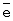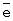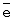Патент на изобретение №2243818
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ МЕТИЛЭТИЛКЕТОНА
(57) Реферат:
Изобретение относится к области основного органического синтеза, а именно к способу получения метилэтилкетона (МЭК) каталитическим окислением н-бутенов кислородом, а также к катализаторам для его осуществления. Описан катализатор для получения метилэтилкетона окислением н-бутенов кислородом и/или кислородсодержащим газом, который состоит из водного раствора ГПК-7 – молибденованадофосфорной гетерополикислоты или смеси гетерополикислоты и/или ее соли, а также палладия с концентрацией 5·10-4-1·10-2 М, стабилизированного фталоцианиновым лигандом Рс при мольном отношении [Pd]:[Рс]=0,5-2. Используют Mo-V-фосфорную ГПК-7 состава H10P3Mo18V7O84, концентрация ванадия в водном растворе ГПК-7 или ее соли составляет 0,4-2,2 грамм-атома на литр. Описан также способ окисления н-бутенов в МЭК, который осуществляют непрерывно в двухстадийном режиме при температуре 15-90°С, а регенерируют катализатор взаимодействием его с кислородом или кислородсодержащим газом при температуре 140-190°С и давлении кислорода 1-10 ата. Технический эффект – увеличение эффективности процесса за счет повышения стабильности компонентов катализатора (Pd+ГПК), что позволяет значительно повысить производительность катализатора и его избирательность в реакциях синтеза. 2 с. и 5 з.п. ф-лы, 1 ил., 6 табл.
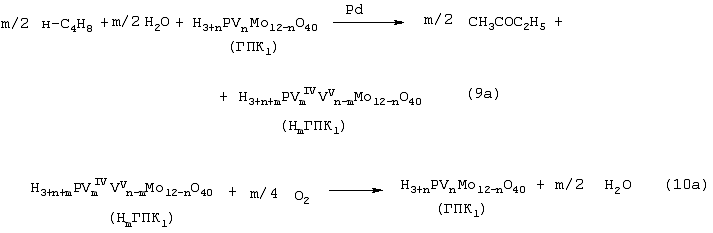
Изобретение относится к области основного органического синтеза, а именно к способу получения метилэтилкетона (МЭК, СН3СОС2Н5) каталитическим окислением н-бутенов кислородом, а также к катализаторам для его осуществления. Вследствие исключительно высокой растворяющей способности метилэтилкетон-МЭК находит широкое промышленное применение [The Chemical Economics Handbook-New York: SRI International (СЕН), 1996]. Он используется в производстве полиуретановых лаков, служащих для покрытия магнитных лент компьютеров, инструмента, аудио- и видеокассет, является лучшим депарафинизатором смазочных масел, обеспечивая их морозостойкость, является растворителем в производстве пенопластов, различных красок, эпоксидных и глифталевых смол, полихлорвинила, искусственной кожи, служит основой чернил для печатания. МЭК используется в качестве сырья при получении метилизопропилкетона, 2,3-бутандиона; оксима метилэтилкетона, предотвращающего образование пленок при хранении красок. Используется для получения этилакриловой и изомерных метилкротоновых кислот, антиоксидантов резин, для пластификации производных нитроцеллюлозы, используемых в производстве бездымных порохов, находит применение как растворитель для пленочных покрытий таблеток и капсул лекарств, как реагент и экстрагент во многих фармацевтических производствах. Пероксид МЭК’а является инициатором полимеризации ненасыщенных полиэфиров в производстве армированных пластиков. В промышленном масштабе МЭК получают несколькими способами. Один из них основан на жидкофазном свободнорадикальном окислении н-бутана по схеме (1), где МЭК является одним из целевых продуктов. В этом способе выход МЭК не превышает 23%, одновременно получается и уксусная кислота. Достоинством этого способа является дешевизна исходного сырья, недостатком – обилие побочных продуктов, сильно осложняющих и удорожающих процесс выделения МЭК. Основным промышленным способом синтеза МЭК является трехстадийная переработка бутиленовой фракции, являющейся побочным продуктом производства дивинила на заводах синтетического каучука. Способ отличается дешевизной сырья; он включает реакции (2)-(4): Из них реакции (2) и (3) – жидкофазные, а реакция (4) – гетерогенная каталитическая, а в некоторых вариантах – жидкофазная. Недостатками этого способа являются: обилие вредных необезвреживаемых отходов в виде загрязненной кислыми