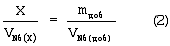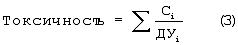Патент на изобретение №2241219
|
||||||||||||||||||||||||||
(54) СПОСОБ ОПРЕДЕЛЕНИЯ ТОКСИЧНОСТИ 1-НИТРОЗОАМИНОВ В ПИЩЕВЫХ ПРОДУКТАХ
(57) Реферат:
Изобретение относится к области анализа гигиенической безопасности пищевых продуктов и продовольственного сырья, а именно к определению токсичности 1-нитрозоаминов в пищевых продуктах методом обращенной газовой хроматографии. Отобранную пробу стерилизуют воздействием рентгеновского излучения, смешивают измельченную и гомогенизированную пробу со стеклянным твердым носителем, пропускают через хроматографическую колонку элюент, состоящий из несмешивающегося с влагой пробы пентана, отгоняют водный азеотроп из экстракта с последующей циркуляцией элюента, подают экстракт в капиллярную хроматографическую колонку, внутренняя поверхность которой модифицирована гидрофобным слоем C16 при постоянной температуре в диапазоне 30-40°С, последовательно вводят смесь насыщенных циклических и ароматических углеводородов в колонку с нанесенной пробой суммы нитрозоаминов и добавками диметил- и диэтилнитрозоаминов с известной массой, определяют величины удерживания ключевой пары углеводородов с одинаковым числом углеродных атомов в пробе и добавках, идентифицируют наличие хроматографической зоны нитрозоаминов по вкладу нитрозоаминогруппы, определяют количество СН-связей в нитрозоаминах пробы пищевых продуктов и находят количественное содержание суммы нитрозоаминов для определения токсичности путем сравнения с гигиеническим нормативом. Достигается повышение достоверности определения токсичности нитрозоаминов в пищевых продуктах. 1 табл.
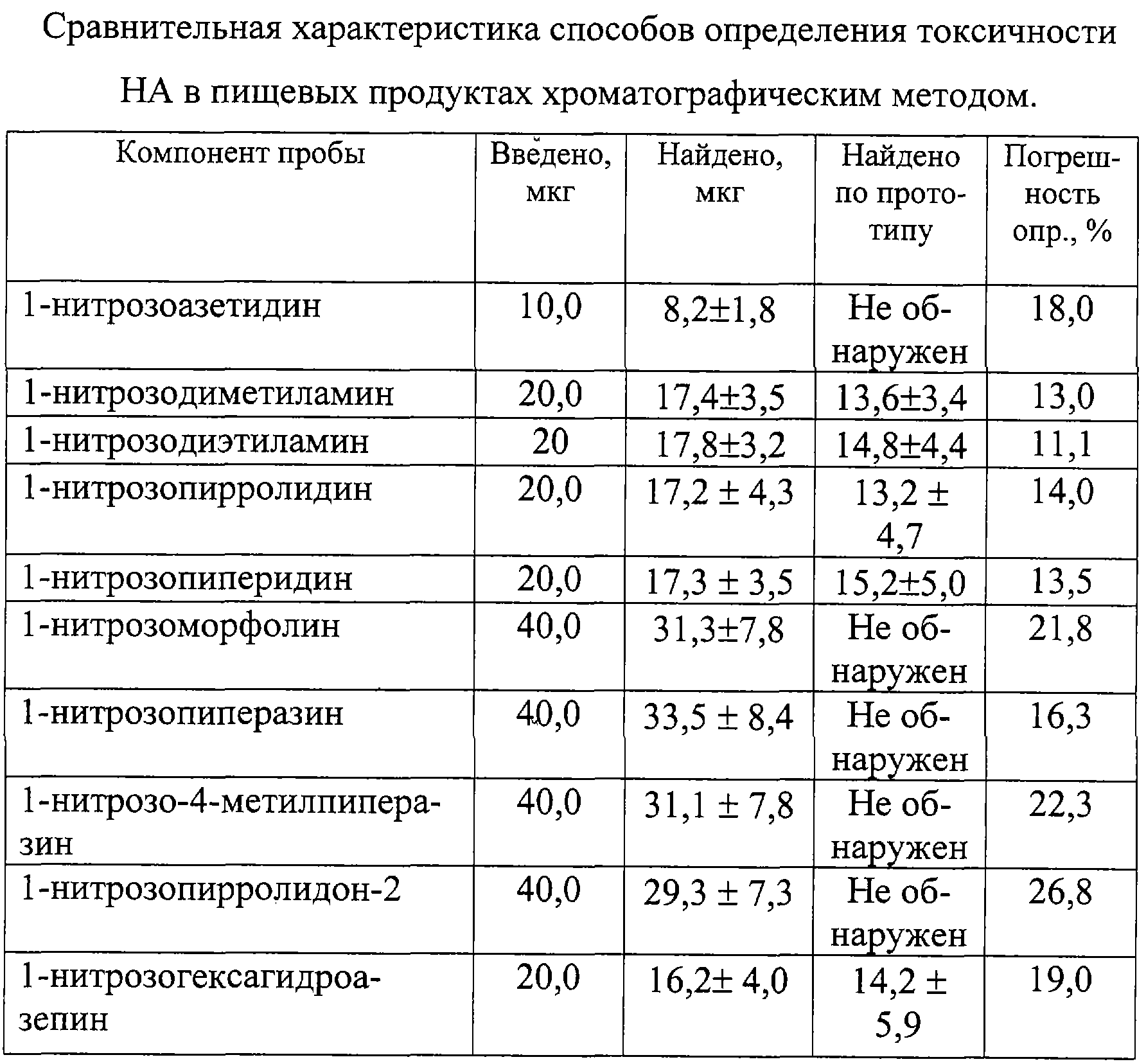
Изобретение относится к области определения безопасности продовольственного сырья и пищевых продуктов, а именно к определению токсичности 1-нитрозоаминов (НА) хроматографическим методом. Одним из известных способов определения токсичности 1-нитрозоаминов является способ, при котором отбирают лабораторную пробу массой до 300 г или объемом до 300 см3, хранят в морозильной камере в диапазоне температур от -8° до -18°С в течение 1-3 суток, после чего навеску измельчают в мясорубке или в гомогенизаторе, переносят в круглодонную колбу, снабженную устройствами для перегонки с паром, к продукту добавляют необходимое количество высаливателя, аликвотный объем раствора сульфаниловой кислоты, аликвотный объем 1 н. раствора серной кислоты для доведения рН пробы, равного 3, добавляют к пробе 1 см3 гексанового раствора внутреннего стандарта, в качестве которого используют 1-нитрозодипропиламин. Затем отгоняют летучие продукты с водяным паром, собирая 200-250 см3 дистиллята, после чего экстрагируют от 4 до 5 раз НА порциями свежеперегнанного дихлорметана. Объединенные экстракты высушивают прокаленным сульфатом магния, отфильтровывают, промывают осушитель для снижения потерь 1-нитрозоаминов, общий фильтрат упаривают в круглодонной колбе емкостью до 150 см3, снабженной дефлегматором до 5-7 см3 при температуре не более 65°С, пропуская небольшой ток инертного газа через концентрируемый объем. Промывают дефлегматор 2 см3 гексана и выпаривают пробу до 1-2 см3. Полученный концентрат подвергают денитрозированию добавлением 1 см3 2-3% раствора бромоводорода в уксусной кислоте и выдержкой пробы в течение 30 мин при комнатной температуре, после чего раствор упаривают досуха на роторном испарителе при температуре не более 50°С и вакууме не менее 15 мм рт.ст. Остаток нейтрализуют раствором гидрокарбоната натрия до рН, равного 8, добавляют аликвотный объем спиртового раствора 8-метокси-5-хинолинсульфонилазиридина (КАЗ) и кипятят 30 мин с обратным холодильником при температуре 100°С, смесь упаривают досуха, а остаток растворяют в 0,3-0,5 см3 этанола. Полученный раствор анализируют методом тонкослойной хроматографии по сравнению со стандартами, в качестве которых используют 8-метокси-5-[N-(2-N-диэтиламино)]хинолинсульфонамидные производные диметиламина, диэтиламина, дипропиламина с концентрацией 2 мг/см3. Для проведения холостой пробы ко второй части дихлорметанового экстракта добавляют 0,1 см3 рабочего раствора КАЗ, 0,2 см3 раствора гидрокарбоната натрия, кипятят 30 мин и обрабатывают аналогично анализируемой пробе. Полученный спиртовой раствор также анализируют методом тонкослойной хроматографии, результаты которого учитывают при расчете содержания НА. Для проведения тонкослойной хроматографии на стартовую линию наносят 10 точек, в которых размещают от 20 до 30 мкдм3 анализируемой пробы и стандартных растворов с различной концентрацией. Пластинку помещают в камеру для хроматографии и элюируют смесью бензол – этилацетат – уксусная кислота в соотношении 100:50:1 до достижения фронтом растворителя верхнего края пластинки, которую вынимают из хроматографической камеры, сушат в течение 15 мин, срезают 2 см пластины ниже фронта растворителя для удаления липофильных примесей. Пластину повторно хроматографируют в смеси ацетонитрил – концентрированный водный раствор аммиака в соотношении 9:1 до достижения фронтом растворителя