Патент на изобретение №2245337
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ 3-АЗИДОПРОИЗВОДНЫХ РИФАМИЦИНА S
(57) Реферат:
Предполагаемое изобретение относится к области химической технологии органических веществ, а именно к способам получения производных 3-азидо-рифамицина S, обладающих свойствами антибиотиков. Задачей предлагаемого технического решения является повышение эффективности способа получения 3-азидо-производных рифамицина S за счет увеличения выхода целевого продукта и упрощения процесса его выделения. Выполнение поставленной задачи достигается тем, что в способе получения 3-азидо-производных рифамицина S, включающем взаимодействие в среде растворителя производного рифамицина S с солью азотистоводородной кислоты путем перемешивания реагентов при температуре от 0°С до 100°С в течение 0,5-2 часов с последующим извлечением целевого продукта, в качестве производного рифамицина S используют 3-галогенпроизводное рифамицина S, в качестве растворителя берут простые алифатические спирты с числом атомов углерода от 1 до 5 или ацетонитрил или смесь воды и несмешивающегося с водой органического растворителя, выбранного из ряда: этилацетат, метилацетат, бензол или его моно- или диметильные аналоги, хлорированные углеводороды с числом атомов углерода от 1 до 3, в качестве соли азотистоводородной кислоты берут азид натрия или азид калия, а извлечение целевого продукта проводят экстракцией несмешивающимся с водой органическим растворителем или разбавлением реакционной смеси водой с последующим фильтрованием. Таким образом, заявляемый способ позволяет на 63-71% повысить выход целевого продукта и упростить процесс его выделения. 1 табл.
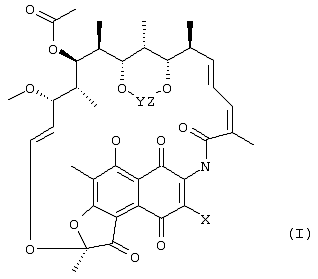
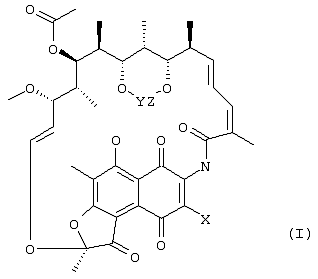
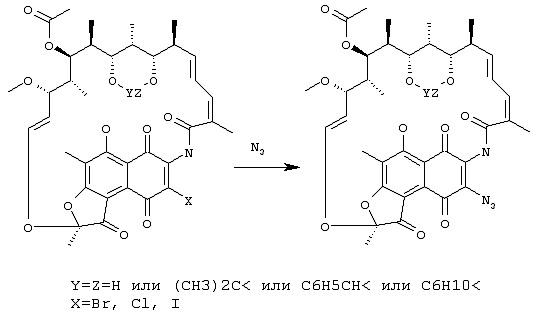
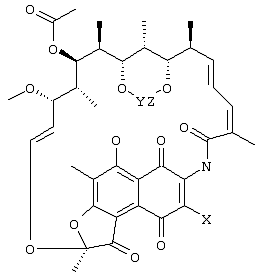
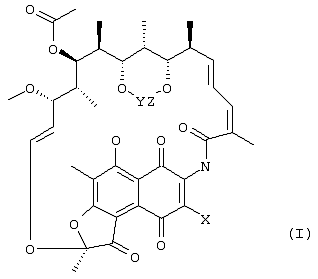
Предлагаемое изобретение относится к области химической технологии органических веществ, а именно к способам получения 3-азидопроизводных рифамицина S, обладающих свойствами антибиотиков. 3-азидо-производные рифамицина S являются важными промежуточными продуктами при производстве 3-аминопроизводных рифамицина S, представляющих интерес и как самостоятельные антибактериальные средства (их антибактериальная активность выше, чем у рифамицина S) и как исходные соединения для получения многочисленных полусинтетических аналогов рифамицина S, находящих применение, например, в качестве эффективных противотуберкулезных препаратов. 3-азидо-производные рифамицина S представляют собой твердые вещества оранжевого цвета, нерастворимые в воде, растворимые в большинстве обычных органических растворителей, особенно хорошо в галогенированных углеводородах. Строение этих соединений может быть представлено общей формулой I: где X=N3, Y=Z=H или (СН3)2С< или С6Н5СН< или С6Н10<. Наиболее близким к заявляемому по технической сущности и достигаемому результату является способ получения 3-азидо-рифамицина S (X=N3, Y=Z=H, пат. США № 4007169, C 07 D 498/08, 1975, прототип), включающий взаимодействие рифамицина S с азидом натрия в растворителе путем перемешивания реагентов при температуре от 0°С до 100°С в течение как минимум 0,5 часа. В качестве растворителей, особенно подходящих для проведения данной реакции, отмечены диметилформамид, метилформамид, диметилсульфоксид, пирролидон и ряд других растворителей, относящихся к группе апротонных биполярных растворителей. После проведения реакции продукт выделяют с использованием либо препаративной колоночной хроматографии, либо дробной кристаллизации. Выход целевого продукта составляет 21%. К недостаткам известного способа получения 3-азидо-рифамицина S следует отнести, в первую очередь, невысокий выход продукта, а также использование высокотоксичных и достаточно дорогостоящих растворителей. Кроме