гудронами серной кислоты; высокая коррозионная активность среды в реакциях (2) и (3); высокая энергоемкость реакции (4); сложность процесса отделения МЭК от многочисленных примесей. Разновидностью этого способа является процесс, в котором вместо H2SО4 используют уксусную кислоту, а промежуточным продуктом является втор-бутилацетат: Катализатором стадий (2а) и (3а) служит сульфокатионит, который быстро забивается смолами, и потому имеет малый срок службы. Использованный сульфокатионит также является вредным отходом производства, серьезно ухудшающим его экологию. В последние годы предпринимались многократные попытки создания новых технологий получения МЭК из н-бутенов. Об этом свидетельствуют многочисленные патенты. Наибольший интерес из них представляют два новых способа: трехстадийный, включающий реакции (5)-(7) с промежуточным получением гидроперекиси втор-бутилбензола [Пат.США 5304684, 1994, фирмы Sumimoto Chem. Ltd.], и прямое окисление н-бутенов по реакции (8). Трехстадийный способ основан на реакциях, подобных используемым в современном промышленном способе получения фенола и ацетона через гидроперекись кумола. Однако получение и разложение гидроперекиси втор-бутилбензола на стадиях (6) и (7) протекает с меньшей избирательностью, чем получение и разложение гидроперекиси кумола. В реакциях (6) и (7), наряду с МЭК и фенолом, образуются карбоновые кислоты, их эфиры, альдегиды, непредельные кетоны, смолы. Побочные продукты удаляют щелочью, что существенно ухудшает экологию производства. Способ (5)-(7) мог бы стать перспективным для промышленности при условии существенного повышения избирательности на стадиях (6) и (7) получения и разложения гидроперекиси втор-бутилбензола. Реакцию (8) можно проводить в одном реакторе, подавая в него стехиометрическую смесь н-С4H8:О2=2:1, которая прежде считалась невзрывоопасной [Справочник химика. 2-е изд. Дополнительный том. – Л.: Химия, 1968. – С.434]. Однако для улучшения селективности и снижения степени риска реакцию следует проводить в две стадии: 1) взаимодействие н-бутилена с раствором промежуточного обратимо действующего окислителя Ох в присутствии соли палладия по уравнению (9); 2) регенерация Ох кислородом по уравнению (10): Первоначально в качестве Ох использовали хлорную медь CuCl2 [Jira R., Freiesleben W., см. выше], восстановленная форма которой – ион CuCl–2 – легко окисляется кислородом. Однако синтез МЭК по реакции (9) в присутствии хлоридной системы (PdCl2+CuCl2 сопровождался образованием большого количества побочных хлорбутанонов (до 25%). В отсутствии Сl–-ионов или при их недостатке Cu(II) вообще не способна служить обратимым окислителем. Поэтому для окисления н-бутиленов такая система оказалась совершенно непригодной. В изобретениях [А.с. СССР 700973, B 01 J 23/44, 1994; а.с. ссср 822417, B 01 J 23/44, 1994] и более поздних [Пат. СССР 1669109, B 01 J 23/44, 1994] впервые было предложено использовать в качестве обратимого окислителя (Ox) Mo-V-фосфорные гетерополикислоты структуры Кеггина, имеющие общую формулу H3+nPVnMo12-nO40, обозначенные нами через ГПK1. С их участием бутиленовая реакция описывалась уравнением (9а), а кислородная – уравнением (10а): В этом способе (Ox = ГПК1) каталитическая система (PdII+ГПК1) не содержала Сl–-ионов и потому обеспечивала полное отсутствие хлорорганических соединений в продуктах реакции. Избирательность системы достигала 95-98%, а активность в бутиленовой реакции (9а) в 100 раз превышала активность хлоридной системы (PdCl2+CuCl2). Каталитическая система (Pd+ГПК1) могла успешно использоваться как для окисления стехиометрической смеси н-C4H8: О2=2:1 в МЭК (одностадийный вариант), так и для проведения реакций (9а) и (10а) в разных реакторах (двухстадийный вариант). Последний вариант исключал контакт кислорода или воздуха с бутенами и с МЭК’ом и обеспечивал безопасность производства. Однако в последнем случае происходило глубокое восстановление молекул