верхнего края пластинки, которую вынимают из хроматографической камеры, сушат в течение 15 мин и вновь помещают в хроматографическую камеру с первоначальной системой растворителя. После подготовки хроматографической пластины ее проявляют в ультрафиолетовом излучении и проводят качественное и количественное определение диметил-, диэтил-, дипропилнитрозоамина путем сравнения критериев удерживания флюоресцирующих производных НА и интенсивности флюоресценции образца, холостой пробы и растворов с различной концентрацией флюоресцирующих производных при помощи флуориметра при длине волны возбуждения 340 нм и флуоресценции 480 нм. Определяют токсичность НА в пищевом продукте путем сравнения суммарного содержания с допустимой суточной дозой (ДСД) в диапазоне 0,002-0,004 мг/кг для сырых и копченых продуктов. Способ характеризуется недостаточной правильностью определения в связи с использованием в качестве внутреннего стандарта дипропилнитрозоамина, поверхностная активность которого более чем в 100 раз превышает поверхностную активность диметилнитрозоамина, и по величине извлечения которого определяется общая степень извлечения НА из образца [Авторское свидетельство СССР №1157449, 1985 г.]. Известен способ определения токсичности летучих НА в пищевых продуктах методом высокоэффективной жидкостной хроматографии с флуоресцентным детектированием, в котором операции выделения летучих НА путем отгонки с водяным паром и экстракции летучих НА из конденсата не отличаются от вышеизложенного аналога, а для упрощения денитрозирования добавляют аликвотный объем смеси 30% бромоводородной и ледяной уксусной кислот, выдерживают 30 мин при комнатной температуре. После этого смесь упаривают досуха на роторном испарителе, к сухому остатку добавляют 200 мкдм3 буферного раствора с рН 10,5 и вводят 100 мкдм3 раствора 5-[диметиламина]нафталин-1-сульфохлорида с концентрацией 1 мг/см3, перемешивают и выдерживают 40 мин при температуре 40°С. После этого добавляют 100 мкдм3 1,5 н. соляной кислоты, перемешивают и хроматографируют на хроматографической колонке (80х2 мм), заполненной сорбентом Зорбакс ODS (5 мкм) с расходом элюента 150 мкдм3/мин, в качестве которого используют смесь ацетонитрил-вода 2:1. Ввод пробы осуществляют при помощи петли типа Реодайн объемом 10 мкдм3, в качестве детектора применяют “Флюорат 0,2-2М” с максимумом возбуждения и эмиссии 350; 530 нм соответственно. Для определения качественного и количественного состава также используют нитрозодипропиламин в качестве внутреннего стандарта, а токсичность определяют путем сравнения общего содержания НА с гигиеническим нормативом [Жур. аналитической химии, 2001, т.56, №4, с.407-411]. Способ характеризуется недостаточной достоверностью определения в связи с недостаточностью разделения диметилнитрозоаминов от циклических и гетероциклических нитрозосоединений, а также низкой степенью извлечения, составляющей для низкомолекулярных НА с содержанием 0,2-0,8 ДСД 72±7,4%. Наиболее близким по технической сущности и достигаемому результату является способ определения токсичности НА в пищевых продуктах методом газовой хроматографии, основанный на выделении НА из анализируемых образцов продовольственного сырья, пищевых продуктов и кормов путем перегонки с водяным паром и последующим извлечением дихлорметаном из водного дистиллята и концентрированием экстракта до 1 см3 в соответствии с вышеприведенным алгоритмом. Анализ экстракта осуществляют методом газожидкостной хроматографии путем ввода пробы объемом 1-10 мкдм3 в испаритель хроматографа, нагретый до температуры 220°С, разделении на стеклянной колонке длиной 3 м и диаметром 2 мм, заполненной хроматоном N-AW-DMCS с нанесенной в качестве неподвижной фазы 15% карбовакса-20М, при этом в качестве газа-носителя используется азот (о.с.ч.) или аргон со скоростью 20 см3/мин при температуре колонки в изотермическом режиме, равной 125°С. В качестве детектора применяют хемилюминесцентный термический анализатор с температурой каталитической печи 450°С, расходом азота 40 см3/мин при давлении кислорода до 0,6 атм. Качественный анализ НА в пробе осуществляют по времени удерживания стандартных веществ, в качестве которых используют 1-нитрозопроизводные диметиламина, диэтиламина, дипропиламина, а количественное определение НА в пробе путем сравнения величины аналитических сигналов, полученных при анализе пробы, и внутреннего стандарта – раствора нитрозодипропиламина с концентрацией 0,2 мкг/см3 в гексане. Токсичность нитрозоаминов в пробе определяют путем сравнения с гигиеническим нормативом [МУК 4.4.1.011-93 хемилюминесцентный метод определения 1-нитрозоаминов в продовольственном сырье и пищевых продуктах (арбитражный метод анализа), ФЦГСЭН, 22.12.1993]. Способ характеризуется высокой систематической погрешностью, связанной с низкой степенью извлечения из анализируемых образцов НА различного строения, достигающей для низкомолекулярных аминов 60-70%. Кроме того, введение малоустойчивых НА в испаритель с высокой температурой приводит к их термическому разложению и окислению до нитроаминов и других неидентифицируемых