидроксифенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 28), МС (C.I.) (М+Н)+ = 997; 13C-ЯМР (50 МГц, CDCl (част./млн) 156,49; 130,00; 128,87; 115,88;(E)-9-[O-[2-[6-[(4-феноксифенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 29), МС (C.I.) (М+Н)+ = 1073; 13C-ЯМР (50 МГц, CDCl (част./млн) 157,43; 156,05; 135,43; 129,69; 129,49; 123,07; 118,92; 118,72; 118,67;(E) -9-[O-[2-[6-[(бифенил-4-ил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 30), МС (C.I.) (М+Н)+ = 1057; 13C-ЯМР (50 МГц, CDCl (част./млн) 140,94; 139,86; 139,40; 128,74; 128,58; 127,13; 127,03;(E)-9-[O-[2-[6-(2-фурилметиламино)гексиламино]этил]оксим] эритромицина A (соединение 31), МС (C.I.) (М+Н)+ = 971; 13C-ЯМР (50 МГц, CDCl (част./млн) 153,92; 141,73; 110,08; 106,81.
Пример 21Получение (E)-9-[O-[2-[6-[(3,5-дихлор-2-гидроксифенил)метиламино] гексиламино]этил]оксима]эритромидина A (соединение 32) Молекулярные сита (6 г; 3  ) и 3,5-дихлор-2-гидроксибензальдегид (0,535 г; 2,8 ммоля) добавляли к раствору (Е)-9-[O-[2-(6-аминогексиламино) этил] оксима]эритромицина A (2,5 г; 2,8 ммоля), полученного согласно примеру 19, в безводном этаноле (100 мл). Реакционную смесь перемешивали при комнатной температуре и через 2 часа молекулярные сита отфильтровывали и к образовавшемуся раствору порциями добавляли борогидрид натрия (0,106 г; 2,89 ммоля). После перемешивания в течение 3 часов растворитель выпаривали при пониженном давлении и остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлорид: метанол: аммиак = 85:15:1,5), получая таким образом (E)-9-[О-[2-[6-[(3,5-дихлор-2-гидроксифенил)метиламино]гексиламино]этил] оксим]эритромицина A (2,2 г). МС (C.I.) (М+Н)+ = 1066; 13C-ЯМР (50 МГц, CDCl част./млн) 153,43; 128,43; 126,42; 124,41; 122,91; 121,61.
По аналогичной методике получали следующие соединения: ) и 3,5-дихлор-2-гидроксибензальдегид (0,535 г; 2,8 ммоля) добавляли к раствору (Е)-9-[O-[2-(6-аминогексиламино) этил] оксима]эритромицина A (2,5 г; 2,8 ммоля), полученного согласно примеру 19, в безводном этаноле (100 мл). Реакционную смесь перемешивали при комнатной температуре и через 2 часа молекулярные сита отфильтровывали и к образовавшемуся раствору порциями добавляли борогидрид натрия (0,106 г; 2,89 ммоля). После перемешивания в течение 3 часов растворитель выпаривали при пониженном давлении и остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлорид: метанол: аммиак = 85:15:1,5), получая таким образом (E)-9-[О-[2-[6-[(3,5-дихлор-2-гидроксифенил)метиламино]гексиламино]этил] оксим]эритромицина A (2,2 г). МС (C.I.) (М+Н)+ = 1066; 13C-ЯМР (50 МГц, CDCl част./млн) 153,43; 128,43; 126,42; 124,41; 122,91; 121,61.