ГПК1. Это приводило к значительным изменениям химического состава и свойств как ионов Pd2+, так и самих молекул ГПК1. Этот вариант (нестационарного) катализа оказался связанным со снижением устойчивости каталитической системы (Pd + ГПК1). Устойчивость катализатора в отношении ионов Pd2+ и молекул ГПК1 по-разному зависела от числа атомов ванадия в молекуле ГПК1 (n) и от отношения m: n (степени восстановления молекулы ГПК1). При малых значениях n (1 За последние 30 лет было предпринято много попыток стабилизировать палладий в катализаторе (Pd + ГПК1), однако все они по разным причинам оказались неприемлемыми для технологии. Простейший способ удержать Pd2+ в растворе введением малых концентраций Сl–-ионов, соответствующих атомным отношениям Сl–:Pd=5-50, был предложен в патенте фирмы Catalytica Inc. (США) [Заявка WO 91/13852, 19.09.1991]. Однако даже при таких концентрациях хлор из раствора катализатора быстро переходил в продукты реакции с образованием хлорорганических соединений. Для поддержания стабильности палладия в катализатор приходилось непрерывно добавлять соляную кислоту и одновременно из продуктов реакции удалять и обезвреживать хлорорганические соединения. Поэтому “малохлоридный катализатор” (PdCl2 + ГПК1) также не нашел промышленного применения. Использование палладия в виде комплексов с пиридинкарбоновыми кислотами ( В изобретении [Пат. Япония 07-149685, 1995] катализаторы (Pd+ ГПКО использовались в смешанных растворителях, содержащих менее 50% воды. В качестве таковых использовались диоксан, этанол, тетрагидрофуран, Эта же причина позволяет понять малую активность в реакции (8) гетерогенизированных катализаторов (Pd + ГПК1 Прототипом предлагаемого способа и катализатора получения МЭК является способ [А.с. СССР 1584200, B 01 J 23/44, 1994], в котором в качестве катализатора используют водный раствор соли палладия, производного пиридина и кислой соли молибдованадофосфорной гетерополикислоты (ГПК1). Реакцию окисления проводят путем взаимодействия катализатора с н-бутиленом при температурах 50-70° С, а после отделения метилэтилкетона окисляют восстановленную форму катализатора кислородом или воздухом при температурах 130-160° С (двухстадийный вариант). В этом способе реакция (8) по-прежнему осуществляется в условиях нестационарного катализа. От глубины и скорости изменений химического состава, окислительных и других физико-химических свойств катализатора зависят его производительность и устойчивость на каждой стадии реакции (8). Оптимизация МЭК-технологии требует, чтобы такие изменения состава и свойств катализатора при протекании обеих стадий (9а) и (10а) были быстрыми, глубокими и обратимыми. Однако из-за выпадения осадков на стадии (9а) требование обратимости нарушается в отношении как палладия, так и молекул ГПК1 структуры Кеггина. Выпадение палладия в бутиленовом реакторе с неполным возвращением его в раствор в воздушном реакторе (нестабильность палладия) является недостатком каталитической системы (Pd + ГПК1). Еще более серьезным ее недостатком является нестабильность молекул ГПК1 обусловленная потерей ими части ванадия, выпадавшего в осадок на стадии (9а) и не возвратившегося в раствор на стадии (10а). Изобретение решает задачу увеличения эффективности процесса за счет повышения стабильности компонентов катализатора (Pd + ГПК1), что позволит значительно повысить производительность катализатора и его активность в обеих реакциях. Задача решается катализатором получения метилэтилкетона окислением н-бутенов кислородом и/или кислородсодержащим газом, который состоит из водного раствора ГПК-7 – молибденованадофосфорной гетерополикислоты (7 – число атомов ванадия в молекуле ГПК-7) или смеси гетерополикислоты и/или ее соли, а также палладия с концентрацией 5· 10-4-1· 10-2 М, стабилизированного фталоцианиновым лигандом Рс при мольном отношении [Pd]: [Рс]=0,5-2. Используют Mo-V-фосфорную ГПК-7 состава H10P3Mo18V7O84 при концентрации ванадия