продуктов, а вследствие этого – низкой достоверностью определения. Введение малой по величине пробы экстракта в хроматографическую систему от 1 до 10 мкдм3 приводит к искажению качественного и количественного состава канцерогенных НА в лабораторной пробе по сравнению с их содержанием в отбираемой пробе продовольственного сырья и пищевых продуктов. Задачей изобретения является повышение достоверности определения токсичности НА в пробе пищевых продуктов. Поставленная задача решается тем, что отбирают пробу пищевого сырья, стерилизуют ее воздействием рентгеновского излучения, измельчают до пастообразного состояния, смешивают с измельченным стеклянным твердым носителем, заполняют полученной смесью хроматографическую колонку для жидкостной хроматографии, снабженную в верхней части водяным обратным холодильником для конденсации летучих растворителей и насадкой для введения компонента подвижной жидкой фазы, а также для разделения воды от углеводородного растворителя. В нижней части колонка содержит впаянную стеклянную мембрану для отделения твердых дисперсных частиц продукта от жидкой дисперсионной среды. Колонку присоединяют к двугорлой круглодонной стеклянной колбе, снабженной в нижней части краном и имеющей широкую трубку для поступления паров в верхнюю часть колонки с водяным обратным холодильником, обеспечивающим конденсацию и поступление жидкости в верхнюю часть хроматографической колонки. В хроматографическую колонку постепенно вносят пентан, объем которого берут исходя из влажности пробы и содержания пентана в азеотропной смеси с водой (1,4 мас.%), при заполнении колбы экстрактом на 1/4 часть ее объема нагревают до начала поступления конденсата в насадку для отделения воды, с последующим переводом углеводородной составляющей конденсата в хроматографическую колонку. После поступления осушенного элюата из хроматографической колонки в колбу его направляют через нижний кран при помощи распылителя в капиллярную колонку диаметром 0,3-0,5 мм и длиной 25 м с модифицированной нанесением углеводородного слоя внутренней поверхностью, размещенную в термостате при температуре протекания процессов жизнедеятельности (30-40°С) хроматографа, снабженного пламенно-ионизационным детектором. Распыление осуществляют подачей гелия в капиллярную колонку с заданной скоростью потока газа-носителя в течение времени, необходимого для прекращения изменения нулевой линии сигнала детектора. Затем периодически вводят в колонку пробу насыщенных циклических и ароматических углеводородов в количестве, необходимом для определения содержания предполагаемых НА массой от 1 до 100 мкг, и определяют время удерживания индивидуальных углеводородов (циклогексана, метилциклогексана, бензола, толуола и этилбензола) при постоянной скорости газа-носителя от 0,1 до 0,2 см3/мин. После чего характеризуют хроматографическую зону по отношению исправленных времен удерживания ароматических углеводородов к насыщенным циклическим углеводородам с соответствующим числом углеродных атомов. При обнаружении зоны с характеристиками, находящимися в диапазоне хроматографических параметров удерживания углеводородов в НА, прекращают подачу элюата при помощи крана в нижней части колбы, а качественное и количественное определение токсичности проводят методом добавок, для чего последовательно вводят в распылитель стандартные растворы 1-нитрозопроизводных диметиламина- и диэтиламина в гексане. Способ осуществляется следующим образом. Навеску пищевого сырья массой до 1 кг, для предотвращения роста микрофлоры и изменения содержания НА, обрабатывают рентгеновским излучением, герметизируют при помощи вакуумной упаковки до момента пробоподготовки и хранят в холодильнике. Для подготовки пробы навеску измельчают в мясорубке в замороженном состоянии и затем гомогенизируют при помощи мельницы. Смешивают диспергированную пробу в виде пасты со стеклянной крошкой или круглыми стеклянными шариками размерами 5-10 мм, заполняют полученной смесью хроматографическую колонку для жидкостной хроматографии, расположенную вертикально на рабочей пластине вибратора. Колонка снабжена в нижней части стеклянной мембраной, а в верхней части водяным обратным холодильником и насадкой, позволяющей как отделять воду от углеводородов, так и при помощи капельной воронки вводить необходимое количество элюента. При этом колонка присоединена нижней частью к двугорлой круглодонной стеклянной колбе с электрообогревом, а также припаянным в нижней части краном и имеющей широкую трубку для поступления паров в верхнюю часть колонки. Капельную воронку заполняют пентаном, причем его количество зависит от влажности пробы и содержания пентана в азеотропной смеси с водой, подают элюент из капельной воронки с такой скоростью, чтобы обеспечить равномерное поступление экстракта в круглодонную колбу. При заполнении колбы экстрактом на 1/4 часть объема ее нагревают до начала поступления конденсата в насадку для отделения воды, с последующим переводом углеводородной составляющей конденсата в верхнюю часть хроматографической колонки. После наступления стационарного режима