По аналогичной методике получали следующие соединения:(E)-9-[О-[2-[6-[(2-нитрофенил)метиламино]гексиламино]этил]оксим] эритромицина A (соединение 33), МС (C.I.) (М+Н)+ = 1027; 13C-ЯМР (50 МГц, CDCl (част./млн) 149,14; 135,79; 133,13; 131,26; 127,87; 124,70;(E)-9-[O-[2-[6-[(3-нитрофенил)метиламино]гексиламино]этил]оксим] эритромицина A (соединение 34), МС (C.I.) (М+Н)+ = 1027; 13C-ЯМР (50 МГц, CDCl (част./млн) 148,37; 142,87; 134,17; 129,22; 122,81; 121,96;(E)-9-[O-[2-[6-[(4-нитрофенил)метиламино]гексиламино]этил]оксим] эритромицина A (соединение 35), МС (C.I.) (М+Н)+ = 1027; 13C-ЯМР (50 МГц, CDCl (част./млн) 148,41; 147,00; 128,59; 123,60;(E)-9-[O-[2-[6-[(4-гидрокси-3-нитрофенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 36), МС (C.I.) (М+Н)+ = 1043; 13C-ЯМР (50 МГц, CDCl (част./млн) 157,29; 137,40; 134,05; 128,01; 125,23; 121,70;(E)-9-[O-[2-[6-[(3-гидрокси-4-нитрофенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 37), МС (C.I.) (М+Н)+ = 1043; 13C-ЯМР (50 МГц, CDCl (част./млн) 155,50; 151,98; 132,51; 125,13; 119,67; 118,59.
Пример 22 Получение (E)-9-[O-[2-[N-метил-6-(N’-метил-N’-фенилметиламино)гексиламино] этил]оксима]эритромицина A (соединение 38)Водный 37%-ный раствор формальдегида (2 мл; 26,6 ммоля) и 10%-ный палладий на угле (0,82 г) добавляли в указанной последовательности к раствору (E)-9-[О-[2-[6-(фенилметиламино)гексиламино]этил]оксима]эритромицина A (2 г; 2 ммоля), полученного согласно примеру 12, в смеси этанол:вода = 1:1 (20 мл) и перемешивали при комнатной температуре. Реакционную смесь помещали в гидрогенизатор Парра, загруженный водородом (1 бар). Через 2 часа реакционную смесь фильтровали для удаления катализатора и образовавшийся раствор упаривали досуха. Образовавшийся остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлорид:метанол:аммиак = 90:10:1), получая (E)-9-[O-[2-[N-метил-6-(N’-метил-N’-фенилметиламино)гексиламино] этил] оксим] эритромицина A (1,8 г). МС (C.I.) (М+Н)+ = 1009; 13C-ЯМР (50 МГц, CDCl3 (част./млн) 139,20; 129,04; 128,17; 126,86.
По аналогичной методике получали следующее соединение:(E)-9-[О-[2-[N-метил-6-[N’-метил-N’-(4-трифторметилфенил)метиламино] гексиламино] этил] оксим] эритромицина A (соединение 39), MC (C.I.) (М+Н)+ = 1078; 13C-ЯМР (50 МГц, CDCl3): (част./млн) 143,65; 129,12; 129,03; 125,10; 124,29.
Пример 23Фармакологическая активность а) Антибактериальная активность in vitro Определение минимальных ингибирующих концентраций (МИК) по отношению к грамположительным и грамотрицательным бактериям осуществляли микробиологическим методом серийных двукратных постепенных разведений жидкой среды [National Committee for Clinical Laboratory Standards, 1990; Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; Approved standards M7-A2-NCCLS, Villanova, Pa] с использованием в качестве среды для культивирования бульона Мюллера-Хинтона (МНВ). В случае “требовательных” бактерий (Streptococcus pneumoniae и Streptococcus pyogenes) в среду добавляли 5%-ную лошадиную сыворотку. В качестве эталонных соединений из класса макролидов применяли рокситромицин и кларитромицин [The Merck Index, XI изд., N 8253 и N 2340 соответственно]. МИК, выраженные в (мкг/мл), определяли после инкубации микропланшетов при 37oC в течение 18 часов, оценивая наименьшую концентрацию антибиотика, обладающую способностью ингибировать развитие бактерий. б) Антибактериальная активность in vivo Терапевтическую эффективность, выраженную в виде средней защищающей дозы (PD50) исследуемых соединений формулы (I), оценивали на примере индуцированной экспериментальным путем легочной