в водном растворе 0,4-2,2 грамм-атома на литр. Задача решается также способом получения метилэтилкетона МЭК путем каталитического окисления н-бутенов с использованием катализатора, который состоит из водного раствора ГПК-7 – молибдованадофосфорной гетерополикислоты (7 – число атомов ванадия в молекуле ГПК-7) или смеси гетерополикислоты и/или ее соли, а также палладия с концентрацией 5· 10-4-1· 10-2 М, стабилизированного фталоцианиновым лигандом Рс при мольном отношении [Pd]:[Рс]=0,5-2. Используют Mo-V-фосфорную ГПК-7 состава H10P3Mo18V7O84 при концентрации ванадия в водном растворе 0,4-2,2 грамм-атома на литр. Способ окисления н-бутенов в МЭК осуществляют непрерывно в двухстадийном режиме, в котором реакцию окисления н-бутенов ведут в интервале температур 15-90° С, а регенерацию катализатора – путем взаимодействия его с кислородом или кислородсодержащим газом проводят в интервале температур 140-190° С при давлении кислорода 1-10 ата. Предлагаемый способ синтеза МЭК, как и прототип, основан на использовании Mo-V-фосфорной ГПК в качестве обратимо действующего окислителя и палладия в качестве сокатализатора. Основным отличием предлагаемого способа синтеза МЭК на катализаторе (Pd+ГПК) является то, что были использованы новые методы стабилизации компонентов катализатора (различные для палладия и ванадия). Они найдены нами в результате анализа причин нестабильности системы (Pd+ГПК1), а также на основе исследований кинетики и механизма реакций (9а) и (10а). Для устранения недостатков известных катализаторов (Pd + ГПК1) и создания катализатора с высокой производительностью и устойчивостью ион палладия был стабилизирован комплексообразованием с фталоцианиновым лигандом Рс, а молекулы ГПК1 кеггиновской структуры заменены молекулами ГПК-7 некеггиновского состава. В результате получен новый катализатор (Pd-Pc+ГПК-7). Предлагаемые катализаторы на основе ГПК-7 готовят способом, описанным в авторском свидетельстве [А.с. СССР 1782934, С 01 В 25/16, 1992]. Такие катализаторы имеют высокую гидролитическую устойчивость и не дают осадков при проведении стадий (9a1), (9a2) и (10а) реакции (8). Активность и производительность катализаторов в широких пределах можно регулировать тремя способами: а) изменениями концентрации ГПК-7; б) вариациями концентраций комплекса Pd+22· Рс; с) регулированием температуры реакции (9а). Для контроля степени восстановления катализатора (или степени его окисленности) впервые использован метод измерения окислительного потенциала (Е) контактного раствора. Найденное значение Е (относительно нормального водородного электрода) сравнивается с полученной заранее кривой зависимости Е от m для используемого катализатора, где m – это степень восстановления ГПК (m=[V(IV)]/[ГПК]). По значению Е находят текущее значение m. В приведенных таблицах примеров имеется отдельная графа, где даны значения Е для окисленных растворов катализаторов. По ним можно судить о том, насколько хорошо регенерирован катализатор, так как от этого будет зависеть его производительность. Отличием предлагаемого способа является создание в растворе высокой концентрации ванадия (до 2,2 грамм-атом на литр), которая приводит к повышению производительности катализатора с обеспечением его стабильности. Следующим отличием этого способа является то, что регенерируют катализатор взаимодействием с кислородом и/или кислородсодержащим газом, а для повышения производительности способа регенерацию ведут при более высоких температурах (до 190° С) и РО2 до 10 ата. Другим отличием является то, что бутиленовую реакцию проводят и при более низких, чем в способе-прототипе, температурах (от +15° С), поскольку катализатор (Pd· Pc+ГПК-7) обладает значительно большей активностью, чем катализатор способа-прототипа, где палладий стабилизирован дипиколиновой кислотой. Сущность изобретения иллюстрируется примерами. Пример 1. Во встряхиваемый стеклянный реактор на 120 мл, закрепленный на качалке, вливают 