хроматографического извлечения безводный экстракт направляют при помощи распылителя через нижний кран в капиллярную колонку, которая термостатирована при температуре 30°С и находится в термостате хроматографа, снабженного пламенно-ионизационным детектором. Распыление экстракта осуществляют подачей гелия в течение времени, необходимого для прекращения дрейфа нулевой линии сигнала детектора. После чего периодически вводят пробу метана, циклогексана, метил-, этилциклогексана, бензола, толуола, этилбензола в капиллярную колонку с делением потока (1/20-1/100), которое позволяет уменьшить величину пробы углеводородов, взаимодействующих с пробой экстракта до величины бесконечного разбавления. Определяют приведенное относительно метана время удерживания компонентов в смеси при постоянной скорости газа-носителя от 0,1 до 0,2 см3/мин. Характеризуют хроматографическую зону нанесенного в капиллярную колонку экстракта отношением приведенных времен удерживания бензола и циклогексана, или толуола и метилциклогексана, или этилбензола и этилциклогексана и при совпадении этих характеристик с предварительно определенными параметрами нитрозопроизводных летучих аминов прекращают подачу элюата при помощи крана в нижней части колбы, вводят последовательно в распылитель стандартные растворы нитрозодиметиламина и нитрозодиэтиламина с концентрацией 500 мкг/см3 в количестве 1 мг аналита и определяют времена удерживания ключевой пары циклического и ароматического углеводорода с оптимальным для определения удерживанием. Строят зависимость логарифма отношения удерживания ключевой пары циклического и ароматического углеводородов от числа СН-связей в стандартных НА и аппроксимацией на ось ординат определяют вклад нитрозоаминогруппы в величину относительного удерживания apeнов, являющийся качественной характеристикой токсичности НА. Определяют количество СН-групп в пробе НА с токсичностью, равной токсичности нитрозопроизводных алифатических аминов, по зависимости логарифма отношения удерживания циклического и ароматического углеводорода в стандартных алифатических НА от числа СН-связей в заместителях. Количественное определение суммы НА в пробе пищевых продуктов осуществляют по величине удерживания ароматического углеводорода в НА, имеющего параметр удерживания ключевой пары, наиболее близкий к анализируемой пробе. Токсичность определяют путем сравнения содержания суммы НА пробы в единице массы пробы в пересчете на нитрозопроизводное амина с одинаковыми параметрами удерживания аренов и гигиенического норматива. Представленная совокупность заявленных признаков обеспечивает решение задачи изобретения: – стерилизация рентгеновским излучением отобранной пробы и ее герметизация предотвращают возможность образования экзогенных НА; – хроматографическое извлечение суммы НА из гомогенизированной пробы пищевого продукта элюентом, состоящим из несмешивающихся друг с другом компонентов (вода и пентан) бинарного растворителя, позволяет отделить более полярные НА от жиров и других труднолетучих компонентов пищевых продуктов; – азеотропная перегонка полученного экстракта постепенно изменяет полярность смешанного экстрагента в процессе хроматографирования путем отделения воды в насадке и обеспечивает рост гидрофобности элюента для вытеснения полярных НА из хроматографической колонки; – распыление безводного экстракта в капиллярную хроматографическую колонку, термостатированную при температуре 30°С, которая является нижней границей процессов жизнедеятельности человеческого организма, позволяет эффективно сорбировать пробу НА на поверхности раздела НЖФ-газ-носитель в колонке, что обеспечивает разделение ключевых пар насыщенных циклических и ароматических углеводородов для оценки токсичности пробы; – нахождение величин удерживания ключевой пары насыщенных циклических и ароматических углеводородов в анализируемой пробе и нитрозопроизводных диметил- и диэтиламинов позволяет определить вклад азонитрозоаминогруппы и количество СН-связей для нитрозоамина, равного по токсичности суммарной токсичности НА в анализируемой пробе пищевых продуктов; – определение величин удерживания аренов в пробе и НА, имеющем логарифм удерживания ключевой пары, наиболее близкий к анализируемой пробе, дает возможность количественно найти содержание суммы НА в пробе пищевых продуктов; – сравнение величин содержания суммы НА в единице массы пищевых продуктов в пересчете на нитрозопроизводное амина с одинаковыми параметрами удерживания аренов и гигиенического норматива позволяет более достоверно определить токсичность НА в анализируемой пробе. Таким образом, в результате стерилизации отобранной пробы воздействием рентгеновского излучения, извлечения и разделения суммы нитрозоаминов из гомогенизированной пробы пищевого продукта, смешанной со стекловидным твердым носителем размерами 5-10 мм, путем пропускания элюента, состоящего из несмешивающегося с влагой пищевого продукта пентаном, азеотропной отгонке воды из экстракта с циркуляцией пентана по длине хроматографической колонки до полной осушки, распылении безводного экстракта в капиллярной