того, при проведении реакции происходит образование значительного числа побочных продуктов. Следует также отметить, что растворы производных рифамицина в таких растворителях как диметилформамид представляют повышенную опасность для работников химических производств из-за того, что диметилформамид сильно увеличивает проницаемость клеточных мембран. К недостаткам относится и использование колоночной хроматографии для выделения целевого продукта, что требует специального оборудования и применения достаточно дорогостоящих сорбентов, а также создает проблему утилизации (регенерации) отработанного сорбента. Задачей предлагаемого технического решения является повышение эффективности способа получения 3-азидо-производных рифамицина S за счет увеличения выхода целевого продукта и упрощения процесса его выделения. Выполнение поставленной задачи достигается тем, что в способе получения 3-азидо-производных рифамицина S формулы I: где X=N3, Y=Z=H или (СН3)2С< или С6Н5СН< или С6Н10<, включающем взаимодействие 3-галогенпроизводного рифамицина S в среде растворителя, в качестве которого используют простые алифатические спирты с числом атомов углерода от 1 до 5 или ацетонитрил или смесь воды и несмешивающегося с водой органического растворителя, выбранного из ряда: этилацетат, метилацетат, бензол или его моно- или диметильные аналоги, хлорированные углеводороды с числом атомов углерода от 1 до 3, с солью азотистоводородной кислоты, в качестве которой используют азид натрия или азид калия, при температуре от 0°С до 100°С в течение 0,5-2 часов, целевой продукт извлекают экстракцией несмешивающимся с водой органическим растворителем или разбавлением реакционной смеси водой с последующим фильтрованием. Ниже приведена схема процесса: Из химической литературы известно, что атом галогена в галогенхинонах, каковыми и являются 3-галогенпроизводные рифамицина S, достаточно подвижен и может быть замещен подходящим заместителем. Нами обнаружено, что взаимодействие 3-галогенпроизводных рифамицина S с азидом натрия либо азидом калия, проводимое в заявляемых условиях, приводит к быстрому образованию 3-азидо-производных рифамицина S с высоким выходом, при этом целевое соединение не требует очистки и может быть непосредственно использовано для последующих превращений. Предлагаемое техническое решение является новым, имеет изобретательский уровень и промышленно применимо. Сущность заявляемого способа поясняется примерами его осуществления. Пример 1. В стеклянный реактор, снабженный термометром и мешалкой, помещают 1500 мл этанола и при перемешивании добавляют 77,5 г (0,1 моль) 21,23-изопропилиденового производного – бром-рифамицина S, полученного по методике, описанной в патенте Бельгии № 893873, а затем приливают раствор 9,7 г (0,15 моль) азида натрия в 30 мл воды. Перемешивают реакционную массу при температуре 20°С в течение 2 часов, после чего ее разбавляют 3000 мл воды, полученный продукт отфильтровывают, промывают водой и сушат. Получают 21,23-изопропилиденового производного 3-азидо-рифамицина S. Выход 21,23-изопропилиденового производного 3-азидо-рифамицина S 71,6 г (92%). Данные CHN анализа: С 62,1, Н 6,1, N 7,15, C40H48N4O12. ИК-спектр: 2165 см-1, что подтверждает наличие азидо-группы. спектр ПМР: отсутствует сигнал для протона при С(3). Высокоэффективная жидкостная хроматография (ВЭЖХ): гомогенен. Пример 2 В стеклянный реактор, снабженный термометром и мешалкой помещают 500 мл этилацетата и при перемешивании добавляют 73,0 г (0,1 моль) 3-хлоррифамицина S, полученного по методике, описанной в патенте Германии № 2548128, а затем приливают раствор 12,1 г (0,15 моль) азида калия в 100 мл воды. Перемешивают реакционную массу при температуре 20°С в течение 2 часов, после чего продукт экстрагируют этилацетатом, органическую фазу промывают водой, сушат безводным сульфатом