20 мл раствора катализатора, имеющего состав: [H10P3Mo18V7O84]=0.3 М, [Рd+2]=6· 10-3 М, мольное отношение [Pd]:[Рс]=2. Реактор термостатируют при 60° С, продувают 10-кратным объемом Пример 2. По примеру 1, отличающемуся тем, что мольное отношение [Pd]: [Рс]=1, за 14 мин окисляют 316 мл Пример 3. По примеру 1, отличающемуся тем, что мольное отношение [Рс]:[Pd]=2 (или [Pd]:[Рс]=0,5), за 24 мин окисляют 335 мл Пример 4. По примеру 1, отличающемуся тем, что на окисление подают промышленную бутиленовую фракцию (БФ) состава, %: бутен-1 – 37.5, цис-2-бутен – 34.9, транс-2-бутен – 25,9 (сумма н-бутенов равна 98,3), дивинил – 0,21, изо-бутилен – 0,1, ацетилены – 0,005, изо-бутан – 1,08, н-бутан – 0,3, за 17 мин окисляют 206 мл БФ (2,77 е) с избирательностью 96,5%. После отпарки МЭК и регенерации катализатора по методике примера 1 с ним проводят 2-й цикл, на котором за 13 мин окисляют 201 мл БФ. Аналогично предыдущим проводят еще 3 цикла. Избирательность и активность катализатора не изменяется: на 5 цикле окисляют 205 мл БФ за 17,5 мин. На этом катализаторе далее проводят циклы 6-10, в которых постепенно поднимают нагрузку на катализатор (т.е. увеличивают его производительность). Катализатор работает стабильно, не изменяя своих показателей (см. табл. 4). На 10-м цикле на нем окисляют за 36 мин 294 мл БФ (m=4,06 е). Осадков в растворе нет. Пример 5. По примеру 4, отличающемуся тем, что мольное отношение [Pd]:[Рс]=1, за 37 мин окисляют 282 мл БФ (m=3,89 е) с избирательностью 97,0%. После отпарки МЭК катализатор окисляют по методике примера 1. С этим катализатором проводят еще 10 циклов (см. табл. 5). Избирательность и активность его практически не изменяются. Так, на 11 цикле раствор за 39 мин окисляет 286 мл БФ (m=3,96 е). Осадков в растворе нет. Пример 6. По примеру 1, отличающемуся тем, что в качестве катализатора используют [H10P3Mo18V7O84]=0.2 М, [Рd+2]=6· 10-3 М при мольном отношении [Pd]: [Рс]=1 и t=18° С, за 40 мин окисляют 64 мл Пример 7. По примеру 4, отличающемуся тем, что в качестве катализатора используют кислую соль [Со0,5Н9Р3Мо18V7O84]=0.3 М, [Рd+2]=6· 10-3 М при мольном отношении [Pd]:[Рс]=4, а также тем, что окисляют БФ (см. пример 4), за 15 мин окисляют 206 мл БФ (m=2,77 е) с избирательностью 96,5%. После отпарки МЭК катализатор окисляют по методике примера 1 в течение 15 мин. С этим катализатором проводят 2-й цикл, на котором за 19 мин окисляют 208 мл БФ. Аналогично предыдущим проводят еще 3 цикла. Активность катализатора стабилизируется на уровне активности 2-го цикла: на 5-м цикле за 19 мин на нем окисляют 207 мл БФ, Избирательность катализатора не изменяется (см. табл.6). Как отмечено в примечании, на стенках реактора к концу испытаний появился незначительный темный налет, свидетельствующий о недостаточной стабилизации палладия при мольном отношении [Pd]: [Рс]=4. Как видно из примеров 4 и 7, это отношение должно быть меньше или равно 2. Все катализаторы, описанные в примерах 1-7, готовят известным способом [А.с. СССР 1782934, С 01 В 25/16, 1992]. Вначале синтезируют раствор ГПК заданного состава и заданной концентрации, исходя из стехиометрических количеств компонентов Н3РO4, МоО3 и V2O5. Затем в полученный раствор вводят навески PdCl2 и фталоцианина. Пример синтеза 250 мл ГПК состава H10P3Mo18V7O84 с концентрацией 0,30 М. Расчет количеств исходных компонентов ведут по уравнению: 3Н3РO4+18МоО3+3,5V2O5+0,5H2O Используют 194,4 г МоО3 х.ч., 47,78 г V2O5 х.ч., 31,3 мл 7,183-молярного раствора Н3РO4 и 30%-ную Н2О2 ос.ч. Растворение реагентов проводят в пять этапов: 1) 194,4 г МоО3+18,3 мл Н3РO4+1500 мл Н2O+2 мл Н2О2 2) 14,78 г V2O5+700 мл Н2O+50 мл Н2O2+4,0 мл Н3РO4 3) 11,00 г V2O5+700 мл Н2O+50 мл Н2O2+3,0 мл Н3РO4 4) 11,00 г V2O5+700 мл Н2O+50 мл Н2О2+3,0 мл Н3РO4 5) 11,00 г V2O5+700 мл Н2O+50 мл H2O2+3,0 мл Н3РO4. Для растворения МоО3 в коническую 3-литровую колбу загружают всю навеску МоО3, 1,5 л дистиллированной воды, 18,3 мл Н3РO4 и 2 мл 30%-ной H2O2 х.