хроматографической колонке с модифицированной нанесением гидрофобного слоя внутренней поверхностью, определении времен удерживания ключевой пары насыщенных циклических и ароматических углеводородов и нитрозопроизводных диметиламина и диэтиламина с известным содержанием, идентификацией хроматографической зоны НА по вкладу нитрозоаминогруппы, определении количества СН-групп для нитрозоамина, равного по токсичности суммарной вредности НА в анализируемой пробе, нахождении количественного содержания суммы НА по величине удерживания арена в пробе и нитрозоамине с наиболее близкими величинами удерживания ключевой пары, можно более достоверно определить токсичность 1-нитрозоаминов в пищевых продуктах путем сравнения содержания НА в единице массы пищевых продуктов с гигиеническим нормативом. Предложенный способ определения токсичности НА в пищевых продуктах иллюстрируется следующими примерами. Пример 1. Определение токсичности НА в субпродуктах. Отбирают из средней пробы, подготовленной в соответствии с технологическим регламентом на данный вид продукции навеску печени крупного рогатого скота массой до 1,0 кг, обрабатывают рентгеновским излучением на рентгеновском аппарате АРИНА-2М, герметизируют при помощи вакуумной упаковки до начала пробоподготовки и хранят в холодильнике. Взвешивают на технических весах (ГОСТ 19491-74) лабораторную навеску субпродукта массой 975,4 г, после чего ее измельчают в мясорубке (ГОСТ 15906-79) в замороженном состоянии и затем гомогенизируют при помощи мельницы. Смешивают диспергированную пробу в виде пасты с твердым носителем, представляющим собой стеклянную крошку размерами 5-10 мм, в объемном соотношении 1:1, заполняют полученной смесью хроматографическую колонку для жидкостной хроматографии длиной 50 см и диаметром 5 см, расположенную вертикально на рабочей пластине вибратора. Установка для жидкостной хроматографии состоит из хроматографической колонки, снабженной в нижней части стеклянной мембраной типа ПОР 16 (ГОСТ 9775-69), верхняя часть которой имеет насадку типа Дина-Старка, позволяющую отделять и удалять воду из смеси с углеводородами, обратный водяной холодильник (ГОСТ 23932-79), двугорлую насадку и капельную воронку (ГОСТ 25336-82) для введения необходимого количества элюента. К нижней части колонки присоединена двугорлая круглодонная стеклянная колба с электрообогревом, имеющая внизу кран, а вверху широкую трубку для поступления паров в верхнюю часть хроматографической колонки с последующей конденсацией. Для выделения НА методом жидкостной хроматографии капельную воронку заполняют необходимым объемом пентана, подают элюент в верхнюю часть хроматографической колонки с такой скоростью, чтобы обеспечить равномерное поступление экстракта в круглодонную колбу. При заполнении колбы экстрактом на 1/4 ее объема его концентрируют путем нагрева до температуры кипения азеотропа пентана с водой, при этом его пары поступают через широкую трубку в конденсатор и стекают в виде флегмы в насадку Дина-Старка, где воду отделяют по мере накопления. Обезвоженный углеводородный слой поступает в хроматографическую колонку для дальнейшего разделения нитрозоаминов от жиров, липоидов и вытеснения нитрозоаминов из колонки. После наступления стационарного режима хроматографического извлечения безводный экстракт направляют через нижний кран в распылитель для получения аэрозоля экстракта с размером частиц не более 10 мкм. Направляют поток аэрозоля в кварцевую капиллярную колонку газового хроматографа типа Цвет-500 с пламенно-ионизационным детектором длиной 25 м и диаметром 0,3-0,5 мм, подготовленной для капиллярной газовой хроматографии нанесением от 5 до 10 мг гексадекана или иммобилизацией внутренней поверхности гидрофобной пленкой С16 со средней толщиной 5 мкм. Колонка термостатирована в изотермическом режиме при 30°С. Режим работы хроматографа: температура испарителя 125°С, детектора 125°С, расход гелия через колонку 0,2 см3/мин, расход гелия, сбрасываемого в испарителе, 5 см3/мин, расход гелия, поддуваемого в пламенно-ионизационный детектор, 40 см3/мин, чувствительность шкалы электрометра 50·10-12 А. Распыление экстракта осуществляют подачей гелия в течение времени, необходимого для прекращения дрейфа нулевой линии сигнала детектора, после чего вводят смесь метана, циклогексана, метил-, этилциклогексана, бензола, толуола и этилциклобензола массой до 100 нг в капиллярную колонку с делением потока. Хроматографируют смесь циклических и ароматических углеводородов в условиях анализа и определяют исправленное (относительно несорбирующегося метана) время удерживания углеводородов и рассчитывают удерживаемые объемы по формуле: где Fo – объемный расход газа-носителя через колонку, см3/с; j – поправка на сжимаемость газа-носителя. Время удерживания бензола в пробе ПА составило 6 мин 37,20 с, а циклогексана 57,15 с. Рассчитанный по формуле 1 объем удерживания бензола в пробе НА составил 1,324 см3, а циклогексана 0,191 см3, хроматографическая зона характеризуется логарифмом отношения бензола и циклогексана, равным 0,8451, который находится в диапазоне изменения этих