натрия и упаривают под вакуумом. Выход 3-азидо-рифамицина S 63,4 г (86%). Данные CHN анализа: С 60,4, Н 6,0, N 7,61, С37H44N4O12. ИК-спектр: 2170 см-1‘, что подтверждает наличие азидо-группы. спектр ПМР: отсутствует сигнал для протона при С(3). Высокоэффективная жидкостная хроматография (ВЭЖХ): гомогенен. Пример 3 В стеклянный реактор, снабженный термометром и мешалкой помещают 1500 мл этанола и при перемешивании добавляют 91 г (0,1 моль) 21,23-бензилиденового производного 3-иод-рифамицина S, полученного по методике, аналогичной описанной в патенте Бельгии № 893873, а затем приливают раствор 9,7 г (0,15 моль) азида натрия в 30 мл воды. Проведение реакции и выделение продукта осуществляют как в примере 1. Выход 21,23-бензилиденового производного 3-азидо-рифамицина S 73,4 г (89%). Данные CHN анализа: С 14,15, Н 5,94, N 6,71, C44H48N4O12. ИК-спектр: 2168 см-1, что подтверждает наличие азидо-группы. спектр ПМР: отсутствует сигнал для протона при С(3). Высокоэффективная жидкостная хроматография (ВЭЖХ): гомогенен. Пример 4 Проводят реакцию, как в примере 1, с 85,5 г (0,1 моль) 21,23-циклогексилиденового производного 3-бром-рифамицина S. Выход 21,23-циклогексилиденового производного 3-азидо-рифамицина S 77 г (90%). Данные СНN анализа: С 63,1, Н 6,47, N 6,79, C43H52N4O12. ИК-спектр: 2172 см-1, что подтверждает наличие азидо-группы. спектр ПМР: отсутствует сигнал для протона при С(3). Высокоэффективная жидкостная хроматография (ВЭЖХ): гомогенен. Аналогично приведенным выше примерам 1-4 осуществлялись примеры 5-7. Процесс получения производных 3-азидо-рифамицина S в примерах 5-7 проводят аналогично примеру 1, а в примерах 8-13 аналогично примеру 2. Реагенты и параметры процесса приведены в таблице. Как видно из таблицы, заявляемый способ позволяет примерно на 65% повысить выход целевого продукта по сравнению со способом-прототипом. Кроме того, как показали полученные результаты проведения реакции между 3-галогенпроизводным рифамицина S и азидом натрия или азидом калия в гомогенных либо гетерогенных условиях, целевой продукт образуется быстро, в мягких условиях и с высоким выходом, что позволят свести к минимуму образование побочных продуктов и получать целевой продукт высокого качества.
Формула изобретения
Способ получения 3-азидопроизводных рифамицина S формулы I где X=N3, Y=Z=H или (СН3)2С< или С6Н5СН< или С6Н10<, включающий взаимодействие 3-галогенпроизводного рифамицина S в среде растворителя, в качестве которого используют простые алифатические спирты с числом атомов углерода от 1 до 5 или ацетонитрил или смесь воды и несмешивающегося с водой органического растворителя, выбранного из ряда: этилацетат, метилацетат, бензол или его моно- или диметильные аналоги, хлорированные углеводороды с числом атомов углерода от 1 до 3, с солью азотистоводородной кислоты, в качестве которой используют азид натрия или азид калия, при температуре от 0 до 100°С в течение 0,5-2 ч, и извлечение целевого продукта экстракцией несмешивающимся с водой органическим растворителем или разбавлением реакционной смеси водой с последующим фильтрованием.
HE4A – Изменение адреса для переписки с обладателем патента Российской Федерации на изобретение
Новый адрес для переписки с патентообладателем:
Извещение опубликовано: 10.04.2006 БИ: 10/2006
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 28.09.2007
Извещение опубликовано: 10.01.2009 БИ: 01/2009
NF4A Восстановление действия патента СССР или патента Российской Федерации на изобретение
Дата, с которой действие патента восстановлено: 10.01.2009
Извещение опубликовано: 10.01.2009 БИ: 01/2009
HE4A – Изменение адреса для переписки с обладателем патента Российской Федерации на изобретение
Новый адрес для переписки с патентообладателем:
Извещение опубликовано: 20.04.2010 БИ: 11/2010
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||