ч. Полученную смесь кипятят (она приобретает соломенно-желтую окраску) и постепенно упаривают до ~ 0,8 л. В кипящую смесь последовательно вводят растворы, полученные растворением V2О5 в воде в присутствии Н2О2 и Н3РO4. Каждую последующую порцию раствора V2O5+Н3РO4 вводят после упаривания смеси в колбе до ~ 1 л. Для растворения 1-й навески V2O5 в 1-литровый стакан загружают 14,78 г V2O5, 0,7 л дистиллированной воды и 50 мл Н2О2. Полученную смесь перемешивают при +15-20° С (не выше) в течение 20-30 минут до получения темнокрасно-коричневого раствора. До полного растворения V2O5 к смеси (при перемешивании) добавляют 4 мл Н3РO4 и ожидают прекращения выделения O2, образующегося за счет разложения перекисных комплексов ванадия. Полученный темно-коричневый (почти черный) раствор переливают в колбу с кипящей суспензией МоО3+Н3РO4 и упаривают до ~ 1 л. Аналогично проводят растворение 2-й, 3-й и 4-й навесок V2O5 и добавление полученного раствора к суспензии МоО3+V2O5+Н3РO4. Эту смесь затем кипятят и упаривают до 400 мл. Полученный раствор ГПК фильтруют через бумажный фильтр с красной полосой и фильтр промывают водой. Если осадок на фильтре превышает 2% от массы исходной V2O5, то его вместе с фильтром обрабатывают в стакане 5-10%-ной Н2О2 (~ 50 мл). Раствор нагревают до полного разложения Н2О2 и присоединяют к объединенному раствору ГПК с промывными водами и упаривают до 250 мл. Катализаторы для опытов, содержащие ГПК, PdCl2 и фталоцианин, обычно готовят в количестве 20 мл. Например, для получения катализатора состава [H10P3Mo18V7O84]=0,30 М, [Pd2+]=6· 10-3 М с мольным соотношением [Рс]:[Pd2+]=1:1 в раствор 0,30 М ГПК вводят навески 0,0213 г PdCl2 и 0,096 г фталоцианина. Раствор нагревают и кипятят 8-10 минут до полного растворения навесок. Затем раствор охлаждают и доводят его объем до 20 мл. Как видно из приведенных примеров и таблиц предлагаемое изобретение позволяет значительно увеличить стабильность работы катализатора и увеличить его производительность.
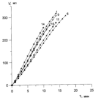
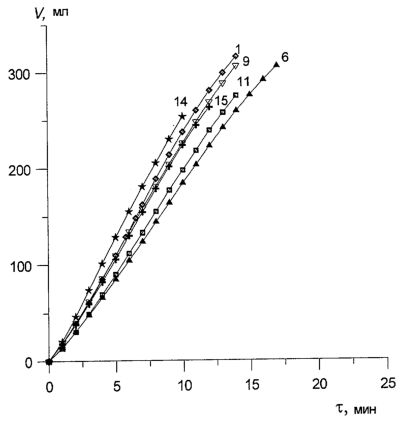
Катализатор испытан на максимальную нагрузку. Вывод: реакция практически останавливается при снижении окислительного потенциала катализатора ниже 0,528-0,530 В. Это соответствует m ~ 6,8-6,9
Формула изобретения
1. Катализатор получения метилэтилкетона каталитическим окислением н-бутенов кислородом и/или кислородсодержащим газом на основе палладия, стабилизированного комплексообразующим лигандом, и гетерополикислоты и/или ее кислых солей, отличающийся тем, что в качестве гетерополикислоты катализатор содержит ГПК-7 – молибдованадофосфорную гетерополикислоту состава H10P3Mo18V7O84 и/или ее кислую соль, а в качестве комплексообразующего лиганда – фталоцианиновый лиганд (Рс). 2. Катализатор по п.1, отличающийся тем, что концентрация ванадия в водном растворе ГПК-7 или ее соли составляет 0,4-2,2 грамм-атома на литр. 3. Катализатор по пп.1 и 2, отличающийся тем, что палладий с концентрацией 5·10-4-1·10-2 М стабилизирован в растворе фталоцианиновым лигандом Рс при мольном отношении [Pd]:[Рс]=0,5-2. 4. Способ получения метилэтилкетона каталитическим окислением н-бутенов кислородом и/или кислородсодержащим газом с использованием катализатора на основе палладия, комплексующего его лиганда и гетерополикислоты и/или ее кислой соли с последующей регенерацией катализатора путем его взаимодействия с кислородом или кислородсодержащим газом, отличающийся тем, что используют катализатор по любому из пп.1-3. 5. Способ по п.4, отличающийся тем, что реакцию окисления н-бутенов ведут при температурах от +15 до +90°С. 6. Способ по пп.4 и 5, отличающийся тем, что регенерацию катализатора проводят путем взаимодействия его с кислородом или кислородсодержащим газом при температуре 140-190°С. 7. Способ по пп.4-6, отличающийся тем, что на стадии регенерации катализатора парциальное давление кислорода равно 1-10 ата.