характеристик для алифатических нитрозоаминов от 0,600 до 0,9105. Прекращают подачу элюата из нижней части колбы при помощи крана, вводят последовательно в распылитель стандартные растворы нитрозодиметил- и диэтиламина с концентрацией 500 мкг/см3 в количестве 1 мг аналита и определяют времена удерживания ключевой пары углеводородов. По зависимости логарифма отношения удерживания ключевой пары от числа СН-связей в нитрозопроизводных диметиламина (6) и диэтиламина (10) определяют аппроксимацией на ось ординат вклад нитрозогруппы, который составил для стандартов 1,27, а для суммы нитрозопроизводных алифатических аминов в анализируемой пробе рассчитывают количество СН-групп, равное 6,5. Поэтому токсичность определяют по содержанию диметилнитрозоамина, имеющего параметр удерживания ключевой пары, наиболее близкий к анализируемой пробе. Количественное определение содержания суммарных НА осуществляли по времени и объему удерживания бензола в пробе и нитрозодиметиламине. Из расчета по методу добавок следует отношение где Х – масса суммарных нитрозоаминов, мкг; mдоб – масса введенного диметилнитрозоамина, мкг; VNб(х) – исправленный объем удерживания бензола в анализируемой пробе, см3; VNб(доб) – исправленный объем удерживания бензола в добавке, см3. Содержание суммы НА в пробе по нитрозодиметиламину составило 1093,5 мкг, которое соответствует 1,121 мг/кг пищевого продукта. Токсичность НА в пробе субпродуктов определяли путем сравнения содержания НА с гигиеническим нормативом, равным для суммы НА 0,003 мг/кг согласно СанПиН 2.3.2.1078-01, что превышает допустимый уровень в 373,7±67,3 раза. Для оценки достоверности указанного метода проводили анализ аналогичной пробы субпродуктов по прототипу. Найдено диметилнитрозоамина 54,0±40,5 мкг; диэтилнитрозоамина 24,7±11,0 мкг; нитрозопирролидина 343,0+257,0 мкг; нитрозодипропиламин, нитрозопиперидин, нитрозопергидроазепин не обнаружены. Расчет токсичности для веществ с одинаковым механизмом биологического действия проводили по формуле суммирующего эффекта где Сi – содержание НА в 1 кг продовольственного сырья или пищевых продуктов, мг/кг; ДУi – максимально допустимый уровень поступления ксенобиотика с пищевыми продуктами, мг/кг. Токсичность пробы равна 140,6±105,4 по сравнению с гигиеническим нормативом по сумме нитрозопроизводных диметиламина и диэтиламина, равным 0,003 мг/кг, что значительно меньше по сравнению с предложенным способом. Пример 2. Определение токсичности НА в субпродуктах животных, проходивших курс лечения при заболевании гельминтозом пиперазин адипинатом. Отбирают из средней пробы, подготовленной в соответствии с технологическим регламентом на данный вид продукции, навеску свиной печени массой до 1,0 кг, обрабатывают рентгеновским излучением на рентгеновском аппарате АРИНА-2М, герметизируют при помощи вакуумной упаковки до начала пробоподготовки и хранят в холодильнике. Взвешивают на технических весах лабораторную навеску субпродукта массой 986,3 г, после чего ее измельчают в мясорубке в замороженном состоянии и затем гомогенизируют при помощи мельницы. Смешивают диспергированную пробу в виде пасты с твердым носителем, представляющим собой круглые стеклянные шарики или стеклянную крошку размером 5-10 мм, в объемном соотношении 1:1, заполняют полученной смесью хроматографическую колонку для жидкостной хроматографии длиной 50 см и диаметром 5 см, расположенную вертикально на рабочей пластине вибратора. Установка для жидкостной хроматографии аналогична описанной в примере 1. Для выделения НА методом жидкостной хроматографии капельную воронку заполняют необходимым объемом пентана, подают элюент в верхнюю часть хроматографической колонки с такой скоростью, чтобы обеспечить равномерное поступление экстракта в круглодонную колбу. При заполнении колбы экстрактом на 1/4 ее объема его концентрируют путем нагрева до температуры кипения азеотропа, при этом его пары поступают через широкую трубку в конденсатор и стекают в виде флегмы в насадку Дина-Старка, где воду отделяют по мере накопления. Обезвоженный углеводородный слой поступает в хроматографическую колонку для дальнейшего разделения НА от жиров, липоидов и вытеснения НА из колонки. После наступления стационарного режима хроматографического извлечения безводный экстракт направляют через нижний кран в ультразвуковое диспергирующее устройство для получения аэрозоля экстракта с размером частиц не более 10 мкм. Направляют поток аэрозоля в кварцевую капиллярную колонку газового хроматографа типа Цвет-500 с пламенно-ионизационным детектором длиной 25 м и диаметром 0,3 мм, подготовленной для капиллярной газовой хроматографии нанесением от 5 до 10 мг гексадекана. Колонка термостатирована в изотермическом режиме при температуре 30°С. Режим работы хроматографа: температура испарителя 125°С, детектора 125°С, расход гелия через колонку 0,1 см3/мин, расход гелия, сбрасываемого в испарителе, 5 см3/мин, расход гелия, поддуваемого в пламенно-ионизационный детектор, 40 см3/мин, чувствительность шкалы электрометра 50·10-12 А. Подачу экстракта осуществляют пропусканием гелия в течение