инфекции, вызванной у мыши Streptococcus pyogenes C 203. Использовали мышей-альбиносов линии Charles River (штамм CD 1) с весом тела 23-35 г, которых помещали в клетку группами по 6 особей, давая стандартный корм и воду по желанию. Суспензию S. pyogenes C 203 (соответствующую приблизительно 108 UFC) в триптозном бульоне (0,05 мл) вводили интраназально каждой мыши, анестезированной смесью этилового эфира и хлороформа. Исследуемые соединения вводили внутрибрюшинно в виде однократной дозы в 0,2%-ной суспензии Твина за 24 часа до или через 1 час после заражения. Наблюдение за гибелью мышей осуществляли в течение периода времени до 10 дней после заражения. Определение PD50, выраженной в виде (мг/кг), проводили методом пробит-анализа. Данные об антибактериальной активности в опытах in vitro по отношению к грамположительным микроорганизмам (таблица 1) и грамотрицательным микроорганизмам (таблица 2) и об антибактериальной активности в опытах in vivo (таблица 3) некоторых характерных соединений формулы (I) приведены ниже. Приведенные в табл. 1 данные явно свидетельствуют о том, что соединения формулы (I), являющиеся предметом настоящего изобретения, обладают антибактериальной активностью по отношению к грамположительным микроорганизмам, практически сопоставимой с таковой рокситромицина и кларитромицина. Антибактериальная активность соединений формулы (I) по отношению к грамотрицательным микроорганизмам, таким, как Escherichia coli и Klebsiella pneumoniae, оказалась существенно выше, чем таковая обоих эталонных соединений. Соединения формулы (I) оказались активными в опытах in vivo, а профили их активности свидетельствуют о том, что эти соединения обладают существенно более высокими продолжительностью действия и периодом полураспада при элиминации из ткани, чем таковые соединений-эталонов. Формула изобретения
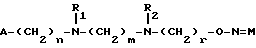 где А обозначает фенильную группу, необязательно замещенную 1 – 3 одинаковыми или различными группами, выбранными из прямых или разветвленных (С1-С4)алкоксигрупп, (С1-С2)алкилендиоксигрупп, (С1-С4)алкилсульфонильных групп, фенильной, фенокси-, гидрокси-, нитрогруппы, галогена и трифторметильной группы, или пиридил, или фурил; R1 и R2, одинаковые или различные, – атом водорода или прямая или разветвленная (С1-С4)алкильная группа; n = 1 или 2; m = 1 – 8, целое число; r = 2 – 6 целое число; М – группа формулы 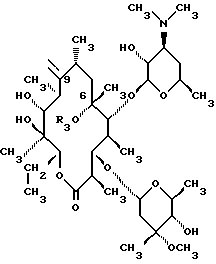 где R3 – атом водорода, и их фармацевтически приемлемые соли. 2. Соединение по п.1, имеющее Е-конфигурацию. 3. Соединение по п.1, где А – пиридил, фурил или фенильная группа, необязательно замещенная 1 – 3 группами, выбранными из гидрокси-, метокси-, метилендиокси-, н-бутокси-, фенокси-, фенильной, метилсульфонильной, нитрогруппы, галогена и трифторметильной группы; R1 и R2, одинаковые или различные, – атом водорода или метильная группа. 4. Соединение по п.1, где А обозначает фенильную группу, необязательно замещенную группой, выбранной из фенокси, нитро и трифторметила; R1 и R2 одинаковые и обозначают атом водорода или метильную группу; n = 1; m = 6; r = 2; 5. Фармацевтическая композиция, обладающая антибиотической активностью и содержащая терапевтически эффективное количество производного эритромицина А в смеси с фармацевтически приемлемым носителем, отличающаяся тем, что в качестве производного эритромицина А она содержит 9-О-оксимовое производное эритромицина А общей формулы I по п.1. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 08.12.2005
Извещение опубликовано: 20.12.2007 БИ: 35/2007
|
||||||||||||||||||||||||||