РИСУНКИ
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
(73) Патентообладатель(и):
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 04.08.2009 № РД0053379
Извещение опубликовано: 10.09.2009 БИ: 25/2009
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
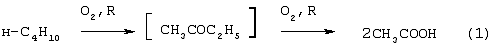
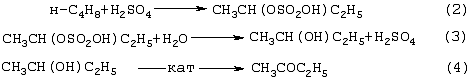
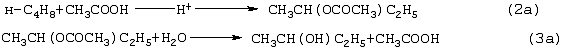
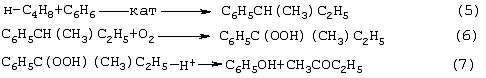
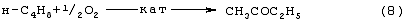
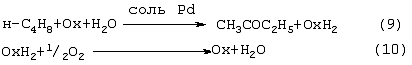
 n
n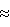 n неустойчивым оказывался палладий, который выделялся на поверхности восстановленного раствора в виде тонких металлических пленок, прилипавших к стенкам реактора. При большом числе атомов ванадия (4
n неустойчивым оказывался палладий, который выделялся на поверхности восстановленного раствора в виде тонких металлических пленок, прилипавших к стенкам реактора. При большом числе атомов ванадия (4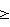 4. Низкая стабильность такого катализатора оказалась его главным недостатком.
4. Низкая стабильность такого катализатора оказалась его главным недостатком. -пиколиновой или дипиколиновой) повышает его стабильность [А.с. СССР 1584200, B 01 J 23/44, 1994; А.с. СССР 1669109, B 01 J 23/44, 1995]. Однако при этом в 10 и более раз снижается активность катализатора в реакции (9а) и падает его производительность. Это происходит потому, что комплекс Pd с производными пиридина оказывается слишком прочным, и он чрезмерно стабилизирует палладий в высшей степени окисления (Pd2+). Поэтому такой способ оказался нетехнологичным.
-пиколиновой или дипиколиновой) повышает его стабильность [А.с. СССР 1584200, B 01 J 23/44, 1994; А.с. СССР 1669109, B 01 J 23/44, 1995]. Однако при этом в 10 и более раз снижается активность катализатора в реакции (9а) и падает его производительность. Это происходит потому, что комплекс Pd с производными пиридина оказывается слишком прочным, и он чрезмерно стабилизирует палладий в высшей степени окисления (Pd2+). Поэтому такой способ оказался нетехнологичным. -бутиролактон или сульфолан. Реакцию (8) проводили одностадийно: растворенный
-бутиролактон или сульфолан. Реакцию (8) проводили одностадийно: растворенный  -С4Н8 – 1,4, н-бутан – 2,2, тяжелые примеси -0,1. Соединяют реактор с бюреткой, заполненной
-С4Н8 – 1,4, н-бутан – 2,2, тяжелые примеси -0,1. Соединяют реактор с бюреткой, заполненной  H10P3Mo18V7O84
H10P3Mo18V7O84 О2=20 мин.
О2=20 мин. m,
m, 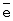
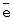
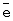
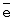 , т.е. практически полному восстановлению ГПК-7. Даже при полном восстановлении в ходе бутиленовой реакции катализатор вполне стабилен.
, т.е. практически полному восстановлению ГПК-7. Даже при полном восстановлении в ходе бутиленовой реакции катализатор вполне стабилен.