времени, необходимого для прекращения дрейфа нулевой линии сигнала детектора, после чего вводят смесь метана, циклогексана, метил-, этилциклогексана, бензола, толуола и этилбензола массой до 100 нг в капиллярную колонку с делением потока. Хроматографируют смесь циклических и ароматических углеводородов в условиях анализа, определяют исправленное (относительно несорбирующегося метана) время удерживания углеводородов и рассчитывают удерживаемые объемы по формуле 1. Время удерживания бензола в пробе ПА составило 1 мин 36,82 с, толуола – 2 мин 57,5 с, циклогексана – 12,76 с, метилциклогексана – 20,6 с. Объем удерживания бензола в пробе НА составил 0,1614 см3, толуола – 0,2959 см3, циклогексана – 0,02137 см3, метилциклогексана – 0,03437 см3. Хроматографическая зона характеризуется логарифмом отношения толуола и метилциклогексана, равным 0,935, который незначительно превышает значение величины удерживания ключевой пары для нитрозодиметиламина (диапазон изменения этих характеристик для алифатических нитрозоаминов 0,583-0,928). Прекращают подачу элюата из нижней части колбы при помощи крана, вводят последовательно в распылитель стандартные растворы нитрозодиметил- и диэтиламина с концентрацией 500 мкг/см3 в количестве 1 мг аналита и определяют времена удерживания толуола и метилциклогексана. По зависимости логарифма отношения удерживания ключевой пары от числа СН-связей в нитрозопроизводных диметиламина (6) и диэтиламина (10) определяют аппроксимацией на ось ординат вклад нитрозоаминогруппы, который составил для стандартов 1,445, а для суммы нитрозопроизводных аминов в анализируемой пробе рассчитывают количество СН-групп, равное 5,9. Поэтому значение гигиенического регламента для определения токсичности пробы выбирают по нитрозодиметиламину, имеющему 6 СН-групп в алкильных заместителях, равное 0,003 мг/кг. Количественное определение осуществляли по формуле 2 методом добавок. Содержание НА в пробе по нитрозодиметиламину составило 111,1 мкг, которое соответствует 0,1126 мг/кг свиной печени. Токсичность НА в пробе субпродуктов животных, проходивших курс лечения пиперазин адипинатом, определяли путем сравнения содержания НА с гигиеническим нормативом по СанПиН 2.3.2.1078-01, из чего следует, что допустимый уровень НА превышен в 37,5±9,4 раза. Для оценки достоверности указанного метода проводили анализ аналогичной пробы субпродуктов по прототипу. Найдено диметилнитрозоамина 5,9±4,4 мкг; диэтилнитрозоамина 4,2±3,2 мкг; нитрозопиперазин, 1-нитрозо-4-метилпиперазин; нитрозопроизводные дипропиламина, пирролидина, пиперидина, пергидроазепина не обнаружены. Расчет токсичности для веществ с одинаковым механизмом биологического действия проводили по формуле 3. Токсичность пробы НА равна 3,4±2,5 по сравнению с гигиеническим нормативом по сумме нитрозопроизводных диметиламина и диэтиламина, что практически на порядок меньше по сравнению с предложенным способом. Пример 3. Определение токсичности НА в копченой рыбе. Отбирают из средней пробы, подготовленной в соответствии с технологическим регламентом на данный вид продукции, навеску съедобной части рыбы холодного или горячего копчения, твердую часть консервов “Рыба копченая в масле” массой до 1,0 кг, обрабатывают рентгеновским излучением на аппарате АРИНА-2М, герметизируют при помощи вакуумной упаковки до начала пробоподготовки и хранят в холодильнике. Взвешивают на технических весах лабораторную навеску рыбного продукта массой 998,6 г, после чего ее измельчают в мясорубке в замороженном состоянии и затем гомогенизируют при помощи мельницы. Смешивают диспергированную пробу в виде пасты со стеклянной крошкой размером 5-10 мм в объемном соотношении 1:1, заполняют полученной смесью хроматографическую колонку для жидкостной хроматографии длиной 50 см и диаметром 5 см, расположенную вертикально на рабочей пластине вибратора. Установка для жидкостной хроматографии состоит из тех же узлов и деталей, описанных в примере 1. Выделение НА методом жидкостной хроматографии осуществляют аналогично операциям, описанных в примерах 1 и 2. После наступления стационарного режима хроматографического извлечения безводный экстракт направляют через нижний кран в распылитель для получения аэрозоля экстракта с размером частиц не более 10 мкм. Направляют поток аэрозоля в кварцевую капиллярную колонку газового хроматографа, подготовленную для капиллярной газовой хроматографии аналогично примеру 1. Колонка термостатирована в изотермическом режиме при температуре 30°С. Режим работы хроматографа Цвет-500 с ПИД такой же, как в примере 2. Вводят смесь метана, циклогексана, метил-, этилциклогексана, бензола, толуола и этилбензола массой до 100 нг в капиллярную колонку с делением потока и хроматографируют смесь циклических и ароматических углеводородов в условиях анализа и определяют исправленное (относительно несорбирующегося метана) время удерживания углеводородов и рассчитывают удерживаемые объемы по формуле 1. Время удерживания этилбензола составило 43,95 с, этилциклогексана – 9,36 с. Объем удерживания этилбензола в пробе НА составил 0,07326 см3, а этилциклогексана 0,0156 см3, хроматографическая зона характеризуется логарифмом отношения этилбензола и этилциклогексана, равным 0,672, который близок к диапазону изменения этих характеристик для алифатических нитрозоаминов (0,5594-0,6324). Прекращают подачу элюата из нижней части колбы при помощи крана, вводят последовательно в распылитель стандартные растворы нитрозодиметил- и диэтиламина с концентрацией 500 мкг/см3 в количестве 1 мг аналита и определяют времена удерживания ключевой пары углеводородов. По зависимости логарифма отношения удерживания этилбензола к этилциклогексану от числа СН-связей в нитрозопроизводных диметиламина (6) и диэтиламина (10) определяют аппроксимацией на ось ординат вклад нитрозоаминогруппы, который составил для стандартов 0,7425, а для суммы нитрозопроизводных алифатических аминов в анализируемой пробе рассчитывают количество СН-групп, равное 3,9. Поэтому токсичность пробы определяют по содержанию НА, имеющего параметр удерживания, наиболее близкий к анализируемой пробе. Количественное определение осуществляли по времени и объему удерживания этилбензола в пробе и нитрозодиметиламине по методу добавок. Содержание НА в пробе по нитрозодиметиламину составило 16,3 мкг, которое соответствует 0,01632 мг/кг. Токсичность НА в пробе рыбных копченых продуктов определяли путем сравнения содержания НА с гигиеническим нормативом согласно СанПиН 2.3.2.1078-01, что превышает допустимый уровень в 5,44±1,36 раза. Для оценки достоверности указанного метода проводили анализ аналогичной пробы субпродуктов по прототипу. Найдено диметилнитрозоамина 2,90±2,05 мкг; диэтилнитрозоамина 2,27±1,70 мкг; нитрозопроизводные дипропиламина, пирролидина, пиперидина, опергидроазепина и морфолина не обнаружены. Расчет токсичности для веществ с одинаковым механизмом биологического действия проводили по формуле суммирующего эффекта 3. Токсичность пробы превышает гигиенический норматив по сумме нитрозопроизводных диметиламина и диэтиламина в 1,72±1,29, что значительно меньше по сравнению с предложенным способом. Как следует из данных, приведенных в таблице, достоверность определения токсичности полярных нитрозопроизводных гетероциклических аминов и амидов в пищевых продуктах и продовольственном сырье предложенным способом значительно выше по сравнению с прототипом. Это позволит более правильно произвести оценку их безопасности или оценить риск возникновения канцерогенных заболеваний. Положительный эффект от предложенного способа определения токсичности НА в пищевых продуктах заключается в увеличении достоверности оценки токсичности вследствие уменьшения многостадийности подготовки пробы к определению, использовании в качестве пробы выделенных методом жидкостной хроматографии нитрозопроизводных аминов, а также стандартов диметил- и диэтилнитрозоаминов и, как следствие, уменьшение погрешности определения.
Формула изобретения
Способ определения токсичности 1-нитрозоаминов в пищевых продуктах, включающий отбор и подготовку проб, хроматографическое определение содержания вредных веществ и оценку их токсичности, отличающийся тем, что стерилизуют отобранную пробу воздействием рентгеновского излучения, смешивают измельченную и гомогенизированную пробу со стеклянным твердым носителем, пропускают элюент, состоящий из несмешивающегося с влагой пробы пентана, отгоняют водный азеотроп из экстракта с последующей циркуляцией элюента по длине колонки до полной осушки пробы, подают распыленный в инертной газовой среде экстракт в капиллярную хроматографическую колонку, внутренняя поверхность которой модифицирована гидрофобным слоем С16 при постоянной температуре в диапазоне 30-40°С, последовательно вводят смесь насыщенных циклических и ароматических углеводородов в колонку с нанесенной пробой нитрозоаминов и добавками диметил- и диэтилнитрозоаминов с известной массой, определяют величины удерживания ключевой пары насыщенных циклических и ароматических углеводородов с одинаковым числом углеродных атомов в анализируемой пробе и добавках нитрозопроизводных диметиламина и диэтиламина, идентифицируют наличие хроматографической зоны нитрозоаминов по вкладу нитрозоаминогруппы, определяют количество СН-связей в нитрозоаминах пробы пищевых продуктов по зависимости логарифма отношения удерживания ключевой пары для диметил- и диэтилнитрозоаминов от их числа, находят количественное содержание суммы нитрозоаминов по величине удерживания арена в алифатическом нитрозоамине, имеющего логарифм удерживания ключевой пары, наиболее близкий к анализируемой пробе, определяют токсичность путем сравнения содержания суммы нитрозоаминов в единице массы продукта с гигиеническим нормативом.
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 21.05.2006
Извещение опубликовано: 10.05.2007 БИ: 13/2007
|
||||||||||||||||||||||||||
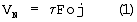
 – время удерживания, измеряемое между выходом несорбируемого компонента (вводом пробы) и максимумом концентрации углеводорода, с;
– время удерживания, измеряемое между выходом несорбируемого компонента (вводом пробы) и максимумом концентрации углеводорода, с;